

Abstract
Starting from α-hydroxymethyl nitroalkenes and various 1,3-dicarbonyl compounds, a one-pot organocatalyzed diastereo- and enantioselective synthesis of polyfunctionalized dihydro- and tetrahydropyran derivatives via a domino Michael-hemiacetalization sequence is reported. The title compounds bearing a variety of functional groups can be synthesized in this way in good yields (59–91%) and with moderate to excellent diastereoselectivities (26–98% de) and enantioselectivities (71–99% ee).
Keywords: organocatalysis, domino reaction, one-pot reaction, tetrahydropyrans, hydrogen bonding
Great progress has been made in the organocatalytic asymmetric synthesis of chiral molecules in a single operation via the concept of domino reactions.1 So far the development of methods for the control of multiple stereocenters was mainly dominated by amino activation modes via Lewis base catalysis or hydrogen bonding activation as another elegant option.2 Owing to the importance of chiral tetrahydropyran structures, which are characteristic structural features of many natural products, these heterocycles have attracted considerable attention in recent years and many methods have been developed for the preparation of this framework.3,4 Recently we and others reported that chromanols 6 can be efficiently synthesized through a Michael addition of 1,3-dicarbonyl compounds 1 to nitrovinylphenol 5 and subsequent intramolecular hemiacetalization.5 The hydroxyl group participates in this process through carbonyl addition and makes this sequence a powerful tool among the existing annulation methods (Scheme 1, a).
Scheme 1.

Domino Michael–hemiacetalization reactions towards chromanols 6 or tetrahydropyranols 3 and dihydropyrans 4
We now would like to report an efficient organocatalytic asymmetric synthesis of functionalized tetrahydropyranols 3 by hydrogen bonding activation of α-hydroxymethyl nitroalkenes 2 as Michael acceptors via a domino Michael–hemiacetalization reaction followed by dehydration to form dihydropyrans 4 (Scheme 1, b).4b,h-j,6
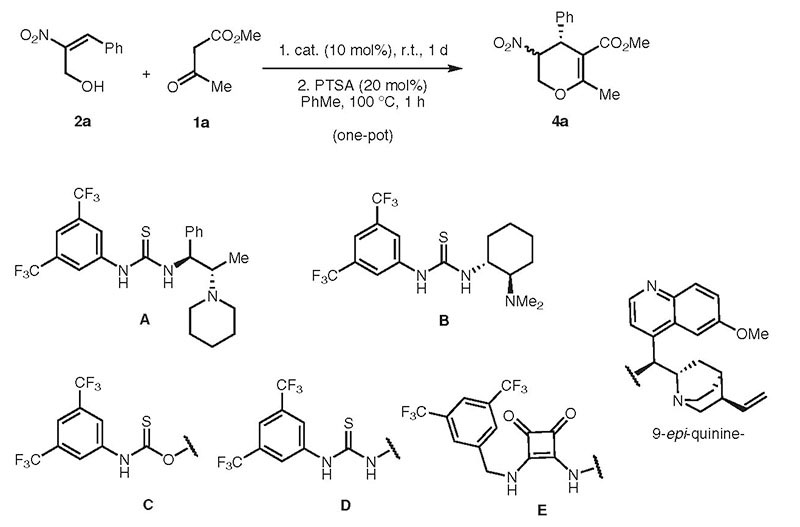
We started our studies on the domino Michael–hemiacetalization reactions by screening the organocatalysts A–E (10 mol%) for the Michael reaction of (E)-2-nitro-3-phenylprop-2-en-1-ol (2a)7 with 1 equivalent of methyl 3-oxobutanoate (1a) in toluene (Table 1, entries 1–5). The intermediate tetrahydropyranol 3a was directly dehydrated to the 3,4-dihydro-2H-pyran 4a in a one-pot procedure (see Table 1). It turned out that 20 mol% PTSA in toluene at 100 °C for one hour is the best reaction condition for the dehydration process, which occurred in good yields and with no competing ring opening and lactonization. With this reaction procedure the diastereo- and enantiomeric excess of 4a were determined by HPLC. While the thiourea derived catalysts A, B, and D showed moderate to good diastereo- and enantioselectivities, the square amide derived catalyst E8 led to the best results (Table 1, entry 5). The monodentate carbamate derived catalyst C9 gave the poorest enantioselectivity but had not much of an impact on the diastereoselectivity (Table 1, entry 3). Solvents like toluene or THF also have only a slight impact on the diastereoselectivity (Table 1, entries 5, 6) while dichloromethane showed the best diastereomeric excess value (Table 1, entry 7). With catalyst E we tried to improve the stereoselectivity as well as the yield of the reaction by decreasing the temperature. The domino Michael-hemiacetalization reaction proceeds at 5 °C as well as at −25 °C, but has only a slight effect with regard to selectivity and longer reaction times were necessary (Table 1, entries 8, 9). Next we decided to change the catalyst loading from 10 mol% over 5 mol% to 1 mol% at ambient temperature (Table 1, entries 10, 11) and this had only an impact on the reaction time and not on the yield or selectivity. In summary, the diastereomeric excess remained in the range of 61–71% in favor of the trans-isomer and could not be improved by catalysts, loading, temperature, or solvents.
Table 1.
Catalyst Screening for the One-Pot Domino Michael–Hemiacetalization and Dehydration Sequence to from 4a
|
|||||
|---|---|---|---|---|---|
|
| |||||
| Entrya | Catalyst | Solvent | Yield (%)b | de (%)c | ee (%)c,d |
| 1 | A | toluene | 86 | 66 | 75 (57) |
| 2 | B | toluene | 83 | 58 | 56 (11)e |
| 3 | C | toluene | 72 | 68 | 27 (27) |
| 4 | D | toluene | 74 | 62 | 51 (28) |
| 5 | E | toluene | 81 | 67 | 87 (61) |
| 6 | E | THF | 79 | 61 | 86 (75) |
| 7 | E | CH2Cl2 | 80 | 71 | 95 (71) |
| 8f | E | CH2Cl2 | 74 | 68 | 91 (71) |
| 9g | E | CH2Cl2 | 80 | 69 | 91 (73) |
| 10h | E | CH2Cl2 | 82 | 65 | 88 (80) |
| 11i | E | CH2Cl2 | 88 | 68 | 91 (69) |
All reactions were performed on a 0.5 mmol scale.
Yield of isolated product 4a.
Determined by HPLC analysis on a chiral stationary phase.
The enantiomeric excess of the minor diastereomer is given in parentheses.
ent-4a was obtained.
Performed with 10 mol% catalyst loading at 5 °C for 2 d.
Performed with 10 mol% catalyst loading at −25 °C for 3 d.
Performed with 5 mol% catalyst loading at r.t. for 7 d.
Performed with 1 mol% catalyst loading at r.t. for 14 d.
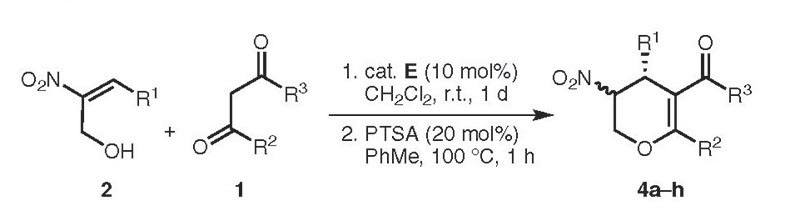
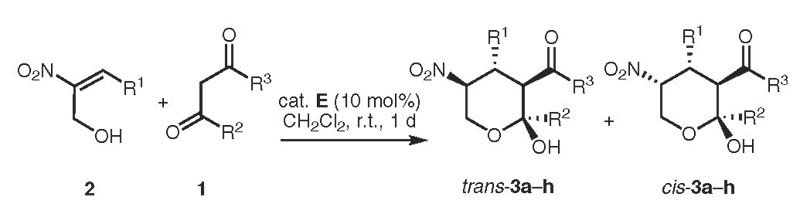
In order to determine the scope of the organocatalytic one-pot reaction a number of (E)- 3-aryl-2-nitroprop-2-en-1-ols 2 were reacted with 1 equivalent of various 1,3 dicarbonyl compounds 1 in the presence of 10 mol% of the catalyst E at ambient temperature in dichloromethane. The dihydropyrans 4a–h were obtained in good yields and stereoselectivities (Table 2). In the cases of different β-keto esters 1, a high increase in stereoselectivity was observed by using the phenyl substituted β-keto ester 1c (Table 2, 4c). In contrast to acetylacetone (Table 2, 4d) a reaction with malonates could not be observed. While using various hydroxymethyl-substituted nitroalkenes 2 good yields (81–86%) and enantioselectivities (78–94% ee) could be achieved. Electronic factors had a great influence on the diastereoselectivity. Both the neutral and the electron-donating group substituted nitrostyrenes 2 led to the expected products with diastereoselectivities of 26–76% de (Table 2, 4e–h). The lowest diastereoselectivity was obtained by using the thienyl-substituted hydroxymethylene nitroolefin (Table 2, product 4g), nevertheless the enantioselectivity was excellent (90/94% ee). In all cases we were unable to separate the diastereomers by column chromatography. Therefore, this protocol was used for the synthesis of the corresponding lactols (Table 3, products 3a–h). While the enantioselectivity of the tetrahydropyranols 3a–h could not be measured by HPLC, the major trans-3a–h isomers were separated from the minor cis- 3a–h isomers by column chromatography. While with four adjacent stereogenic centers eight diastereomers of tetrahydropyran derivatives were possible, only two diastereomers could be isolated.
Table 2.
Scope of the Asymmetric Synthesis of the Dihydropyran Derivates 4 via a One-Pot Domino Michael–Hemiacetalization and Dehydration Reaction
|
||||||
|---|---|---|---|---|---|---|
|
| ||||||
| 4a | R1 | R2 | R3 | Yield (%)b | de (%)c (trans) | ee (%)d,e |
| 4a | Ph | Me | OMe | 80 | 71 | 95 (71) |
| 4b | Ph | Me | OEt | 85 | 64 | 88 (73) |
| 4c | Ph | Ph | OEt | 81 | 98 | 99 (99) |
| 4d | Ph | Me | Me | 77 | 87 | 92 (99) |
| 4e | 4-MeC6H4 | Me | OMe | 81 | 45 | 88 (89) |
| 4f | 3-BrC6H4 | Me | OMe | 86 | 76 | 88 (78) |
| 4g | 2-thienyl | Me | OMe | 82 | 26 | 90 (94) |
| 4h | 3,4-(OCH2O)C6H3 | Me | OMe | 81 | 43 | 91 (83) |
All reactions were performed on a 1.0 mmol scale.
Yield of isolated product.
Determined by HPLC analysis.
Determined by HPLC analysis on a chiral stationary phase.
The enantiomeric excess of the minor cis-diastereomer is given in parentheses.
Table 3.
Scope of the Asymmetric Synthesis of the Tetrahydropyranols 3 via Domino Michael–Hemiacetalization Reaction
| |||||
|---|---|---|---|---|---|
|
| |||||
| 3a | R1 | R2 | R3 | Yield (%)b trans-3 | Yield (%)b cis-3 |
| 3a | Ph | Me | OMe | 80 | 14 |
| 3b | Ph | Me | OEt | 77 | 18 |
| 3c | Ph | Ph | OEt | 91 | 2 |
| 3d | Ph | Me | Me | 80 | 6 |
| 3e | 4-MeC6H4 | Me | OMe | 66 | 25 |
| 3f | 3-BrC6H4 | Me | OMe | 85 | 9 |
| 3g | 2-thienyl | Me | OMe | 59 | 34 |
| 3h | 3,4-(OCH2O)C6H3 | Me | OMe | 65 | 26 |
All reactions were performed on a 1.0 mmol scale.
Yield of isolated product after column chromatography.
This can be explained by stereochemical outcome of the quinine square amide catalyst-based Michael additions of β-keto esters to nitroolefins, which is well known in this case. The relative and absolute configuration of trans-3g was determined as 2S,3R,4R,5S by X-ray crystal-structure analysis (Figure 1)10 showing a cis orientation between the ester and hydroxyl group and a trans relation between the ester and aryl substituent. No thermodynamic equilibrium between the hemiacetal and the open hydroxyl ketone was observed in NMR experiments. The relative configuration of all tetrahydropyranols 3a–h was unambiguously determined by NOE measurements. They are all stable solids and can be easily recrystallized from benzene or methanol.
Figure 1.

Absolute configuration of trans-3g confirmed by X-ray crystal-structure analysis10
Next various transformations of the enantiomerically pure domino product trans-3a were investigated to emphasize the value of this approach, for example, towards the synthesis of bioactive compounds such as pharmaceuticals (Scheme 2). It is well known that the hemiacetal unit can be reduced, methylated, or dehydrated to the corresponding tetrahydro- and dihydropyran derivatives. Therefore, 3a was synthesized, the epimers separated, and the trans-3a was recrystallized from benzene in good yield (73%). Then trans-3a was dehydrated to 4a in good yield (91%) to obtain the virtually diastereo- and enantiomerically pure dihydropyran indicating that no epimerization or racemization had occurred. By treatment with 20 mol% PTSA in MeOH for eight hours at room temperature trans-3a was converted to the acetal 7 in moderate yield (62%), but again with excellent diastereo- (98%) and enantiomeric excess (99%). Compound trans-3a could be reduced to 8 with three equivalents Et3SiH in dichloromethane at 0 °C in moderate yield (72%) and again with excellent diastereo- (97% de) and enantiomeric excess (98% ee). The high stereoselectivity of our approaches is evident from the fact that only two diastereomers are formed while building up four contiguous stereocenters. The various transformations performed with the hemiacetal underpin the stereochemical outcome of the first Michael reaction.
Scheme 2.

Various synthetic transformations of enantiopure tetrahydropyranol trans-3a
In conclusion, we have developed an efficient diastereo- and enantioselective asymmetric synthesis of polyfunctionalized dihydro- and tetrahydropyrans by employing a square amide-catalyzed domino Michael–hemiacetalization reaction. The new protocol allows several functional groups, such as the nitro, ester, ketone, and hydroxyl functions as well as substituents at the aromatic ring, attached to the hydropyran core. Subsequent transformations of the intermediate tetrahydropyranols gave acetals, dihydro-2H-pyrans, and tetrahydro-2H-pyrans bearing two to four contiguous stereocenters. The title compounds are important heterocycles due to their widespread occurrence in nature and as privileged scaffolds in medicinal chemistry. They were obtained in very good yields and with good to excellent stereoselectivities.
Materials and Methods
Starting materials and reagents were purchased from commercial suppliers and used without further purification, unless stated otherwise. All solvents were dried by conventional methods. Preparative column chromatography: Merck silica gel 60, particle size 0.040–0.063 mm (230–240 mesh, flash). Analytical TLC: silica gel 60 F254 plates from Merck, Darmstadt. Visualization of the developed TLC plates was performed by ultraviolet irradiation (254 nm) or by staining with a solution of KMnO4. Analytical HPLC was carried out on a Hewlett-Packard 1100 Series instrument using chiral stationary phases. 1H and 13C NMR spectra were recorded at r.t. on Varian Mercury 300, Varian Inova 400, or Varian Inova 600 instruments. Mass spectra were acquired on a Finnigan SSQ7000 (EI 70 eV) spectrometer, high-resolution mass spectra on a Finnigan MAT 95 and high-resolution ESI spectra on a Thermo Fisher Scientific LTQ-Orbitrap XL. IR spectra were recorded on a PerkinElmer 100 FT-IR Spectrum instrument. Microanalyses were performed with a Vario EL element analyzer. Melting points were determined with a Büchi melting point B-540 apparatus. Optical rotation values were measured on a Perkin-Elmer 241 polarimeter.
Domino Michael-Hemiacetalization Reaction; General Procedure 1 (GP1)
In a glass vial equipped with a magnetic stirring bar, catalyst E (10 mol%) was added to a mixture of 1,3 dicarbonyl compound 1 (1.0 mmol) and (E)-3-aryl-2-nitroprop-2-en-1-ol 2 (1.0 mmol) in CH2Cl2 (4.0 mL) at r.t. The reaction was monitored by TLC. After complete conversion of the starting material, the solvent was evaporated and the crude reaction mixture was purified by column chromatography affording products trans- and cis-3a–h, respectively, as pale yellow to colorless solids (Table 3).
One-Pot Domino Michael-Hemiacetalization and Dehydration Reaction; General Procedure 2 (GP2)
The tetrahydropyranols trans-/cis-3a–h were synthesized according GP1. After complete conversion of the starting material, the solvent was evaporated and toluene (10 mL) and PTSA (20 mol%) were added, and stirred for 1 h at 100 °C. After complete conversion, the solvent was evaporated and the crude reaction mixture was purified by column chromatography affording products 4a–h as pale yellow to colorless oils and as a cis/trans mixture (Table 2).
Methyl (2S,3R,4S,5S)-2-Hydroxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-carboxylate (trans-3a)
Compound trans-3a was synthesized according to GP 1; yield: 238 mg (80%); colorless solid; mp 148 °C (benzene); Rf = 0.42 (n-pentane–Et2O, 1:1); [α]D20–83.2 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3386 (vs), 1729 (vs), 1545 (vs), 1446 (s), 1355 (s), 1262 (m), 1180 (m), 1115 (vs), 1066 (vs), 887 (m), 763 (s), 695 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 1.48 (s, 3 H, CH3), 2.96 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 3.44 (s, 3 H, CO2CH3), 3.87 (s, 1 H, OH), 4.01 (dd, J = 11.9, 11.9 Hz, 1 H, CHPh), 4.11 (dd, J = 10.9, 5.0 Hz, 1 H, CH2), 4.49 (dd, J = 10.9, 10.9 Hz, 1 H, CH2), 4.92 (ddd, J = 11.9, 10.9, 5.0 Hz, 1 H, CHNO2), 7.22–7.32 (m, 5 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 27.9 (CH3), 43.8 (CHCO2CH3), 52.3 (CO2CH3), 55.2 (CHPh), 61.5 (CH2), 85.6 (CHNO2), 94.8 (CqOH), 127.8 (2 CHAr), 128.4 (CHAr), 129.0 (2 CHAr), 135.7 (CAr), 171.7 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 295 ([M]+, 1), 278 (28), 205 (19), 189 (27), 177 (54), 163 (70), 145 (56), 131 (100), 117 (51), 91 (27), 77 (23), 59 (23).
Anal. Calcd for C14H17NO6: C, 56.94; H, 5.80; N, 4.74. Found: C, 57.19; H, 5.84; N, 4.62.
Methyl (2S,3R,4S,5R)-2-Hydroxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-carboxylate (cis-3a)
Compound cis-3a was synthesized according to GP 1; yield: 41 mg (14%); colorless solid; mp 56 °C (MeOH); Rf = 0.12 (n-pentane–Et2O, 1:1); [α]D20–11.0 (c 0.5, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 2.24 (s, 3 H, CH3), 3.35 (s, 1 H, OH), 3.55 (s, 3 H, CO2CH3), 3.87 (dd, J = 12.4, 4.0 Hz, 1 H, CHPh), 4.10 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 4.14 (dd, J = 13.4, 3.5 Hz, 1 H, CH2), 4.58 (dd, J = 13.4, 3.0 Hz, 1 H, CH2), 4.79 (ddd, J = 4.0, 3.5, 3.0 Hz, 1 H, CHNO2), 7.22–7.34 (m, 5 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 28.6 (CH3), 41.1 (CHCO2CH3), 48.0 (CO2CH3), 52.4 (CHPh), 61.8 (CH2), 84.7 (CHNO2), 94.2 (CqOH), 127.8 (2 CHAr), 128.2 (CHAr), 128.9 (2 CHAr), 136.1 (CAr), 172.9 (CO2CH3).
Ethyl (2S,3R,4S,5S)-2-Hydroxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-carboxylate (trans-3b)
Compound trans-3b was synthesized according to GP 1; yield: 238 mg (77%); colorless solid (benzene); mp 112 °C; Rf = 0.39 (n-pentane–Et2O, 1:1); [α]D20–65.8 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3449 (s), 1718 (vs), 1547 (vs), 1456 (m), 1381 (vs), 1261 (m), 1179 (m), 1111 (vs), 1069 (vs), 881 (vs), 762 (s), 700 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 0.83 (t, J = 7.4 Hz, 3 H, CH2CH3), 1.47 (s, 3 H, CH3), 2.92 (d, J = 12.4 Hz, 1 H, CHCO2Et), 3.89 (dq, J = 7.4, 2.5 Hz, 2 H, CH2CH3), 3.98 (dd, J = 11.9, 11.9 Hz, 1 H, CHPh), 4.09 (s, 1 H, OH), 4.11 (dd, J = 10.9, 5.0 Hz, 1 H, CH2), 4.49 (dd, J = 10.9, 10.9 Hz, 1 H, CH2), 4.92 (ddd, J = 11.9, 10.9, 5.0 Hz, 1 H, CHNO2), 7.20–7.32 (m, 5 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 13.6 (CH2CH3), 27.8 (CH3), 44.1 (CHCO2Et), 55.0 (CHPh), 61.5 (CH2), 61.6 (CH2), 85.6 (CHNO2), 94.7 (CqOH), 128.0 (2 CHAr), 128.4 (CHAr), 128.9 (2 CHAr), 135.6 (CAr), 171.8 (CO2Et).
MS (EI, 70 eV): m/z (%) = 292 ([M – H2O]+, 6), 219 (10), 203 (10), 191 (31), 177 (89), 173 (23), 157 (21), 149 (16), 145 (51), 131 (100), 117 (52), 103 (37), 91 (39), 77 (24), 51 (23).
HRMS: m/z calcd for C15H19NO6 + Na: 332.1110; found: 332.1095.
Ethyl (2S,3R,4S,5R)-2-Hydroxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-carboxylate (cis-3b)
Compound cis-3b was synthesized according to GP 1; yield: 56 mg (18%); colorless solid; mp 42 °C (benzene); Rf = 0.21 (n-pentane–Et2O, 1:1); [α]D20–9.1 (c 0.5, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 0.97 (t, J = 6.9 Hz, 3 H, CH2CH3), 1.62 (s, 3 H, CH3), 3.59 (s, 1 H, OH), 3.84 (dd, J = 12.9, 4.5 Hz, 1 H, CHPh), 4.01 (q, J = 6.9 Hz, 2 H, CH2CH3), 4.06 (d, J = 12.9 Hz, 1 H, CHCO2Et), 4.13 (dd, J = 13.4, 1.0 Hz, 1 H, CH2), 4.59 (dd, J = 13.4, 3.0 Hz, 1 H, CH2), 4.78 (ddd, J = 4.5, 3.0, 1.0 Hz, 1 H, CHNO2), 7.20–7.32 (m, 5 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 13.7 (CH2CH3), 28.5 (CH3), 41.3 (CHPh), 48.0 (CHCO2Et), 61.4 (CH2CH3), 61.8 (CH2), 84.6 (CH-NO2), 95.1 (CqOH), 128.1 (CHAr), 128.2 (CHAr), 128.3 (CHAr), 128.8 (2 CHAr), 136.0 (CAr), 172.7 (CO2Et).
Ethyl (2S,3R,4S,5S)-2-Hydroxy-5-nitro-2,4-diphenyltetrahydro-2H-pyran-3-carboxylate (trans-3c)
Compound trans-3c was synthesized according to GP 1 to yield 338 mg (91%) of a colorless solid; mp 156 °C (benzene); Rf = 0.44 (n-pentane–Et2O, 1:1); [α]D20–113.3 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3432 (s), 1727 (vs), 1546 (vs), 1453 (m), 1356 (vs), 1275 (m), 1177 (s), 1110 (s), 1055 (vs), 981 (vs), 760 (s), 701 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 0.57 (t, J = 6.9 Hz, 3 H, CH2CH3), 3.16 (d, J = 12.4 Hz, 1 H, CHCO2Et), 3.62 (q, J = 6.9 Hz, 2 H, CH2CH3), 4.15 (dd, J = 12.4, 11.4 Hz, 1 H, CHPh), 4.32 (dd, J = 10.9, 5.0 Hz, 1 H, CH2), 4.73 (dd, J = 10.9, 10.9 Hz, 1 H, CH2), 5.00 (s, 1 H, OH), 5.12 (ddd, J = 11.4, 10.9, 5.0 Hz, 1 H, CHNO2), 7.23–7.55 (m, 10 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 13.3 (CH2CH3), 44.8 (CHCO2Et), 56.6 (CHPh), 61.4 (CH2), 62.1 (CH2), 85.5 (CHNO2), 96.0 (CqOH), 125.4 (2 CHAr), 127.9 (CHAr), 128.4 (CHAr), 128.6 (2 CHAr), 128.7 (2 CHAr), 128.9 (2 CHAr), 135.2 (CAr), 141.1 (CAr), 171.6 (CO2Et).
MS (EI, 70 eV): m/z (%) = 354 ([M – H2O]+, 2), 131 (8), 105 (100), 77 (26).
Anal. Calcd for C20H21NO6: C, 64.68; H, 5.70; N, 3.77. Found: C, 64.77; H, 5.62; N, 3.66.
1-[(2S,3S,4S,5S)-2-Hydroxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-yl]ethanone (trans-3d)
Compound trans-3d was synthesized according to GP 1; yield; 223 mg (80%); colorless solid; mp 98 °C (benzene); Rf = 0.21 (n-pentane–Et2O, 1:1); [α]D20–54.4 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3502 (s), 3413 (s), 1695 (vs), 15498 (vs), 1361 (vs), 1233 (w), 1123 (vs), 1051 (vs), 929 (m), 885 (m), 752 (s), 702 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 1.41 (s, 3 H, CH3), 1.77 (s, 3 H, CH3), 3.12 (d, J = 12.4 Hz, 1 H, CHCOCH3), 3.92 (dd, J = 11.9, 11.9 Hz, 1 H, CHPh), 4.11 (dd, J = 10.4, 5.0 Hz, 1 H, CH2), 4.32 (s, 1 H, OH), 4.49 (dd, J = 10.9, 10.4 Hz, 1 H, CH2), 4.95 (ddd, J = 11.9, 10.9, 5.0 Hz, 1 H, CHNO2), 7.19–7.36 (m, 5 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 27.9 CH3), 32.7 (COCH3), 44.5 (CHPh), 60.3 (CHCOMe), 61.5 (CH2), 85.6 (CHNO2), 94.8 (CqOH), 127.8 (2 CHAr), 128.7 (CHAr), 129.4 (2 CHAr), 135.5 (CAr), 210.9 (COMe).
MS (CI, 70 eV): m/z (%) = 262 ([M – H2O]+, 100), 220 (50), 189 (15), 173 (64), 147 (48), 131 (21), 91 (13).
Anal. Calcd for C14H17NO5: C, 60.21; H, 6.14; N, 5.02. Found: C, 60.27; H, 6.09; N, 4.90.
1-[(2S,3S,4S,5R)-2-Hydroxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-yl]ethanone (cis-3d)
Compound cis-3d was synthesized according to GP 1; yield: 17 mg (6%); colorless solid (MeOH); mp 40 °C; Rf = 0.23 (n-pentane–Et2O, 1:2); [α]D20–6.2 (c 0.2, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 1.56 (s, 3 H, CH3), 2.08 (s, 3 H, CH3), 3.68 (s, 1 H, OH), 3.79 (dd, J = 12.9, 4.0 Hz, 1 H, CHPh), 4.13 (d, J = 13.4 Hz, 1 H, CH2), 4.27 (d, J = 12.4 Hz, 1 H, CHCOMe), 4.58 (dd, J = 13.4, 3.0 Hz, 1 H, CH2), 4.76 (m, 1 H, CHNO2), 7.20–7.24 (m, 2 H, CHAr), 7.29–7.34 (m, 3 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 28.7 (CH3), 32.5 (COCH3), 42.0 (CHPh), 54.0 (CHCOMe), 61.7 (CH2), 84.8 (CHNO2), 95.0 (CqOH), 128.1 (2 CHAr), 128.6 (CHAr), 129.2 (2 CHAr), 135.7 (CAr), 211.5 (COMe).
Methyl (2S,3R,4S,5S)-2-Hydroxy-2-methyl-5-nitro-4-(p-tolyl)tetrahydro-2H-pyran-3-carboxylate (trans-3e)
Compound trans-3e was synthesized according to GP 1; yield: 204 mg (66%); colorless solid; (benzene); mp 151 °C; Rf = 0.38 (n-pentane–Et2O, 1:1); [α]D20–85.3 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3508 (vs), 2998 (w), 1716 (vs), 1543 (vs), 1442 (m), 1365 (vs), 1217 (m), 1120 (vs), 1066 (vs), 879 (m), 809 (m), 727 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 1.48 (s, 3 H, CH3), 2.29 (s, 3 H, p-CH3), 2.94 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 3.44 (s, 3 H, CO2CH3), 3.85 (s, 1 H, OH), 3.97 (dd, J = 11.9, 11.9 Hz, 1 H, CHAr), 4.09 (dd, J = 10.4, 5.0 Hz, 1 H, CH2), 4.47 (dd, J = 10.9, 10.4 Hz, 1 H, CH2), 4.88 (ddd, J = 11.9, 10.9, 5.0 Hz, 1 H, CHNO2), 7.07–7.13 (m, 4 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 21.1 (p-CH3), 28.0 (CH3), 43.5 (CHCO2CH3), 52.3 (CO2CH3), 55.2 (CHAr), 61.6 (CH2), 85.7 (CHNO2), 94.8 (CqOH), 127.6 (2 CHAr), 129.7 (2 CHAr), 132.6 (CAr), 138.1 (CAr), 172.0 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 309 ([M+], 14), 292 (100), 219 (18), 203 (20), 191 (18), 185 (22), 176 (26), 159 (10), 145 (19).
HRMS: m/z calcd for C15H19NO6 + Na: 332.1105; found: 332.1105.
Anal. Calcd for C15H19NO6: C, 58.25; H, 6.19; N, 4.53. Found: C, 58.17; H, 6.28; N, 4.82.
Methyl (2S,3R,4S,5R)-2-Hydroxy-2-methyl-5-nitro-4-(p-tolyl)tetrahydro-2H-pyran-3-carboxylate (cis-3e)
Compound cis-3e was synthesized according to GP 1; yield: 77 mg (25%); colorless solid (benzene); mp 71 C; Rf = 0.31 (n-pentane–Et2O, 1:2); [α]D20–7.1 (c 0.5, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 1.61 (s, 3 H, CH3), 2.29 (s, 3 H, p-CH3), 3.36 (s, 1 H, OH), 3.56 (s, 3 H, CO2CH3), 3.83 (dd, J = 12.9, 4.5 Hz, 1 H, CHAr), 4.07 (d, J = 12.9 Hz, 1 H, CHCO2CH3), 4.13 (dd, J = 13.4, 1.0 Hz, 1 H, CH2), 4.57 (dd, J = 13.4, 3.0 Hz, 1 H, CH2), 4.76 (ddd, J = 4.5, 3.0, 1.0 Hz, 1 H, CHNO2), 7.08–7.12 (m, 4 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 21.0 (p-CH3), 28.6 (CH3), 40.7 (CHCO2CH3), 48.0 (CHAr), 52.3 (CO2CH3), 61.7 (CH2), 84.8 (CHNO2), 95.1 (CqOH), 127.6 (2 CHAr), 129.6 (2 CHAr), 133.0 (CAr), 137.8 (CAr), 173.0 (CO2CH3).
Methyl (2S,3R,4S,5S)-4-(3-Bromophenyl)-2-hydroxy-2-methyl-5-nitrotetrahydro-2H-pyran-3-carboxylate (trans-3f)
Compound trans-3f was synthesized according to GP 1; yield: 318 mg (85%); colorless solid; mp 158 °C (benzene); Rf = 0.33 (n-pentane–Et2O, 1:1); [α]D20–48.4 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3556 (s), 1725 (vs), 1547 (vs), 1436 (m), 1363 (vs), 1263 (m), 1120 (vs), 1066 (vs), 885 (m), 842 (m), 787 (s), 691 (s), 576 (s), 536 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 1.48 (s, 3 H, CH3), 2.92 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 3.48 (s, 3 H, CO2CH3), 3.72 (s, 1 H, OH), 4.09 (dd, J = 12.4, 11.9 Hz, 1H, CHAr), 4.11 (dd, J = 10.4, 5.0 Hz, 1 H, CH2), 4.45 (dd, J = 10.9, 10.4 Hz, 1 H, CH2), 4.88 (ddd, J = 11.9, 10.9, 5.0 Hz, 1 H, CHNO2), 7.16–7.20 (m, 2 H, CHAr), 7.35–7.42 (m, 1 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 27.9 (CH3), 43.2 (CHAr), 52.5 (CO2CH3), 55.0 (CHCO2CH3), 61.4 (CH2), 85.2 (CHNO2), 94.8 (CqOH), 123.0 (CAr), 126.7 (CHAr), 130.5 (CHAr), 130.8 (CHAr), 131.6 (CHAr), 138.8 (CAr), 171.3 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 356 ([M]+, 7), 267 (20), 257 (26), 255 (30), 241 (35) 225 (43), 213 (39), 211 (62), 195 (25), 183 (15), 155 (34), 128 (100), 116 (55), 102 (48), 77 (15), 59 (28).
HRMS: m/z calcd for C14H16BrNO6 + Na: 398.0038; found: 398.0718.
Methyl (2S,3R,4S,5R)-4-(3-Bromophenyl)-2-hydroxy-2-methyl-5-nitrotetrahydro-2H-pyran-3-carboxylate (cis-3f)
Compound cis-3f was synthesized according to GP 1; yield: 34 mg (9%); colorless solid (benzene); mp 88 °C; Rf = 0.29 (n-pentane–Et2O, 1:2); [α]D20–5.1 (c 0.2, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 1.63 (s, 3 H, CH3), 3.22 (s, 1 H, OH), 3.60 (s, 3 H, CO2CH3), 3.87 (dd, J = 12.9, 4.5 Hz, 1 H, CHAr), 4.05 (d, J = 12.9 Hz, 1 H, CHCO2Me), 4.15 (d, J = 12.9, 1.0 Hz, 1 H, CH2), 4.58 (dd, J = 12.9, 3.0 Hz, 1 H, CH2), 4.78 (m, 1 H, CHNO2), 7.19–7.20 (m, 2 H, CHAr), 7.36 (s, 1 H, CHAr), 7.39–7.42 (m, 1 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 28.6 (CH3), 40.6 (CHAr), 47.9 (CO2CH3), 52.5 (CHCO2CH3), 61.8 (CH2), 84.4 (CHNO2), 95.2 (CqOH), 122.9 (CAr), 126.3 (CHAr), 128.3 (CHAr), 130.4 (CHAr), 131.4 (CHAr), 138.6 (CAr), 172.3 (CO2CH3).
Methyl (2S,3R,4R,5S)-2-Hydroxy-2-methyl-5-nitro-4-(thiophen-2-yl)tetrahydro-2H-pyran-3-carboxylate (trans-3g)
Compound trans-3g was synthesized according to GP 1; yield: 178 mg (59%); colorless solid; mp 111 °C (benzene); Rf = 0.45 (n-pentane–Et2O, 1:1); [α]D20–99.4 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3497 (s), 2315 (m), 2094 (m), 1734 (vs), 1547 (vs), 1439 (m), 1361 (vs), 1219 (m), 1135 (vs), 1060 (vs), 879 (m), 840 (m), 722 cm−1 (vs).
1H NMR (600 MHz, CDCl3): δ = 1.49 (s, 3 H, CH3), 2.94 (d, J = 11.9 Hz, 1 H, CHCO2CH3), 3.49 (s, 1 H, OH), 3.56 (s, 3 H, CO2CH3), 4.09 (dd, J = 10.9, 5.0 Hz, 1 H, CH2), 4.39 (dd, J = 11.9, 11.9 Hz, 1 H, CHAr), 4.45 (dd, J = 10.9, 10.9 Hz, 1 H, CH2), 4.85 (ddd, J = 11.4, 10.9, 5.0 Hz, 1 H, CHNO2), 6.90–6.92 (m, 2 H, CHAr), 7.20–7.23 (m, 1 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 27.9 (CH3), 39.3 (CHCO2CH3), 52.6 (CO2CH3), 56.4 (CHAr), 61.5 (CH2), 86.7 (CHNO2), 94.9 (CqOH), 125.4 (CHAr), 126.7 (CHAr), 127.1 (CHAr), 138.8 (Cq), 171.5 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 301 ([M]+, 11), 236 (14), 211 (27), 195 (39), 179 (38) 177 (100), 168 (23), 163 (38), 151 (14), 137 (58), 123 (15), 111 (11), 109 (16), 97 (13), 65 (8).
Anal. Calcd for C12H15NO6S: C, 47.83; H, 5.02; N, 4.65. Found: C, 47.86; H, 5.18; N, 4.65.
Methyl (2S,3R,4R,5R)-2-Hydroxy-2-methyl-5-nitro-4-(thiophen-2-yl)tetrahydro-2H-pyran-3-carboxylate (cis-3g)
Compound cis-3g was synthesized according to GP 1; yield: 102 mg (34%); colorless solid; mp 63 °C (benzene); [α]D20–38.3 (c 0.5, CHCl3); de >95%; Rf = 0.25 (n-pentane–Et2O, 1:1). The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 1.60 (s, 3 H, CH3), 3.37 (s, 1 H, OH), 3.60 (s, 3 H, CO2CH3), 4.03 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 4.12 (dd, J = 13.4, 1.0 Hz, 1 H, CH2), 4.14 (dd, J = 12.4, 4.5 Hz, 1 H, CHAr), 4.56 (dd, J = 13.4, 3.0 Hz, 1.0 Hz, 1 H, CH2), 4.83 (ddd, J = 4.5, 3.0, 1.0 Hz 1 H, CHNO2), 6.90–6.94 (m, 2 H, CHAr), 7.18–7.20 (m, 1 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 28.5 (CH3), 37.0 (CHAr), 50.0 (CO2CH3), 52.5 (CHCO2CH3), 61.6 (CH2), 84.6 (CHNO2), 95.2 (CqOH), 125.3 (CHAr), 126.3 (CHAr), 127.0 (CHAr), 138.6 (CAr), 172.5 (CO2CH3).
Methyl (2S,3R,4S,5S)-4-(Benzo[d][1,3]dioxol-5-yl)-2-hydroxy-2-methyl-5-nitrotetrahydro-2H-pyran-3-carboxylate (trans-3h)
Compound trans-3h was synthesized according to GP 1; yield: 221 mg (65%); colorless solid; mp 166 °C (benzene); Rf = 0.22 (n-pentane–Et2O, 1:1); [α]D20–85.0 (c 1.0, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
IR (film): 3503 (s), 3148 (s), 1733 (vs), 1552 (vs), 1493 (m), 1442 (vs), 1365 (vs), 1245 (vs), 1155 (vs), 1114 (vs), 1047 (vs), 933 (m), 886 (s), 807 (vs), 734 cm−1 (s).
1H NMR (400 MHz, CDCl3): δ = 1.47 (s, 3 H, CH3), 2.88 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 3.49 (s, 1 H, OH), 3.50 (s, 3 H, CO2CH3), 3.93 (dd, J = 11.9, 11.9 Hz, 1 H, CHAr), 4.08 (dd, J = 10.4, 5.0 Hz, 1 H, CH2), 4.45 (dd, J = 10.9, 10.4 Hz, 1 H, CH2), 4.82 (ddd, J = 11.9, 10.9, 5.0 Hz, 1 H, CHNO2), 5.94 (m, 2 H, OCH2O), 6.65–6.72 (m, 3 H, CHAr).
13C NMR (100 MHz, CDCl3): δ = 27.9 (CH3), 43.5 (CHCO2CH3), 52.5 (CO2CH3), 55.3 (CHAr), 61.5 (CH2), 85.9 (CHNO2), 94.8 (CqOH), 101.3 (CH2), 107.8 (CHAr), 108.6 (CHAr), 121.5 (CHAr), 129.4 (Cq), 147.6 (Cq), 148.0 (Cq), 171.7 (CO2CH3).
MS (CI, 70 eV): m/z (%) = 339 ([M]+, 100), 249 (17), 233 (41), 215 (84), 206 (41) 201 (31), 189 (11), 175 (74), 144 (21), 122 (12), 115 (20), 89 (12).
HRMS: m/z calcd for C15H17NO8 + K: 378.0591; found: 378.0592.
Methyl (2S,3R,4S,5R)-4-(Benzo[d][1,3]dioxol-5-yl)-2-hydroxy-2-methyl-5-nitrotetrahydro-2H-pyran-3-carboxylate (cis-3h)
Compound cis-3h was synthesized according to GP 1; yield: 88 mg (26%); colorless solid; mp 87 °C (MeOH); Rf = 0.19 (n-pentane–Et2O, 1:2); [α]D20–26.5 (c 0.5, CHCl3); de >95%. The diastereomeric excess was determined by NMR spectroscopy.
1H NMR (600 MHz, CDCl3): δ = 1.61 (s, 3 H, CH3), 3.33 (br s, 1 H, OH), 3.59 (s, 3 H, CO2CH3), 3.79 (dd, J = 12.9, 4.0 Hz, 1 H, CHAr), 3.99 (d, J = 12.9 Hz, 1 H, CHCO2CH3), 4.11 (d, J = 13.4 Hz, 1 H, CH2), 4.54 (dd, J = 13.4, 3.0 Hz, 1 H, CH2), 4.75 (dd, J = 4.0, 3.0 Hz, 1 H, CHNO2), 5.93 (d, J = 2 Hz, 2 H, OCH2O), 6.67–6.73 (m, 3 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 28.6 (CH3), 40.8 (CHAr), 52.4 (CO2CH3), 48.3 (CHCO2CH3), 61.7 (CH2), 84.9 (CHNO2), 95.2 (CqOH), 101.2 (CH2), 108.2 (CHAr), 108.6 (CHAr), 121.2 (CHAr), 129.8 (Cq), 147.4 (Cq), 148.0 (Cq), 172.8 (CO2CH3).
Methyl (4R)-6-Methyl-3-nitro-4-phenyl-3,4-dihydro-2H-pyran-5-carboxylate (4a)
Compound 4a was synthesized according to GP 2; yield: 222 mg (80%); colorless oil; Rf = 0.41 (n-pentane–Et2O, 6:1); [α]D20–171.3 (c 0.9, CHCl3); ee (trans) = 95%; ee (cis) = 71%; de = 71%. The enantiomeric excess was determined by chiral stationary phase HPLC using a Chiralcel OD column (n-heptane–i-PrOH, 7:3, flow rate 1.0 mL/min, λ = 254 nm); tR = 7.38 min (minor), tR = 15.36 min (major).
IR (film): 2945 (vs), 1708 (vs), 1627 (vs), 1552 (vs), 1449 (vs), 1377 (s), 1262 (vs), 1189 (w), 1097 (vs), 941 (m), 883 (m), 763 (s), 706 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 2.37 (s, 3 H, CH3), 3.53 (s, 3 H, CO2CH3), 4.03 (dd, J = 12.9, 2.5 Hz, 1 H, CH2), 4.64 (dd, J = 2.5, 2.0 Hz, 1 H, CHNO2), 4.69 (dd, J = 12.9, 2.0 Hz, 1 H, CH2), 4.83 (br s, 1H, CHPh), 7.23–7.29 (m, 3 H, CHAr), 7.33–7.36 (m, 2 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 20.0 (CH3), 39.9 (CHPh), 51.9 (CO2CH3), 61.8 (CH2), 82.4 (CHNO2), 100.4 (Cq), 127.63 (2 CHAr), 128.4 (CHAr), 129.0 (2 CHAr), 141.0 (CAr), 165.2 (CqO), 167.2 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 278 ([M + 1]+, 27), 246 (22), 230 (100), 198 (48), 171 (17), 157 (14), 128 (13), 91 (13).
HRMS: m/z calcd for C14H15NO5 + Na: 300.0842; found: 300.0844.
Ethyl (4R)-6-Methyl-3-nitro-4-phenyl-3,4-dihydro-2H-pyran-5-carboxylate (4b)
Compound 4b was synthesized according to GP 2; yield: 248 mg (85%); colorless oil; Rf = 0.35 (n-pentane–Et2O, 8:1); [α]D20–266.9 (c 1.1, CHCl3); ee (trans) = 88%; ee (cis) = 73%; de = 64%. The enantiomeric excess was determined by chiral stationary phase HPLC using a (S,S) Whelk column (n-heptane–EtOH, 97:3, flow rate 0.5 mL/min, λ = 254 nm); tR = 12.54 min (major), tR = 13.68 min (minor), tR = 15.77 min (major), tR = 18.46 min (minor).
IR (film): 2979 (m), 1701 (vs), 1625 (vs), 1550 (vs), 1451 (m), 1371 (s), 1253 (vs), 1154 (w), 1091 (vs), 942 (w), 899 (w), 844 (w), 761 (m), 703 cm−1 (s).
1H NMR (600 MHz, CDCl3): δ = 0.98 (t, J = 7.2 Hz, 3 H, CH2CH3), 2.36 (s, 3 H, CH3), 3.90–4.10 (ddq, J = 12.5, 7.2, 2.3 Hz, 3 H, CH2), 4.60 (dd, J = 2.3, 2.3 Hz, 1 H, CHNO2), 4.68 (dd, J = 12.5, 2.3 Hz, 1 H, CH2), 4.81 (br s, 1 H, CHPh), 7.31–7.35 (m, 2 H, CHAr), 7.22–7.28 (m, 3 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 13.9 (CH2CH3), 19.8 (CH3), 40.1 (CHPh), 60.0 (CH2CH3), 61.9 (CH2), 82.6 (CHNO2), 101.0 (Cq), 127.6 (CHAr), 127.8 (2 CHAr), 129.0 (2 CHAr), 141.3 (CAr), 164.9 (CqO), 166.7 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 292 ([M + 1]+, 12), 244 (100), 215 (32), 198 (66), 171 (30), 157 (37), 128 (24), 115 (12), 91 (44).
HRMS: m/z calcd for C15H17NO5 + Na: 314.0999; found: 314.0999.
Ethyl (4R)-3-Nitro-4,6-diphenyl-3,4-dihydro-2H-pyran-5-carboxylate (4c)
Compound 4c was synthesized according to GP 2; yield: 286 mg (81%); colorless oil; Rf = 0.39 (n-pentane–Et2O 6:1); [α]D20–267.8 (c 1.3, CHCl3); ee (trans) = 99%; ee (cis) = 99%; de = 98%. The enantiomeric excess was determined by chiral stationary phase HPLC using a Chiralcel OD column (n-heptane–EtOH, 9:1, flow rate 1.0 mL/min, λ = 254 nm); tR = 10.44 min (minor), tR = 19.70 min (major).
IR (film): 2317 (m), 2109 (m), 1888 (m), 1676 (vs), 1550 (vs), 1451 (s), 1357 (vs), 1290 (s), 1251 (s), 1212 (m), 1150 (m), 1098 (vs), 1052 (vs), 931 (m), 873 (m), 762 (s), 703 cm−1 (vs).
1H NMR (600 MHz, CDCl3): δ = 0.75 (t, J = 6.9 Hz, 3 H, CH3), 3.78–3.89 (m, 2 H, CH2CH3), 4.25 (dd, J = 12.4, 2.0 Hz, 1 H, CH2), 4.75 (dd, J = 2.5, 2.0 Hz, 1 H, CHNO2), 4.86 (dd, J = 12.4, 2.5 Hz, 1 H, CH2), 5.20 (br s, 1 H, CHPh), 7.24–7.27 (m, 2 H, CHAr), 7.29–7.49 (m, 5 H, C6H5).
13C NMR (151 MHz, CDCl3): δ = 13.4 (CH3), 40.8 (CHPh), 60.3 (CH2), 63.0 (CH2), 82.9 (CHNO2), 103.4 (Cq), 127.9 (2 CHAr), 128.1 (2 CHAr), 128.6 (CHAr), 129.1 (2 CHAr), 129.3 (2 CHAr), 129.8 (CHAr), 135.1 (CAr), 140.5 (CAr), 163.1 (CqO), 166.9 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 353 ([M]+, 3), 306 (100), 277 (12), 261 (17), 233 (29), 204 (11), 128 (10), 105 (67), 91 (24), 77 (30).
HRMS: m/z calcd for C20H19NO5 + Na: 376.1155; found: 376.1158.
1-[(4R)-6-Methyl-3-nitro-4-phenyl-3,4-dihydro-2H-pyran-5-yl]ethanone (4d)
Compound 4d was synthesized according to GP 2; yield: 201 mg (77%); colorless oil; Rf = 0.40 (n-pentane–Et2O, 8:1); [α]D20–298.7 (c 1.4, CHCl3); ee (trans) = 92%; ee (cis) = 99%; de = 87%. The enantiomeric excess was determined by chiral stationary phase HPLC using a (S,S) Whelk column (n-heptane–EtOH, 97:3, flow rate 1.0 mL/min, λ = 254 nm); tR = 13.12 min (major), tR = 18.59 min (minor), tR = 15.97 min (major), tR = 19.80 min (minor).
IR (film): 1689 (vs), 1622 (m), 1545 (vs), 1448 (m), 1353 (vs), 1277 (vs), 1122 (vs), 1081 (vs), 1021 (s), 916 (w), 846 (m), 765 (vs), 696 cm−1 (vs).
1H NMR (600 MHz, CDCl3): δ = 2.06 (s, 3 H, CH3), 2.31 (s, 3 H, COCH3), 4.03 (dd, J = 12.9, 2.5 Hz, 1 H, CH2), 4.64 (dd, J = 2.5, 2.0 Hz, 1 H, CHNO2), 4.73 (dd, J = 12.9, 2.0 Hz, 1 H, CH2), 4.83 (br s, 1 H, CHPh), 7.24–7.27 (m, 2 H, CHAr), 7.29–7.32 (m, 1 H, CHAr), 7.37–7.41 (m, 2 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 20.5 (CH3), 29.2 (COCH3), 40.7 (CHPh), 61.7 (CH2), 82.5 (CHNO2), 109.1 (Cq), 128.2 (2 CHAr), 128.2 (CHAr), 129.5 (2 CHAr), 140.3 (CAr), 164.1 (CqO), 198.2 (COCH3).
MS (EI, 70 eV): m/z (%) = 261 ([M]+, 8), 215 (100), 199 (46), 171 (58), 155 (27), 128 (32), 115 (19), 91 (68), 77 (15), 59 (22).
HRMS: m/z calcd for C14H16NO4: 262.1074; found: 262.1073.
Methyl (4R)-6-Methyl-3-nitro-4-(p-tolyl)-3,4-dihydro-2H-pyran-5-carboxylate (4e)
Compound 4e was synthesized according to GP 2; yield: 236 mg (81%); colorless oil; Rf = 0.34 (n-pentane–Et2O, 8:1); [α]D20–226.5 (c 0.9, CHCl3); ee (trans) = 88%; ee (cis) = 89%; de = 45%. The enantiomeric excess was determined by chiral stationary phase HPLC using a (S,S) Whelk column (n-heptane–EtOH, 97:3, flow rate 0.5 mL/min, λ = 254 nm); tR = 13.00 min (major), tR = 14.78 min (minor), tR = 16.63 min (major), tR = 20.29 min (minor).
IR (film): 2940 (vs), 1704 (vs), 1624 (vs), 1549 (vs), 1441 (s), 1362 (s), 1256 (vs), 1191 (w), 1095 (vs), 936 (m), 881 (m), 800 (m), 724 cm−1 (m).
1H NMR (600 MHz, CDCl3): δ = 2.30 (s, 3 H, CH3), 2.34 (s, 3 H, CH3), 3.55 (s, 3 H, CO2CH3), 4.04 (dd, J = 12.9, 2.0 Hz, 1 H, CH2), 4.58 (dd, J = 2.0, 2.0 Hz, 1 H, CHNO2), 4.69 (dd, J = 12.9, 2.0 Hz, 1 H, CH2), 4.79 (br s, 1 H, CHAr), 7.04–7.08 (m, 4 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 19.8 (CH3), 21.0 (CH3), 39.5 (CHAr), 51.5 (CO2CH3), 61.8 (CH2), 82.6 (CHNO2), 100.6 (Cq), 127.7 (2 CHAr), 129.8 (2 CHAr), 137.4 (CAr), 138.0 (CAr), 165.0 (CqO), 167.4 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 291 ([M]+, 15), 244 (100), 229 (36), 213 (80), 171 (47), 143 (19), 128 (25), 115 (19), 105 (63), 91 (10).
HRMS: m/z calcd for C15H17NO5 + Na: 314.0999; found: 314.0999.
Methyl (4R)-4-(3-Bromophenyl)-6-methyl-3-nitro-3,4-dihydro-2H-pyran-5-carboxylate (4f)
Compound 4f was synthesized according to GP 2; yield: 306 mg (86%); colorless oil; Rf = 0.26 (n-pentane–Et2O, 8:1); [α]D20–271.3 (c 1.1, CHCl3); ee (trans) = 88%; ee (cis) = 78%; de = 76%. The enantiomeric excess was determined by chiral stationary phase HPLC using a Chiralcel OJ column (n-heptane–EtOH, 80:20, flow rate 1.0 mL/min, λ = 254 nm); tR = 4.68 min (major), tR = 7.69 min (minor), tR = 13.50 min (major), tR = 20.80 min (minor).
IR (film): 2951 (vs), 1710 (vs), 1626 (vs), 1555 (vs), 1435 (vs), 1382 (vs), 1359 (vs), 1259 (vs), 1189 (s), 1150 (m), 1102 (vs), 1051 (s), 941 (s), 890 (s), 784 (s), 699 cm−1 (s).
1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3 H, CH3), 3.53 (s, 3 H, CO2CH3), 3.99 (dd, J = 12.8, 2.0 Hz, 1 H, CH2), 4.57 (dd, J = 2.0, 2.0 Hz, 1 H, CHNO2), 4.69 (dd, J = 12.8, 2.0 Hz, 1 H, CH2), 4.79 (br s, 1 H, CHAr), 7.10–7.24 (m, 3 H, CHAr), 7.36–7.41 (m, 1 H, CHAr).
13C NMR (100 MHz, CDCl3): δ = 20.1 (CH3), 39.6 (CHAr), 51.5 (CO2CH3), 61.8 (CH2), 82.0 (CHNO2), 99.8 (Cq), 123.3 (CAr), 126.5 (CHAr), 130.7 (CHAr), 130.9 (CHAr), 131.0 (CHAr), 143.5 (CAr), 165.8 (CqO), 166.9 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 355 ([M]+, 8), 324 (11), 310 (100), 276 (33), 251 (20), 235 (18), 229 (28), 197 (25), 169 (31), 155 (18), 128 (33), 115 (15).
HRMS: m/z calcd for C14H14BrNO5 + Na: 377.9948; found: 377.9951.
Methyl (4R)-6-Methyl-3-nitro-4-(thiophen-2-yl)-3,4-dihydro-2H-pyran-5-carboxylate (4g)
Compound 4g was synthesized according to GP 2; yield: 232 mg (82%); colorless oil; Rf = 0.23 (n-pentane–Et2O, 8:1); [α]D20–384.3 (c 1.3, CHCl3); ee (trans) = 90%; ee (cis) = 94%; de = 26%. The enantiomeric excess was determined by chiral stationary phase HPLC using a Chiralpak IA column (n-heptane–EtOH, 95:5, flow rate 0.7 mL/min, λ = 254 nm); tR = 13.64 min (minor), tR = 15.20 min (major), tR = 14.63 min (major), tR = 16.40 min (minor).
IR (film): 2950 (vs), 1704 (vs), 1621 (vs), 1551 (vs), 1441 (s), 1361 (s), 1258 (vs), 1203 (s), 1094 (vs), 933 (m), 883 (m), 845 (m), 789 (w), 706 cm−1 (vs).
1H NMR (600 MHz, CDCl3): δ = 2.33 (s, 3 H, CH3), 3.61 (s, 3 H, CO2CH3), 4.20 (dd, J = 12.9, 2.0 Hz, 1 H, CH2), 4.70 (dd, J = 2.0, 2.0 Hz, 1 H, CHNO2), 4.88 (dd, J = 12.9, 2.0 Hz, 1 H, CH2), 5.05 (br s, 1 H, CHAr), 6.88 (m, 1 H, CHAr), 6.95 (m, 1 H, CHAr), 7.24 (m, 1 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 20.0 (CH3), 34.9 (CHPh), 51.5 (CO2CH3), 62.3 (CH2), 81.9 (CHNO2), 101.4 (Cq), 125.31 (CHAr), 125.9 (CHAr), 127.4 (CHAr), 144.9 (CAr), 165.0 (CqO), 167.0 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 283 ([M]+, 11), 236 (69), 205 (100), 176 (16), 163 (72), 135 (21), 97 (57), 91 (13).
HRMS: m/z calcd for C12H13NO5S + Na: 306.0407; found: 306.0409.
Methyl (4R)-4-(Benzo[d][1,3]dioxol-5-yl)-6-methyl-3-nitro 3,4-dihydro-2H-pyran-5-carboxylate (4h)
Compound 4h was synthesized according to GP 2; yield: 260 mg (81%); colorless oil; Rf = 0.36 (n-pentane–Et2O, 6:1); [α]D20–311.8 (c 1.2, CHCl3); ee (trans) = 91%; ee (cis) = 83%; de = 43%. The enantiomeric excess was determined by chiral stationary phase HPLC using a (S,S) Whelk column (n-heptane–EtOH, 97:3, flow rate 1.0 mL/min, λ = 254 nm); tR = 12.36 min (major), tR = 15.20 min (minor), tR = 17.75 min (major), tR = 23.79 min (minor).
IR (film): 2952 (vs), 1713 (vs), 1633 (vs), 1555 (vs), 1488 (vs), 1446 (vs), 1383 (vs), 1235 (s), 1189 (m), 1036 (vs), 930 (vs), 872 (vs), 818 (s), 762 (m), 735 (s), 762 cm−1 (m).
1H NMR (600 MHz, CDCl3): δ = 2.34 (s, 3 H, CH3), 3.57 (s, 3 H, CO2CH3), 4.01 (dd, J = 12.4, 2.0 Hz, 1 H, CH2), 4.52 (dd, J = 2.0, 2.0 Hz, 1 H, CHNO2), 4.64 (dd, J = 12.4, 12.4 Hz, 1 H, CH2), 4.73 (br s, 1 H, CHAr), 5.94 (s, 2 H, CH2), 6.60–6.69 (m, 3 H, CHAr).
13C NMR (151 MHz, CDCl3): δ = 20.0 (CH3), 39.6 (CHAr), 51.5 (CO2CH3), 61.9 (CH2), 82.5 (CHNO2), 100.7 (Cq), 101.3 (CH2), 108.2 (CHAr), 108.7 (CHAr), 121.1 (CHAr), 134.9 (CAr), 147.1 (CAr), 148.3 (CAr), 165.0 (CqO), 167.2 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 321 ([M]+, 50), 274 (23), 243 (100), 214 (28), 201 (57), 173 (11), 143 (10), 135 (68), 115 (26), 77 (12).
HRMS: m/z calcd for C15H15NO7 + Na: 344.0741; found: 344.0744.
Methyl (2S,3R,4S,5S)-2-Methoxy-2-methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-carboxylate (7)
In a glass vial equipped with a magnetic stirring bar, trans-3a (295 mg, 1 mmol) was added to a mixture of PTSA (38 mg, 20 mol%) in MeOH (6 mL) and stirred for 8 h at r.t. The reaction was monitored by TLC. After completion, the solvent was evaporated and the crude reaction mixture was purified by column chromatography (silica gel, n-pentane–Et2O, 10:1) to yield 7 in 257 mg (62%) as a colorless solid; mp 83 °C; Rf = 0.40 (n-pentane–Et2O, 4:1); [α]D20–71.3 (c 1.0, CHCl3); ee = 99%; de = 98%. The enantiomeric excess was determined by chiral stationary phase HPLC using a Chiralcel OD column (n-heptane–i-PrOH, 97:3, flow rate 0.7 mL/min, λ = 254 nm); tR = 11.45 min (minor), tR = 14.89 min (major).
IR (film): 1731 (vs), 1548 (vs), 1454 (m), 1386 (m), 1267 (vs), 1173 (vs), 1118 (s), 1039 (vs), 869 (s), 760 (vs), 699 cm−1 (vs).
1H NMR (600 MHz, CDCl3): δ = 1.51 (s, 3 H, CH3), 2.99 (d, J = 12.4 Hz, 1 H, CHCO2CH3), 3.33 (s, 3 H, OCH3), 3.47 (s, 3 H, CO2CH3), 4.04, (dd, J = 10.9, 5.5 Hz, 1 H, CH2), 4.08 (dd, J = 10.9, 10.4 Hz, 1 H, CH2), 4.16 (dd, J = 11.9, 11.9 Hz, 1 H, CHPh), 4.83 (ddd, J = 11.9, 10.4, 5.5 Hz, 1 H, CHNO2), 7.21–7.29 (m, 5 H, C6H5)
13C NMR (151 MHz, CDCl3): δ = 22.1 (CH3), 42.6 (CHCO2CH3), 48.7 (COCH3), 51.9 (CO2CH3), 56.3 (CHPh), 61.2 (CH2), 86.5 (CHNO2), 98.2 (CqOMe), 128.0 (2 CHAr), 128.1 (CHAr), 128.8 (2 CHAr), 136.7 (Cq), 198.7 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 309 ([M]+, 1), 128 (29), 248 (11), 235 (14), 188 (100), 173 (15), 157 (26), 129 (87), 117 (62), 103 (28), 91 (19), 77 (10), 59 (19).
HRMS: m/z calcd for C15H19NO6 + Na: 332.1105; found: 332.1104.
Methyl (2S,3R,4S,5S)-2-Methyl-5-nitro-4-phenyltetrahydro-2H-pyran-3-carboxylate (8)
In a glass vial equipped with a magnetic stirring bar, trans-3a (295 mg, 1 mmol) was added to a mixture Et3SiH (240 μL, 1.5 mmol), trifluoroborane diethyl etherate (130 μL, 1.0 mmol) in CH2Cl2 (6 mL) and stirred for 6 h at 0 °C. The reaction was monitored by TLC. The reaction mixture was warmed up to r.t. and quenched with sat. aq NaHCO3 (5 mL), the organic phase was separated, and the aqueous phase was extracted with EtOAc (3 × 5 mL). The organic extracts were combined, dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by chromatography (silica gel, n-pentane–EtOAc, 6:1) to yield 8 in 211 mg (72%); mp 131 °C; Rf = 0.33 (n-pentane–Et2O, 4:1); [α]D20–64.4 (c 1.0, CHCl3); ee = 98%; de = 97%. The enantiomeric excess was determined by chiral stationary phase HPLC using a Chiralcel OJ column (n-heptane–EtOH, 7:3, flow rate 0.7 mL/min, λ = 254 nm); tR = 9.35 min (minor), tR = 12.50 min (major).
IR (film): 1729 (vs), 1542 (vs), 1450 (s), 1344 (vs), 1270 (m), 1196 (s), 1121 (vs), 1077 (vs), 903 (m), 767 (vs), 701 cm−1 (vs).
1H NMR (400 MHz, CDCl3): δ = 1.24 (d, J = 6.0 Hz, 3 H, CH3), 2.65 (dd, J = 11.9, 9.9 Hz, 1 H, CHCO2CH3), 3.43 (s, 3 H, CO2CH3), 3.75 (dd, J = 11.9, 11.4 Hz, 1 H, CHPh), 3.82 (m, 1 H, CHCH3), 3.91 (dd, J = 10.9, 10.4 Hz, 1 H, CH2), 4.41 (dd, J = 10.4, 4.5 Hz, 1 H, CH2), 4.90 (ddd, J = 11.4, 10.9 4.5 Hz, 1 H, CHNO2), 7.19–7.32 (m, 5 H, C6H5).
13C NMR (100 MHz, CDCl3): δ = 19.5 (CH3), 48.1 (CHCO2CH3), 51.9 (CO2CH3), 55.3 (CHPh), 69.2 (CH2), 75.7 (CHCH3), 85.8 (CHNO2), 127.6 (2 CHAr), 128.4 (CHAr), 129.0 (2 CHAr), 135.9 (Cq), 170.8 (CO2CH3).
MS (EI, 70 eV): m/z (%) = 279 ([M]+, 15), 232 (25), 189 (34), 172 (100), 157 (73), 129 (63), 115 (31), 103 (18), 91 (27), 77 (15), 59 (22).
HRMS: m/z calcd for C14H17NO5 + Na: 302.1004; found: 302.1003.
Supplementary Material
Acknowledgment
D. E. thanks the European Research Council for an ERC Advanced Grant (DOMINOCAT). We thank the BASE SE for donation of chemicals.
References
- (1).For reviews on organocatalytic domino reactions, see: ; (a) Sheffler U, Mahrwald R. Chem. Eur. J. 2013;19:14346. doi: 10.1002/chem.201301996. [DOI] [PubMed] [Google Scholar]; (b) Bonne D, Constanieux T, Coquerel Y, Rodriguez J. Chem. Eur. J. 2013;19:2218. doi: 10.1002/chem.201204018. [DOI] [PubMed] [Google Scholar]; (c) Pellissier H. Chem. Rev. 2013;113:442. doi: 10.1021/cr300271k. [DOI] [PubMed] [Google Scholar]; (d) Grossmann A, Enders D. Angew. Chem. Int. Ed. 2011;51:314. doi: 10.1002/anie.201105415. [DOI] [PubMed] [Google Scholar]; (e) Enders D, Jeanty M, Grondal C. Nat. Chem. 2010;2:167. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]; (f) Alba A-N, Companyo X, Viciano M, Rios R. Curr. Org. Chem. 2009;13:1432. [Google Scholar]; (g) Yu X, Wang W. Org. Biomol. Chem. 2008;6:2037. doi: 10.1039/b800245m. [DOI] [PubMed] [Google Scholar]; (h) Enders D, Grondal C, Hüttl MRM. Angew. Chem. Int. Ed. 2007;46:1570. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]
- (2).For selected general reviews on hydrogen bonding, see: ; (a) Lu T, Wheeler SE. Chem. Eur. J. 2013;19:15141. doi: 10.1002/chem.201302990. [DOI] [PubMed] [Google Scholar]; (b) Wurm FR, Klok H-A. Chem. Soc. Rev. 2013;42:8220. doi: 10.1039/c3cs60153f. [DOI] [PubMed] [Google Scholar]; (c) Brière J-F, Oudeyer S, Dalla V, Levacher V. Chem. Soc. Rev. 2012;41:1696. doi: 10.1039/c1cs15200a. [DOI] [PubMed] [Google Scholar]; (d) Alemán J, Parra A, Jiang H, Jørgensen KA. Chem. Eur. J. 2011;17:6890. doi: 10.1002/chem.201003694. [DOI] [PubMed] [Google Scholar]; (e) Knowles RR, Jacobsen EN. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20678. doi: 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Etzenbach-Effers K, Berkessel A. Top. Curr. Chem. 2009;291:1. doi: 10.1007/978-3-642-02815-1_3. [DOI] [PubMed] [Google Scholar]; (g) Takemoto Y. Chem. Pharm. Bull. 2010;58:593. doi: 10.1248/cpb.58.593. [DOI] [PubMed] [Google Scholar]; (h) Kotke M, Schreiner PR. In: Hydrogen Bonding in Organic Synthesis. Pihko PM, editor. Wiley-VCH; Weinheim: 2009. p. 141. [Google Scholar]; (i) Zhang Z, Schreiner PR. Chem. Soc. Rev. 2009;38:1187. doi: 10.1039/b801793j. [DOI] [PubMed] [Google Scholar]; (j) Doyle AG, Jacobsen EN. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; (k) Taylor MS, Jacobsen EN. Angew. Chem. Int. Ed. 2006;45:1520. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]; (l) Schreiner PR. Chem. Soc. Rev. 2003;32:289. doi: 10.1039/b107298f. [DOI] [PubMed] [Google Scholar]
- (3).For reviews on tetrahydropyran syntheses, see: ; (a) Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron. 2010;66:413. [Google Scholar]; (b) Larrosa I, Romea P, Urpí F. Tetrahedron. 2008;64:2683. [Google Scholar]; (c) Clarke PA, Santos S. Eur. J. Org. Chem. 2006:2045. [Google Scholar]; (d) Shindo M. Top. Heterocycl. Chem. 2006;5:179. [Google Scholar]; (e) Lee E. Pure Appl. Chem. 2005;77:2073. [Google Scholar]; (f) Yeung K-S, Paterson I. Chem. Rev. 2005;105:4237. doi: 10.1021/cr040614c. [DOI] [PubMed] [Google Scholar]; (g) Boivin TLB. Tetrahedron. 1987;43:3309. [Google Scholar]
- (4).For selected examples of tetrahydropyran syntheses, see: ; (a) Dange NS, Hong B-C, Lan D-J, Liao J-H, Lee G-H. Eur. J. Org. Chem. 2013;12:2472. [Google Scholar]; (b) Wang Y, Zhu S, Ma D. Org. Lett. 2011;13:1602. doi: 10.1021/ol200004s. [DOI] [PubMed] [Google Scholar]; (c) Lee K, Kim H, Hong J. Eur. J. Org. Chem. 2012:1025. [Google Scholar]; (d) Ishikawa H, Sawano S, Yasui Y, Shibata Y, Hayashi Y. Angew. Chem. Int. Ed. 2011;50:3774. doi: 10.1002/anie.201005386. [DOI] [PubMed] [Google Scholar]; (e) Liu L, Floreancig PE. Angew. Chem. Int. Ed. 2010;49:3069. doi: 10.1002/anie.201000033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fuwa H, Noto K, Sasaki M. Org. Lett. 2010;12:1636. doi: 10.1021/ol100431m. [DOI] [PubMed] [Google Scholar]; (g) Trost BM, Gutierrez AC, Livingston RC. Org. Lett. 2009;11:2539. doi: 10.1021/ol9007876. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Chandrasekhar S, Mallikarjun K, Pavankumarreddy G, Veeramohana Rao K, Jagadeesh B. Chem. Commun. 2009:4985. doi: 10.1039/b904662c. [DOI] [PubMed] [Google Scholar]; (i) Cao C-L, Zhou Y-Y, Sun X-L, Tang Y, Li Y-X, Li G-Y, Sun J. Chem. Eur. J. 2009;15:11384. doi: 10.1002/chem.200900696. [DOI] [PubMed] [Google Scholar]; (j) Gotoh H, Okamura D, Ishikawa H, Hayashi Y. Org. Lett. 2009;11:4056. doi: 10.1021/ol901483x. [DOI] [PubMed] [Google Scholar]; (k) De Brab JK, Liu B, Qian M. Org. Lett. 2008;10:2533. doi: 10.1021/ol8008107. [DOI] [PubMed] [Google Scholar]; (l) Hiebel M-A, Pelotier B, Goekjian P, Piva O. Eur. J. Org. Chem. 2008:713. [Google Scholar]
- (5).(a) Enders D, Urbanietz G, Raabe G. Synthesis. 2011:1905. [Google Scholar]; (b) Enders D, Urbanietz G, Hahn R, Raabe G. Synthesis. 2012;44:773. [Google Scholar]; (c) Ramachary DB, Madhavachary R, Prasad MS. Org. Biomol. Chem. 2012;10:5825. doi: 10.1039/c2ob07122c. [DOI] [PubMed] [Google Scholar]
- (6).For some selected examples of (E)-2-nitro-3-phenylprop-2-en-1-ol in synthesis, see: ; (a) Beck AK, Duschmale J, Ebert M-O, Purkayastha N, Schweizer WB, Seebach D, Sun X, Wennemers H, Benohoud M, Hayashi Y, Mukaiyama T, Reiher M. Helv. Chim. Acta. 2013;96:799. [Google Scholar]; (b) An J, Lu L-Q, Wang T, Xiao W-J, Yang Q-Q. Org. Lett. 2013;15:542. doi: 10.1021/ol303363r. [DOI] [PubMed] [Google Scholar]; (c) Talavera G, Reyes E, Vicario JL, Carrillo L. Angew. Chem. Int. Ed. 2012;51:4104. doi: 10.1002/anie.201200269. [DOI] [PubMed] [Google Scholar]; (d) Parra A, Reboredo S, Alemán J. Angew. Chem. Int. Ed. 2012;51:9734. doi: 10.1002/anie.201203489. [DOI] [PubMed] [Google Scholar]; (e) Fioravanti S, Pellacani L, Vergari MC. Org. Biomol. Chem. 2012;10:524. doi: 10.1039/c1ob06260c. [DOI] [PubMed] [Google Scholar]; (f) Guo Z-W, Xie J-W, Chen C, Zhu W-D. Org. Biomol. Chem. 2012;10:8471. doi: 10.1039/c2ob26165k. [DOI] [PubMed] [Google Scholar]; (g) Rai A, Yadav LDS. Org. Biomol. Chem. 2011;9:8058. doi: 10.1039/c1ob06274c. [DOI] [PubMed] [Google Scholar]; (h) Bakthadoss M, Sivakumar N. Synlett. 2011:1296. [Google Scholar]; (i) Bakthadoss M, Shivakumar N, Deraraj A. Synthesis. 2011:611. [Google Scholar]
- (7).The (E)-3-aryl-2-nitroprop-2-en-1-ols were prepared from the corresponding (E)-β-nitrostyrenes and formaldehyde in THF according to a literature procedure: see ref. 6i.
- (8).Malerich JP, Hagihara K, Rawal VH. J. Am. Chem. Soc. 2008;130:14416. doi: 10.1021/ja805693p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tripathi CB, Mukherjee S. Angew. Chem. Int. Ed. 2013;52:8450. doi: 10.1002/anie.201304173. [DOI] [PubMed] [Google Scholar]
- 10.CCDC 965195 (trans-3g) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif or by writing to the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44(1223)336033; deposit@ccdc.cam.ac.uk.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.