

Abstract
The role of p63 in cancer has been an area of intense debate and controversy. Is TP63 (which encodes p63) a tumour suppressor gene or an oncogene? This debate is partly due to the complexity of the gene. There are several p63 isoforms — some with tumour suppressive functions and others with oncogenic functions. In this Opinion article, we focus on the recent advances in understanding p63 biology and its roles in cancer. In this regard, we discuss the role of p63 in multiple stem cell compartments, ageing, in the response to DNA damage and in DNA repair. Finally, we highlight the importance of understanding the interactions between all three p53 family members and the potential impact of this knowledge on cancer therapy and regenerative medicine.
TP53 (which encodes p53) is commonly mutated in human cancer and is well known to be an important tumour suppressor gene1,2; however, the tumour suppressive roles of p63 and p73, which are p53 family members, have been less clear. More than 10 years after the discovery of TP63 and TP73 (refs 3–6), specific isoforms of p63 and p73 have now been widely accepted as having tumour suppressive functions7–11. The road leading to this discovery has not been an easy one. Studies of the expression patterns of p63 in human cancer in the late 1990s and early 2000s were confusing and led to contradictory results (Supplementary information S1 (table)). These results left researchers perplexed as to whether p63 suppresses or promotes tumorigenesis. This confusion was mostly due to the complex structure of the TP63 gene and also due to antibodies that did not distinguish between the different isoforms of p63 (fig. 1). p63 is encoded by the ancestral gene of the p53 family3,12 and is of particular interest owing to its essential role and high expression in epithelial tissues13,14. TP63 encodes multiple isoforms that can be placed in two categories: isoforms with an acidic transactivation domain, which are known as the TA isoforms; and isoforms that lack this domain, which are known as the ΔN isoforms (fig. 1). Data from in vitro experiments have provided evidence that the TA isoforms of TP63 are tumour suppressor genes and that the ΔN isoforms of TP63 are oncogenes, which antagonize p53, TAp63 and TAp73 by inhibiting their ability to transactivate downstream target genes8,9,12,15. Further in vivo work has recently demonstrated that the TAp63 and TAp73 isoforms are tumour suppressors in mice8–10. Both TP63 and TP73 also contain several carboxy-terminal spliced isoforms (fig. 1). These isoforms transactivate target genes to varying degrees, which then induce cellular functions, such as apoptosis, cell cycle arrest and senescence8,12,15,16. The TAp63γ and TAp73β isoforms, which most closely resemble p53, are the most transcriptionally active isoforms to induce apoptosis12,15,17. The activity of each isoform in other biological processes is still not completely understood but is an area of intense investigation.
Figure 1. p53, p63 and p73 sequence and structural similarity.
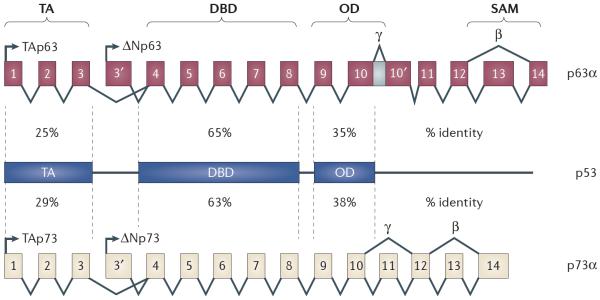
TP63 and TP73 are composed of multiple isoforms that can be placed in two categories: the TA isoforms, which contain an acidic transactivation (TA) domain that is encoded by the first three exons; and the ΔN isoforms, which lack this amino-terminal domain. Shown for both TP63 and TP73 are the α-, β- and γ-isoforms, which are determined by alternative splicing of the carboxyl terminus. TP53, TP63 and TP73 share sequence nucleotide similarity in three regions: the TA domain, the DNA binding domain (DBD) and the oligomerization domain (OD). Numbers shown represent shared percentage identity between TP53, TP63 and TP73. Only the p63α and p73α isoforms contain a sterile alpha motif (SAM) domain in both the TA and the ΔN isoforms.
Much of the research carried out on p63 has focused on its role in the morphogenesis of epidermal tissues. Mice lacking all isoforms of Trp63 (which encodes p63 in mice) do not have complete stratified epithelia13,14, and the mechanisms used by p63 to regulate epithelialization have important implications for cancer and metastasis. In this Opinion article, we summarize the current literature on the p53 family, with an emphasis on p63 and its roles in cancer, metastasis, DNA damage, ageing and stem cell biology. We also discuss the intricate interplay of p63 with the other p53 family members.
Functions of p63: in vitro studies
Transcriptional activity
p53 induces multiple cellular processes, including apoptosis and cell cycle arrest in response to DNA damage, by transactivating a large network of genes, including BCL-2-associated X protein (BAX), BCL-2 binding component 3 (BBC3; also known as PUMA), PMAIP1 (also known as NOXA), PERP and cyclin-dependent kinase inhibitor 1A (CDKN1A; which encodes p21)12,15. These biological activities are important for the function of p53 in the DNA damage response and for its function as a tumour suppressor. New mouse models with novel p53 mutations have revealed the crucial activities and transcriptional targets of p53 that are necessary for its function in tumour suppression18,19. Mice with mutations in one of the transactivation domains of p53 (refs 25,26) retained the ability to transactivate genes that are involved in senescence and were still able to suppress tumorigenesis. This form of p53 is unable to induce G1 phase arrest and apoptosis, indicating that these functions of p53 are dispensable for tumour suppression18. More recently, the role of p53 in metabolism was revealed to be key to its tumour suppressive function. This was demonstrated using a mouse model harbouring three mutations at acetylation sites of p53 (p53 3KR)19. This mutant form of p53 fails to induce target genes that are involved in cell cycle arrest, apoptosis and senescence, but mice expressing this p53 mutant do not succumb to the rapid development of thymic lymphomas that is seen in p53-deficient mice20,21. This form of p53 retains its tumour suppressive function through its ability to transactivate glutaminase 2 (GLS2), which encodes an enzyme that regulates glutamine use and promotes mitochondrial respiration and ATP production.
TAp63 shares structural and sequence homology with p53 (fig. 1) and TAp73. Consequently, TAp63, p53 and TAp73 bind as tetramers to similar consensus sequences to transactivate downstream target genes12,17,22,23. Each can form homotetramers, and TAp63 and TAp73 can also form heterotetramers22. The TA isoforms of p63 are potent transactivators of the well-known p53 target genes CDKN1A, BAX and MDM2 (fig. 2), whereas the ΔNp63 isoforms do not transactivate these targets to high levels12. The ΔNp63 isoforms also have the ability to transactivate downstream target genes such as CDKN1A, resulting in the inhibition of cellular proliferation24. This transcriptional activation is possible through a proline-rich domain that is located at the amino terminus of ΔNp63 isoforms24, and the mechanisms of p53 family target selectivity and transcriptional activity is an area of much investigation in the field.
Figure 2. Extensive interaction between p53 and TAp63 in response to DNA damage.

a Subsequent to DNA damage, which can be induced through treatment with ionizing radiation, doxorubicin or cisplatin, p53, TAp63 and TAp73 activate downstream transcriptional targets to induce multiple cell fates. p53, TAp63 and TAp73 can transcriptionally activate cyclin-dependent kinase inhibitor 1A (CDKN1A; which encodes p21) individually to induce cell cycle arrest. b | Subsequent to DNA damage, TAp63 and TAp73 are required in complex with p53 to transcriptionally activate genes that are involved in apoptosis, including BCL-2 binding component 3 (BBC3; also known as PUMA), BCL-2-associated X protein (BAX), PMAIP1 (also known as NOXA) and PERP. c | p53, TAp63 and TAp73 transactivate genes that are involved in metabolism. TAp63 transactivates sirtuin 1 (SIRT1), STK11 (also known as LKB1) and PRKAA2 (also known as AMPKA2) to regulate glucose and lipid metabolism. TAp73 transactivates cytochrome c oxidase subunit IV isoform 1 (COX4I1), which is a subunit of the mitochondrial multimeric enzyme that executes the last step in aerobic respiration. p53 transactivates C12ORF5 (also known as TIGAR) and glutaminase 2 (GLS2), which are crucial for its function as a tumour suppressor.
Differences between p53 and p63 binding to target sequences have been found, and these are due to differences in the DNA binding motif and in the three-dimensional (3D) structures of p53 and p63. The p53 consensus binding site contains two repeats of ten base pairs known as half-sites (RRRC(A/T) (A/T)GYYY) that are separated by a spacer sequence of 0–13 base pairs25. Tetramerization of p53 is dependent on the oligomerization domain that is present in the C terminus. p53 tetramers are composed of symmetric dimers of dimers that each binds a half-site. Similarly, both p63 and p73 form homotetramers, and in some cases hetero tetramers, with each other22,23. The optimal p63 DNA binding consensus motif was found to differ slightly from that of p53: it is enriched for CATG in the cores of both half sites (RRRCATGYYY)17. These data suggest that the p63 isoforms may have distinct transcriptional targets from p53 (refs 9,16,17,26,27). Indeed, several unique targets of p63 have been identified using Trp63−/− mouse embryonic fibroblasts (MEFs) or immortalized human keratinocytes (HaCaT cells) and high-throughput technologies, such as cDNA and microRNA (miRNA) microarrays and chromatin immunoprecipitation–microarray (ChIP–chip)17,23,27,28. TAp63 can transactivate genes that are involved in miRNA biogenesis (such as DICER1)9, in the cell cycle (Cdkn1c; which encodes p57)16 and in epidermal stratification (transcription factor AP2γ (TFAP2C))29. ΔNp63 isoforms transactivate genes that are involved in epidermal morphogenesis17,28,30. Interestingly, the ΔNp63 and ΔNp73 isoforms were also found to be potent transcriptional activators of genes that are involved in DNA repair, including Brca2, Rad51 and Mre11a27. Moreover, ΔNp63 cooperates with ΔNp73 for full transactivation of these genes27. These data indicate that the ΔN isoforms of both p63 and p73 may also have tumour suppressive functions through their ability to transactivate genes that are crucial for DNA repair. Although data from in vitro analyses of tissue culture cells support this conclusion, further in vivo data using mouse models are required to fully understand the potential tumour suppressive activities of ΔNp63. Such in vivo analyses have been carried out to understand the functions of the TAp63 isoform using mouse models, but the potential tumour phenotypes of ΔNp63- and ΔNp73-deficient mice have not been fully studied.
In addition to the transactivation activity of the ΔNp63 isoforms, they can also antagonize p53 and the TA isoforms of p63 and p73. This is because interactions between isoforms of p63, p73 and p53 result in complex biological responses. ΔNp63 can inhibit p53, TAp63 and TAp73 transcriptional activity in a dose-dependent manner12 by competing for DNA consensus sites23. The inhibition of p53, TAp63 and TAp73 suggests that the ΔNp63 and ΔNp73 isoforms have oncogenic potential. Indeed, ΔNp63 and ΔNp73 can inhibit apoptosis in cells in vitro12,31,32. The TAp63 and TAp73 isoforms can also be inhibited through interactions with cancer-associated p53 mutants33,34. These interactions result in the inability of the TA isoforms to induce tumour suppressive cellular processes, such as apoptosis, through the formation of protein complexes that are unable to bind to DNA at the promoters of target genes23,33,34. This complex interplay between the p53 family members indicates the need for a clear mechanistic understanding of the activities of this family in DNA damage responses.
The 3D structure formed by p53 and p63 monomers affects DNA binding affinity and downstream transcriptional target selectivity. The p63 monomer structure is similar to that of p53 with the exception of the LB-2 and L1 loops, which are necessary for dimer–dimer interface interactions35. Complex regulation by the conversion of TAp63α dimers to tetramers was recently found to be crucial for the response to DNA damage in mouse oocytes36. In addition to DNA binding sequences and 3D structure, cofactors, such as p300, which bind to p63, p53 and p73, determine the specificity and the selectivity of downstream transcriptional targets37,38.
Post-translational modifications
Post-translational modifications of p63, p73 and p53 have important roles in the induction and the stabilization of the proteins. In response to genotoxic stress, both p53 and TAp63 rapidly accumulate1,15. The accumulation of p53 is due to the inability of MDM2 to bind and degrade phosphorylated p53 (ref. 39). p63 and p73 can also interact with MDM2 and MDM4 (also known as MDMX). However, MDM2 and MDM4 do not target either p63 or p73 for degradation40,41. The interaction of TAp63 with MDM2 results in the translocation of TAp63 to the cytoplasm, which in turn inhibits apoptosis42. Biological roles for TAp63 in the cytoplasm have not yet been identified. Like p53, TAp63 and TAp73 are also phosphorylated in response to DNA damage43–47. For example, TAp63γ is phosphorylated by inhibitor of nuclear factor-κB kinase-β (IKKβ) in response to ionizing radiation, resulting in the stabilization of TAp63γ45. TAp73 is phosphorylated by the tyrosine kinase ABL (also known as ABL1) in response to genotoxic stress that is induced by cisplatin43,44,46,47. The full complement of post-translational modifications for p63, p73 and their various isoforms is still an area that has not been extensively investigated.
Although MDM2 and MDM4 are not crucial for p63 turnover, other E3 ubiquitin ligases have been shown to target p63 for ubiquitylation and subsequent degradation. Both TAp63α and ΔNp63α are targeted for ubiquitylation by the E3 ubiquitin ligase ITCH48. Isoforms of p73 are regulated in a similar manner49. The regulation of ΔNp63 seems to be coordinated in a more sophisticated manner by two scaffold proteins, syntaxin-binding protein 4 (STXBP4) and receptor of activated kinase C1 (RACK1; also known as GNB2L1), which bind to ΔNp63 (ref. 50). On genotoxic stress, STXBP4 is downregulated and this results in ΔNp63 destabilization through RACK1, a scaffold protein that adopts a seven-bladed β-propeller structure to facilitate binding to E3 ubiquitin ligases. This binding in turn targets ΔNp63 for proteasomal degradation50.
Tissue and cellular specificity
The complexity of p53 family member interactions in response to DNA damage and apoptosis was revealed using mice deficient for Trp53, Trp63 and Trp73 (ref. 15) (fig. 2). p53 was found to require the expression of p63 and p73 to induce target genes that are involved in apoptosis in the developing brain and in MEFs in response to DNA damage15. However, the requirement of the whole family for the induction of apoptosis seems to be tissue dependent. Thymocytes that were deficient for Trp63 and Trp73 were as sensitive to genotoxic stress as wild-type thymocytes and they underwent apoptosis, indicating that p53 is functional in this context51. The differing results of the two studies are probably due to differences in tissue expression between the p53 and the p63 isoforms. Although p53 is ubiquitously expressed, p63 is mostly expressed in epithelial tissues13,14, neurons52 and the germ line53,54. Another group of researchers found that p63 is not detectable in neurons and that p73 is essential for the development of the central nervous system (CNS)55. Although this result indicates that p63 is not necessary for CNS development, it does not exclude the possibility that p63 may have a role in the response to stress in this tissue. In addition, TAp63 isoforms are expressed in response to DNA damage or other stresses in the epithelium, neurons and the germ line15,53,54,56, whereas ΔNp63 isoforms are constitutively expressed in the basal compartment of the epidermis and in other epithelial tissues and are not induced by DNA damage13,14,30. Conversely, ΔNp63 is degraded in response to genotoxic stress50. These spatial and temporal differences in the expression of the p53 family members and the p63 isoforms lead to differences in the transcriptional programme that is activated by each.
Examples of target gene specificity and selectivity by distinct p63 isoforms have been best demonstrated in the skin and germ line16,30,54. In the skin, TAp63 is expressed in stem cells known as skin-derived precursors (SKPs), which are derived from SOX2-positive hair follicle dermal cells16. Conversely, ΔNp63 is expressed in transiently amplifying cells in the basal layer of the epidermis30. This expression pattern results in the transactivation of specific target genes, Cdkn1c for TAp63 in SKPs16 and keratin 14 (Krt14) by ΔNp63 in basal cells of the epidermis30. An additional example of tissue specificity is in the female germ line: p63 has a crucial role in maintaining the genome of mouse oocytes, and these functions are independent of p53 (ref. 54). In this regard, TAp63 induces apoptosis through transcriptional activation of Bbc3 and Pmaip1 in a p53-independent manner54. More recently, a unique isoform of TAp63 that is present only in human and ape testes was found to protect the male germ line by inducing apoptosis53. Thus, TAp63 has now been dubbed the `guardian' of the germ line.
Similarly, p73 is expressed in specific tissues, including in neurons, lung, kidney and pancreas7,10,57. Studies of mice expressing mutant p73 have revealed that p73 has important roles in the development of the CNS58, neurodegeneration59 and in cancer7,11. More specifically, TAp73 has been found to be crucial for the maintenance of neural stem cells57,60–63. Additionally, mice lacking TAp73 are prone to the development of lung adenocarcinoma10. Collectively, these studies highlight the importance of understanding the p53 family as a whole in response to various DNA-damaging agents in different tissue and cellular contexts.
Functions of p63: in vivo mouse models
The in vitro experiments carried out in tissue culture cells have provided crucial information about the function of the different isoforms of p63. The TAp63 isoforms have been shown to activate transcriptional target genes that induce apoptosis, which is consistent with a role for these isoforms as tumour suppressors12,15. By contrast, the roles of the ΔNp63 isoforms seem to be more complex. The ΔNp63 isoforms can inhibit p53, TAp63 and TAp73 function, which is consistent with a role for the ΔNp63 isoforms as oncogenes12. The oncogenic potential of ΔNp63α was demonstrated in a cellular model in which this p63 isoform was shown to bypass HRAS-G12V-induced senescence to promote the proliferation of a KRT15-positive stem cell population in the skin64. The ΔNp63 isoforms can transactivate genes that are involved in DNA repair27, which is consistent with a role for these isoforms as tumour suppressors. Additionally, some human tumours have been reported to overexpress ΔNp63 isoforms (Supplementary information S1 (table)), consistent with oncogenic roles, and others have been shown to lose expression of ΔNp63 isoforms, further indicating that these isoforms may also function as tumour suppressor genes65. Recent mouse models that were engineered to express p63 mutations have revealed important and unique p53-independent functions of p63 that are associated with cancer. TAp63 has crucial roles in maintaining stem cells in quiescence and in preventing premature ageing16, as well as in senescence8,9,16. These activities are crucial for TAp63 to function as a tumour suppressor. Mice engineered with knocked down30 or total loss of expression66 of ΔNp63 revealed that ΔNp63 is crucial for the terminal differentiation of cells in the epidermis. The cancer-associated phenotypes of these mouse models of ΔNp63 have not yet been reported.
p63 in the suppression of tumorigenesis and metastasis
Mouse models with p63 mutations have revealed that TAp63 is a suppressor of tumorigenesis and metastasis7–9. This was first demonstrated in double-heterozygous Trp53+/−(tm1Tyj)Trp63+/−(tm1Fmc) mice. Of the tumours that developed in these mice, 90% metastasized in contrast to the 5% frequency of metastatic disease in Trp53+/−(tm1Tyj) mice7. Isoforms of p63 can transcriptionally activate genes that are involved in cell cycle arrest and apoptosis. It is thought that the loss of these cellular functions is partially responsible for the tumour phenotype of the Trp53+/− (tm1Tyj)Trp63+/−(tm1Fmc) mice. Importantly, mice engineered to carry mutations of p53 (Trp53+/R515A(tm3.1Glo), Trp53R172H/+(tm2.1Tyj) and Trp53R270H/+(tm3.1Tyj)) in the DNA binding domain that are frequently detected in patients with Li–Fraumeni syndrome, a rare autosomal dominant hereditary disorder that results in tumour predisposition, had a similar tumour and metastatic phenotype to the Trp53+/−(tm1Tyj)Trp63+/−(tm1Fmc) mice67–69 (table 1), indicating that these mutations of p53 are gain of function. The increased metastasis was ascribed to the ability of mutant p53 to bind and inactivate the transcriptional activation of target genes such as Cdkn1a by p63 (refs 67–69). MEFs derived from mutant p53 mice (Trp53R172H/R172H or Trp53R270H/R270H) are similarly transformed to MEFs that are deficient for all three p53 family members. These MEFs were generated from Trp53-deficient embryos, and small interfering RNA (siRNA) was used to knock down p63 and p73 expression7,67–69. These data further suggest that the p53 family as a whole is involved in the suppression of tumorigenesis. More recently, p53 and p63 were shown to extensively interact to suppress metastasis. Transforming growth factor-β (TGFβ)-dependent migration, invasion and metastasis of breast cancer cells were driven by mutant p53 through the inhibition of p63 (ref. 70) (fig. 3). Moreover, mutant p53 was found to drive invasion by promoting integrin recycling through the inhibition of TAp63 (ref. 71) (fig. 3). This mechanistic insight into p53–TAp63 interplay in the suppression of metastasis highlights the importance of understanding the mechanistic roles of TAp63 in the suppression of tumorigenesis and metastasis.
Table 1.
Mouse models of p63 in cancer
| Genotype* | Phenotype | Refs | |
|---|---|---|---|
| Cancer | Development | ||
| Trp63+/−(tm1Fmc) | Tumour prone; develops squamous cell hyperplasia, sarcomas and carcinomas | Premature ageing | 7 |
| Trp63+/−(tm1Brd) | Not tumour prone; develops epithelial hyperplasia | Premature ageing | 73 |
| Trp63−/−(tm1Brd) | Unknown | No limbs, no epidermis, cleft palate and perinatal death | 13 |
| Trp63−/−(tm1Fmc) | Unknown | No limbs, no epidermis, cleft palate and perinatal death | 14 |
|
Trp53+/−(tm1Tyj) Trp63+/−(tm1Fmc) |
Tumour prone; develops multiple carcinomas and metastatic sarcomas and carcinomas | Premature ageing | 7 |
|
Trp53+/−(tm1Brd) Trp63+/−(tm1Brd) |
Reduced tumorigenesis | Premature ageing | 73 |
| TAp63+/−(tm1.1Elrf) | Tumour prone; develops multiple carcinomas and metastatic tumours | None | 9 |
| TAp63−/−(tm1.1Elrf) | Tumour prone; develops multiple carcinomas and metastatic tumours | Develops skin blisters, has a wound-healing defect and exhibits premature ageing, diabetes and obesity | 9, 16, 96 |
| TAp63−/−(tm2Fmc) | Unknown | Oocytes are sensitive to DNA damage | 54 |
|
TAp63fl/fl(tm1Elrf) Krt14Cre+ |
Unknown | No phenotype | 16 |
|
TAp63+/−(tm1.1Elrf) Trp53+/−(tm1Tyj) |
Tumour prone; develops metastatic sarcomas and carcinomas | No phenotype | 9 |
|
TAp63−/−(tm1.1Elrf) Trp53+/−(tm1Tyj) |
Tumour prone; develops sarcomas, carcinomas and metastatic carcinomas | Develops skin blisters, has a wound-healing defect and exhibits premature ageing | 9 |
|
Trp53+/R515A(R172H) (tm3.1Glo) |
Tumour prone; develops sarcomas and metastatic sarcomas | No phenotype | 68 |
|
Trp53R172H/+(tm2.1Tyj) or Trp53R270H/+(tm3.1Tyj) |
Tumour prone; develops sarcomas and carcinomas, and metastatic sarcomas and carcinomas | No phenotype | 69 |
Krt14, keratin 14.
Mouse nomenclature of mouse genome informatics (MGI) is indicated: Trp63+/− (tm1Fmc), Trp63-knockout mouse generated by F. McKeon (Fmc); Trp63+/−(tm1Brd), Trp63-knockout mouse generated by A. Bradley (Brd); Trp53+/−(tm1Tyj), Trp53-knockout mouse generated by T.Jacks (Tyj); Trp53+/−(tm1Brd), Trp53-knockout mouse generated by Brd; TAp63−/−(tm1.1Elrf), TAp63-knockout mouse generated by Elsa R. Flores (Elrf); TAp63−/−(tm2Fmc), TAp63-knockout mouse generated by Fmc; TAp63fl/fl(tm1Elrf), TAp63-conditional-knockout mouse generated by Elrf; Trp53+/R515A(R172H)(tm3.1Glo), p53-R172H knock-in mouse generated byG. Lozano(Glo); Trp53R172H/+(tm2.1Tyj), p53-R172H knock-in mouse generated by Tyj; Trp53R270H/+(tm3.1Tyj), p53-R270H knock-in mouse generated by Tyj.
Figure 3. Mechanisms used by TAp63 to suppress metastasis.
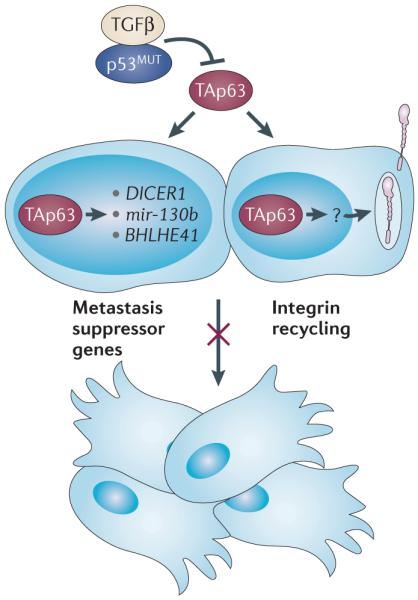
TAp63 suppresses metastasis by transcriptionally activating metastasis suppressor genes or microRNAs, including DICER1, mir-130b and basic helix–loop–helix family, member e41 (BHLHE41; also known as SHARP1). TAp63 is also crucial to induce other, as yet unknown, target genes (indicated by a question mark) that suppress metastasis and that are involved in integrin recycling. Mutant p53 (p53MUT) and transforming growth factor-β (TGFβ) can inhibit the metastasis-suppressive activities of the TAp63 isoforms.
To further investigate the mechanisms of tumour suppression by TAp63, the creation of TAp63−/− mice was essential. Indeed, TAp63−/− mice developed metastatic mammary and lung adenocarcinoma, and squamous cell carcinoma, with metastases to the lung, liver and brain9. TAp63 was found to suppress metastasis through the transcriptional activation of Dicer1 and the miRNA mir-130b9 (fig. 3). Given the extensive network of genes regulated by miRNAs, the finding that TAp63 transcriptionally regulates Dicer1 and mir-130b has many important implications for the role of TAp63 isoforms in multiple biological processes. Understanding the miRNA network that is regulated by TAp63 isoforms in individual cancers could be important for designing targeted therapy for metastatic cancers.
p63 in longevity
Ageing is a risk factor for the development of cancer. The incidence of cancer rises exponentially after the age of 50 years. Although ageing is known to accelerate the frequency of tumorigenesis, the cellular activities that are associated with ageing, such as senescence, can also have a tumour suppressive role in certain tissues9,18,72,73. This dichotomy of senescence and ageing in tumour suppression has also been demonstrated using knockout mouse models of Trp63. Several of these mouse models have revealed that premature senescence or ageing leads to reduced tumour incidence (table 1). In another Trp53+/−(tm1Brd)Trp63+/−(tm1Brd) mouse model, fewer tumours developed compared with Trp53+/−(tm1Brd) mice72,73. The reason cited was the premature ageing of Trp53+/−(tm1Brd)Trp63+/−(tm1Brd) mice72,73. Although this result is at odds with the previously reported Trp53+/−(tm1Tyj) Trp63+/−(tm1Fmc) mice7, which had an increase in tumour incidence and metastasis, more recently published data help to shed light on the differing phenotypes of these mouse models that harbour different alleles of Trp53 and Trp63. First, it has been reported that the Trp63−/−(tm1Brd) mice express some isoforms of p63 (ref. 74). This complicates the original interpretation of the phenotypes of the Trp53+/−(tm1Brd)Trp63+/−(tm1Brd) mouse model, which showed a diminution of tumorigenesis on a Trp53+− background73. Expression of some p63 isoforms in Trp53+/− (tm1Brd)Trp63+/−(tm1Brd) mice may have led to the tumour resistance observed in these mice, suggesting that these p63 isoforms suppress tumorigenesis on a Trp53+/− background. However, recent data indicate that the premature ageing phenotype that is detected in TAp63−/− mice can lead to a decrease in the aggressiveness of osteosarcomas9, and this is in agreement with the reduction in tumour development that is observed in Trp53+/−(tm1Brd)Trp63+/−(tm1Brd) mice73 in which premature ageing has also been observed. Paradoxically, the TAp63−/−Trp53+/− mice exhibited phenotypes of the Trp53+/− (tm1Tyj)Trp63+/−(tm1Fmc) mice, which were reported to have a more aggressive phenotype than Trp53+/−(tm1Tyj) mice, and phenotypes of the Trp53+/−(tm1Brd)Trp63+/−(tm1Brd) mice, which were reported to have a less aggressive phenotype than Trp53+/−(tm1Brd) mice. Analysis of the TAp63−/−Trp53+/− mice has shed some light on this issue. On the one hand, tissues with high levels of senescence, such as those from which sarcomas are derived, were less aggressive — in agreement with the tumour spectrum of the Trp53+/− (tm1Brd)Trp63+/−(tm1Brd) mice. On the other hand, epithelial tissues from TAp63−/−Trp53+/− mice did not have a senescent phenotype and had a marked increase in genomic instability9. Consequently, carcinomas derived from these tissues were aggressive and metastatic, similar to the tumour phenotypes of Trp53+/−(tm1Tyj) Trp63+/−(tm1Fmc) mice. Taken together, these data indicate that premature ageing that is induced by the total loss of TAp63 results in the suppression of metastatic osteosarcomas but also in the acceleration of aggressive and metastatic carcinomas. Therefore, ageing tissues seem to have differing sensitivities to tumour formation, and understanding the molecular mechanisms of tumour suppression by p63 and the other p53 family members in different tissue contexts is crucial to treating cancer effectively.
Further studies are required to fully understand the complex function of TAp63 isoforms in tumour suppression in various tissue and cellular contexts. Additionally, the mammary adenocarcinoma phenotypes of the Trp53+/−(tm1Tyj)Trp63+/−(tm1Fmc) mice suggest that ΔNp63 isoforms may also have a tumour suppressive role, perhaps through the ability of ΔNp63 isoforms to transactivate genes in the DNA repair pathway (Brca2, Mre11a and Rad51)27. Further experiments using ΔNp63-deficient mice are needed to test this possibility.
p63 in the maintenance of stem cells in epithelial tissues
The role of p63 in skin stem cells has been another controversial area. p63 is highly expressed in the basal layer of the epithelium where stem cells and transient-amplifying cells reside13,14,56. The analysis of multiple mouse models that lacked all isoforms of p63 led to the conclusion that p63 has crucial roles in epithelial morphogenesis and that it is necessary either for stem cell renewal14,56,75 or for terminal differentiation13,30. Some researchers have further demonstrated, using keratinocytes that were deficient for p63, that it is crucial for stem cell proliferation75, and others have argued that p63 is dispensable for this function but that it is crucial for terminal differentiation30. The most highly expressed isoform of p63 in the basal layer of epithelial tissues is ΔNp63α (fig. 4). Studies have shown that ΔNp63 transcriptionally activates genes that are required for terminal differentiation, such as KRT14, suppressor of fused homologue (SUFU), homeobox C4 (HOXC4) and myelin protein zero-like 2 (MPZL2; also known as EVA1)17,28,30. Importantly, TAp63 and ΔNp63 also transcriptionally activate PERP, which is crucial for the assembly of desmosomal adhesive complexes and epidermal integrity15,76. Recently, the function of TAp63 in stem cells in the skin has been more carefully characterized. Using TAp63−/− mice, TAp63 was found to maintain dermal stem cells in quiescence through the transcriptional activation of Cdkn1c (which encodes p57)16 (fig. 4). SOX2-positive hair follicle dermal stem cells (SKPs) deficient for TAp63 are hyperproliferative in vitro and in vivo and have high levels of genomic instability16. These data provide at least one mechanism by which TAp63 suppresses tumorigenesis: through the suppression of the aberrant proliferation of adult stem cells, such as SKPs. Another mechanism for tumour suppression by TAp63 is through its ability to induce senescence in response to oncogenic stress8. Indeed, TAp63−/− mice are prone to developing metastatic cutaneous squamous cell carcinomas9, and these may arise from hyperproliferating SKPs, in which TAp63 has an essential role in maintaining quiescence.
Figure 4. TAp63 and ΔNp63 are expressed in discrete areas of the dermis and epidermis.

A cross-section through a hair follicle and the surrounding epidermis is shown. TAp63 is expressed in the dermal sheath and the dermal papilla of the hair follicle in dermal stem cells, which are known as skin-derived precursor cells (SKPs). TAp63 transcriptionally regulates cyclin-dependent kinase inhibitor 1C (CDKN1C; which encodes p57) to maintain SKPs in quiescence. ΔNp63 is not expressed in SKPs but is expressed in the basal cells of the epidermis and is crucial for the proliferation and terminal differentiation of these cells.
Taken together, the roles of the p63 isoforms in epithelial stem cells have important implications for cancers arising from epithelial tissues. Moreover, the roles of the p63 isoforms in cancer stem cells will probably be an area of further research.
Functions of p63 in human cancer
Multiple studies using samples from human patient-derived tumours have been carried out to determine the status of p63 expression. The results from these studies are summarized in Supplementary information S1 (table). In many studies, researchers have used antibodies raised against the DNA binding domain of p63, and these antibodies do not distinguish between the TA and the ΔN isoforms. Consequently, early studies indicated that p63 is expressed in tumours (Supplementary information S1 (table)), which is consistent with an oncogenic role for p63. The examination of large sets of paraffin-embedded patient samples indicated that p63 is expressed in the nucleus of cells from many tumour types, including multiple types of head and neck cancers9,77–79, diffuse large B cell lymphoma80 and bladder carcinoma81–86. Although p63 expression was typically located in the nucleus, some cases of prostate carcinoma expressed p63 in the cytoplasm87, suggesting that p63 may not be functional in these tumours or that p63 may have cytoplasmic roles, as has been reported for p53 (ref. 88). Interestingly, in a study of 2,158 oestrogen receptor (ER)-positive breast cancers, low-grade tumours expressed p63, whereas high-grade tumours were devoid of p63 expression89, suggesting that p63 is involved in suppressing tumour progression in these tumours.
Studies using antibodies or PCR primers that distinguish between the TA and the ΔN isoforms of p63 in human patient tumour samples showed that the TA isoforms of p63 either are not expressed or are expressed at very low levels9,77. Conversely, the ΔNp63 isoforms are expressed at high levels78,83,90. These expression patterns are consistent with TAp63 functioning as a tumour suppressor gene and ΔNp63 functioning as an oncogene. Importantly, there are several examples of tumours in which the loss of TAp63 expression has been found in invasive and metastatic lesions, including bladder carcinoma, mammary and lung adenocarcinoma, and head and neck squamous cell carcinoma, which is consistent with its function as a metastasis suppressor (Supplementary information S1 (table)). Low expression of TAp63 frequently coincides with high expression of ΔNp63 in lesions that have progressed.
Although p53 is commonly mutated in human cancer, the mutation status of p63 has not been well studied. Some p63 mutations, which are located in the DNA binding domain or in the transactivation domain and that inhibit the transactivation of p63 target genes, are associated with various syndromes, including ectrodactyly, ectodermal dysplasia and cleft lip/palate syndrome (EEC), ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC), limb mammary syndrome (LMS), acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome and Rapp–Hodgkin syndrome (RHS). Interestingly, these patients do not seem to be prone to developing tumours91, suggesting that mutations of p63 are not driver mutations in cancer. However, some mutations of p63 have been identified in patient tumour samples in the DNA binding domain in exon 4 of TP63 (Supplementary information S1 (table)), which affect the transactivation of both the TA and the ΔN isoforms of p63 (ref. 92). With the advent of low-cost high -throughput sequencing, additional mutations and alterations in p63 are likely to be unveiled throughout the gene in a similar manner to TP53. Much like p53, there will probably be mutations that affect the ability of p63 isoforms to bind to DNA and there could also be others in regions that disrupt protein–protein interactions that are crucial for the downstream transcriptional activation of target genes that function in cellular processes that suppress tumorigenesis. Furthermore, it will be important to generate mutations in p63 in mice that will allow further dissection of the regions and activities of the p63 protein, which will allow us to determine the biological functions of p63 that are crucial for tumour suppression, as has been done for p53 (refs 18,19).
Conclusions and future directions
It is clear from the multiple cellular processes regulated by members of the p53 family through the transcriptional regulation of downstream target genes that they are important suppressors of tumorigenesis. The ability of the family members to induce apoptosis, cell cycle arrest, senescence and metabolic regulation in response to environmental stresses is key to the suppression of tumour formation. The complex interplay between the p53 family impinges on the activity of individual isoforms of the p53 family to suppress tumorigenesis. Although p53, TAp63 and TAp73 can all induce apoptosis and cell cycle arrest through the transactivation of target genes, the ΔN isoforms of p63 and p73 can inhibit these activities12. These activities suggest that p53, TAp63 and TAp73 are tumour suppressors, whereas the ΔNp63 and ΔNp73 isoforms are oncogenic. Although there is still much left to understand about the complexes of p53 family members that are formed in different contexts and the downstream consequences for tumour suppression, it has become clear from mouse models that are deficient in Trp73 and Trp63, as well as from human tumour samples, that TAp73 and especially TAp63 are suppressors of metastasis7,9,70,71. Moreover, recent studies have shown that both TAp73 (refs 93,94) and TAp63 (refs 63,95,96) have important roles in metabolism. In the future, it will be interesting to understand how their functions in metabolism impinge on their tumour suppressive activities.
How does p63 suppress metastasis? We know that p63 has crucial roles in epithelial biology. Functions for p63 have been found in epithelial differentiation13, adhesion13,14,76, stem cell maintenance16 and proliferation56,75. These functions in epithelial tissues have important consequences for the formation of a carcinoma and the cellular movements that are required for the multiple steps in metastasis. It is important to be conscious of the TAp63 and ΔNp63 isoforms and their known activities in the skin. TAp63 maintains stem cells in the dermis in quiescence, but ΔNp63 is not expressed and has no role in these cells16. At the same time, ΔNp63 induces terminal differentiation of transiently amplifying cells in the epidermis, whereas TAp63 is dispensable for this function30,66. This exemplifies how p63 isoforms have different functions in the same tissue, and there is a need to understand how these activities impinge on the cancer phenotypes that are observed in Trp53+/− (tm1Tyj)Trp63+/−(tm1Fmc) mice. For example, the regulation of cellular activities by each TAp63 and ΔNp63 isoform in these adult skin stem cells could be key to understanding the mechanisms at work in the progression of the squamous cell carcinomas that are observed in Trp53+/−(tm1Tyj)Trp63+/−(tm1Fmc) mice and the TAp63 and ΔNp63 expression patterns observed in human SCC. Additionally, mounting evidence indicates that stem cells and the epithelial -to -mesenchymal transition (EMT) have important roles in tumour progression and metastasis97, making it important to understand how each p63 isoform may regulate processes in tumour progression, particularly in carcinomas, in which TAp63 and ΔNp63 expression is altered. Last, key p63 target genes that have important roles in epithelial adhesion and integrity, such as PERP, have been shown to be involved in the suppression of tumorigenesis and metastasis98. PERP is primarily transactivated by TAp63 isoforms, revealing further complex regulation of target genes by individual p63 isoforms. As PERP is a desmosome -associated protein, this provides further evidence that understanding the roles of both TAp63 and ΔNp63 in cellular adhesion is key to unlocking the functions of p63 in metastasis.
Future work in the p63 field should aim to understand the molecular mechanisms of each individual isoform in regulating epithelial biology and the roles of these mechanisms in cancer development and metastasis. It will be crucial to identify the protein complexes that are formed in the family and the downstream consequences of such interactions in various tissues and contexts. Moreover, it will be crucial to integrate the pathways that are regulated by p63 with those that are regulated by p53 and p73. Such information can now be easily obtained by high -throughput sequencing using RNA–seq and ChIP–seq technologies coupled with antibodies for p53 family member isoforms in various cellular and biological contexts. A complete picture of the molecular biology that is regulated by the whole p53 family is emerging and is crucial for targeting this pathway therapeutically in human cancer.
Supplementary Material
Acknowledgements
E.R.F. is a scholar of the Leukemia and Lymphoma Society of America, the Rita Allen Foundation and the V Foundation for Cancer Research. E.R.F. is supported by grants from NCI (R01CA160394), NCI (R01CA134796), CPRIT (RP120124), the Mel Klein Foundation, and the Hildegardo E. and Olga M. Flores Foundation. D.C. is a CPRIT Scholar (RP101502).
Footnotes
Competing interests statement The authors declare no competing financial interests.
References
- 1.Lane DP. Cancer p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 3.Kaghad M, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 4.Osada M, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nature Med. 1998;4:839–843. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- 5.Schmale H, Bamberger C. A novel protein with strong homology to the tumor suppressor p53. Oncogene. 1997;15:1363–1367. doi: 10.1038/sj.onc.1201500. [DOI] [PubMed] [Google Scholar]
- 6.Trink B, et al. A new human p53 homologue. Nature Med. 1998;4:747–748. doi: 10.1038/nm0798-747. [DOI] [PubMed] [Google Scholar]
- 7.Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Guo X, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nature Cell Biol. 2009;11:1451–1457. doi: 10.1038/ncb1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su X, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–990. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomasini R, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–2691. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemajerova A, Petrenko O, Trumper L, Palacios G, Moll UM. Loss of p73 promotes dissemination of Myc-induced B cell lymphomas in mice. J. Clin. Invest. 2010;120:2070–2080. doi: 10.1172/JCI40331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang A, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 13.Mills AA, et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 14.Yang A, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 15.Flores ER, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 16.Su X, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64–75. doi: 10.1016/j.stem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang A, et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Brady CA, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li T, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 21.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 22.Davison TS, et al. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J. Biol. Chem. 1999;274:18709–18714. doi: 10.1074/jbc.274.26.18709. [DOI] [PubMed] [Google Scholar]
- 23.Yang A, et al. Genome-wide mapping indicates that p73 and p63 co-occupy target sites and have similar dna-binding profiles in vivo. PLoS ONE. 2010;5:e11572. doi: 10.1371/journal.pone.0011572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helton ES, Zhu J, Chen X. The unique NH2-terminally deleted (ΔN) residues, the PXXP motif, and the PPXY motif are required for the transcriptional activity of the ΔN variant of p63. J. Biol. Chem. 2006;281:2533–2542. doi: 10.1074/jbc.M507964200. [DOI] [PubMed] [Google Scholar]
- 25.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nature Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 26.Cho MS, Chan IL, Flores ER. ΔNp63 transcriptionally regulates brachyury, a gene with diverse roles in limb development, tumorigenesis and metastasis. Cell Cycle. 2010;9:2434–2441. doi: 10.4161/cc.9.12.12051. [DOI] [PubMed] [Google Scholar]
- 27.Lin YL, et al. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 2009;5:e1000680. doi: 10.1371/journal.pgen.1000680. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Vigano MA, et al. New p63 targets in keratinocytes identified by a genome-wide approach. EMBO J. 2006;25:5105–5116. doi: 10.1038/sj.emboj.7601375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koster MI, Kim S, Huang J, Williams T, Roop DR. TAp63α induces AP-2γ as an early event in epidermal morphogenesis. Dev. Biol. 2006;289:253–261. doi: 10.1016/j.ydbio.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 30.Koster MI, et al. p63 induces key target genes required for epidermal morphogenesis. Proc. Natl Acad. Sci. USA. 2007;104:3255–3260. doi: 10.1073/pnas.0611376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belloni L, et al. DNp73α protects myogenic cells from apoptosis. Oncogene. 2006;25:3606–3612. doi: 10.1038/sj.onc.1209321. [DOI] [PubMed] [Google Scholar]
- 32.Jost CA, Marin MC, Kaelin WG., Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 33.Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999;19:1438–1449. doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001;21:1874–1887. doi: 10.1128/MCB.21.5.1874-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Gorlatova N, Kelman Z, Herzberg O. Structures of p63 DNA binding domain in complexes with half-site and with spacer-containing full response elements. Proc. Natl Acad. Sci. USA. 2011;108:6456–6461. doi: 10.1073/pnas.1013657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deutsch GB, et al. DNA damage in oocytes induces a switch of the quality control factor TAp63α from dimer to tetramer. Cell. 2011;144:566–576. doi: 10.1016/j.cell.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 38.MacPartlin M, et al. p300 regulates p63 transcriptional activity. J. Biol. Chem. 2005;280:30604–30610. doi: 10.1074/jbc.M503352200. [DOI] [PubMed] [Google Scholar]
- 39.Olsson A, Manzl C, Strasser A, Villunger A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 2007;14:1561–1575. doi: 10.1038/sj.cdd.4402196. [DOI] [PubMed] [Google Scholar]
- 40.Zdzalik M, et al. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle. 2010;9:4584–4591. doi: 10.4161/cc.9.22.13871. [DOI] [PubMed] [Google Scholar]
- 41.Zeng X, et al. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999;19:3257–3266. doi: 10.1128/mcb.19.5.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadakia M, Slader C, Berberich SJ. Regulation of p63 function by Mdm2 and MdmX. DNA Cell Biol. 2001;20:321–330. doi: 10.1089/10445490152122433. [DOI] [PubMed] [Google Scholar]
- 43.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature. 1999;399:809–813. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 44.Gong JG, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 45.MacPartlin M, Zeng SX, Lu H. Phosphorylation and stabilization of TAp63γ by IκB kinase-β. J. Biol. Chem. 2008;283:15754–15761. doi: 10.1074/jbc.M801394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White E, Prives C. DNA damage enables p73. Nature. 1999;399:734–737. doi: 10.1038/21539. [DOI] [PubMed] [Google Scholar]
- 47.Yuan ZM, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814–817. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- 48.Rossi M, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc. Natl Acad. Sci. USA. 2006;103:12753–12758. doi: 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossi M, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Peart MJ, Prives C. Stxbp4 regulates ΔNp63 stability by suppression of RACK1-dependent degradation. Mol. Cell. Biol. 2009;29:3953–3963. doi: 10.1128/MCB.00449-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Senoo M, Manis JP, Alt FW, McKeon F. p63 and p73 are not required for the development and p53-dependent apoptosis of T cells. Cancer Cell. 2004;6:85–89. doi: 10.1016/j.ccr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 52.Jacobs WB, et al. p63 is an essential proapoptotic protein during neural development. Neuron. 2005;48:743–756. doi: 10.1016/j.neuron.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 53.Beyer U, Moll-Rocek J, Moll UM, Dobbelstein M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc. Natl Acad. Sci. USA. 2011;108:3624–3629. doi: 10.1073/pnas.1016201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suh EK, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624–628. doi: 10.1038/nature05337. [DOI] [PubMed] [Google Scholar]
- 55.Holembowski L, et al. While p73 is essential, p63 is completely dispensable for the development of the central nervous system. Cell Cycle. 2011;10:680–689. doi: 10.4161/cc.10.4.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su X, et al. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28:1904–1915. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flores ER. p73 is critical for the persistence of memory. Cell Death Differ. 2011;18:381–382. doi: 10.1038/cdd.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 59.Wetzel MK, et al. p73 regulates neurodegeneration and phospho-tau accumulation during aging and Alzheimer's disease. Neuron. 2008;59:708–721. doi: 10.1016/j.neuron.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 60.Agostini M, et al. p73 regulates maintenance of neural stem cell. Biochem. Biophys. Res. Commun. 2010;403:13–17. doi: 10.1016/j.bbrc.2010.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujitani M, et al. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr. Biol. 2010;20:2058–2065. doi: 10.1016/j.cub.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 62.Gonzalez-Cano L, et al. p73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of p53. Cell Death Dis. 2010;1:e109. doi: 10.1038/cddis.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Talos F, et al. p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ. 2010;17:1816–1829. doi: 10.1038/cdd.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keyes WM, et al. ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164–176. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di Como CJ, et al. p63 expression profiles in human normal and tumor tissues. Clin. Cancer Res. 2002;8:494–501. [PubMed] [Google Scholar]
- 66.Romano RA, et al. ΔNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–782. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iwakuma T, Lozano G, Flores ER. Li-Fraumeni syndrome: a p53 family affair. Cell Cycle. 2005;4:865–867. doi: 10.4161/cc.4.7.1800. [DOI] [PubMed] [Google Scholar]
- 68.Lang GA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 69.Olive KP, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 70.Adorno M, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 71.Muller PA, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 72.Keyes WM, Mills AA. p63: a new link between senescence and aging. Cell Cycle. 2006;5:260–265. doi: 10.4161/cc.5.3.2415. [DOI] [PubMed] [Google Scholar]
- 73.Keyes WM, et al. p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc. Natl Acad. Sci. USA. 2006;103:8435–8440. doi: 10.1073/pnas.0602477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Talos F, Wolff S, Beyer U, Dobbelstein M, Moll UM. Brdm2 - an aberrant hypomorphic p63 allele. Cell Death Differ. 2010;17:184–186. doi: 10.1038/cdd.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Senoo M, Pinto F, Crum CP, McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- 76.Ihrie RA, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120:843–856. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Nylander K, et al. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J. Pathol. 2002;198:417–427. doi: 10.1002/path.1231. [DOI] [PubMed] [Google Scholar]
- 78.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 79.Sniezek JC, Matheny KE, Westfall MD, Pietenpol JA. Dominant negative p63 isoform expression in head and neck squamous cell carcinoma. Laryngoscope. 2004;114:2063–2072. doi: 10.1097/01.mlg.0000149437.35855.4b. [DOI] [PubMed] [Google Scholar]
- 80.Hedvat CV, et al. Expression of p63 in diffuse large B-cell lymphoma. Appl. Immunohistochem. Mol. Morphol. 2005;13:237–242. doi: 10.1097/01.pai.0000142160.52670.ce. [DOI] [PubMed] [Google Scholar]
- 81.Buza N, Cohen PJ, Pei H, Parkash V. Inverse p16 and p63 expression in small cell carcinoma and high-grade urothelial cell carcinoma of the urinary bladder. Int. J. Surg. Pathol. 2010;18:94–102. doi: 10.1177/1066896909359914. [DOI] [PubMed] [Google Scholar]
- 82.Castillo-Martin M, Domingo-Domenech J, Karni-Schmidt O, Matos T, Cordon-Cardo C. Molecular pathways of urothelial development and bladder tumorigenesis. Urol. Oncol. 2010;28:401–408. doi: 10.1016/j.urolonc.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 83.Comperat E, et al. p63 gene expression study and early bladder carcinogenesis. Urology. 2007;70:459–462. doi: 10.1016/j.urology.2007.04.030. [DOI] [PubMed] [Google Scholar]
- 84.Comperat E, et al. Immunohistochemical expression of p63, p53 and MIB-1 in urinary bladder carcinoma. A tissue microarray study of 158 cases. Virchows Arch. 2006;448:319–324. doi: 10.1007/s00428-005-0092-2. [DOI] [PubMed] [Google Scholar]
- 85.Karni-Schmidt O, et al. Distinct expression profiles of p63 variants during urothelial development and bladder cancer progression. Am. J. Pathol. 2011;178:1350–1360. doi: 10.1016/j.ajpath.2010.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stepan A, Margaritescu C, Simionescu C, Ciurea R. E-cadherin and p63 immunoexpression in dysplastic lesions and urothelial carcinomas of the bladder. Rom. J. Morphol. Embryol. 2009;50:461–465. [PubMed] [Google Scholar]
- 87.Dhillon PK, et al. Aberrant cytoplasmic expression of p63 and prostate cancer mortality. Cancer Epidemiol. Biomarkers Prev. 2009;18:595–600. doi: 10.1158/1055-9965.EPI-08-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanker L, et al. Clinical relevance of the putative stem cell marker p63 in breast cancer. Breast Cancer Res. Treat. 2010;122:765–775. doi: 10.1007/s10549-009-0608-6. [DOI] [PubMed] [Google Scholar]
- 90.Crook T, Nicholls JM, Brooks L, O'Nions J, Allday MJ. High level expression of ΔN-p63: a mechanism for the inactivation of p53 in undifferentiated nasopharyngeal carcinoma (NPC)? Oncogene. 2000;19:3439–3444. doi: 10.1038/sj.onc.1203656. [DOI] [PubMed] [Google Scholar]
- 91.Rinne T, Brunner HG, van Bokhoven H. p63-associated disorders. Cell Cycle. 2007;6:262–268. doi: 10.4161/cc.6.3.3796. [DOI] [PubMed] [Google Scholar]
- 92.Cabanillas M, et al. A novel heterozygous point mutation in the p63 gene in a patient with ectodermal dysplasia associated with B-cell leukemia. Pediatr. Dermatol. 2011;28:707–710. doi: 10.1111/j.1525-1470.2011.01474.x. [DOI] [PubMed] [Google Scholar]
- 93.Flores ER, Lozano G. The p53 family grows old. Genes Dev. 2012;26:1997–2000. doi: 10.1101/gad.202648.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rufini A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012;26:2009–2014. doi: 10.1101/gad.197640.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burgess DJ. Metabolism: TAp63 tips the energy balance? Nature Rev. Cancer. 2012;12:736–737. doi: 10.1038/nrc3386. [DOI] [PubMed] [Google Scholar]
- 96.Su X, et al. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012;16:511–525. doi: 10.1016/j.cmet.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beaudry VG, et al. Loss of the p53/p63 regulated desmosomal protein Perp promotes tumorigenesis. PLoS Genet. 2010;6:e1001168. doi: 10.1371/journal.pgen.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.