

Abstract
Lung cancer, mostly nonsmall cell lung cancer, continues to be the leading cause of cancer-related death worldwide. With the development of tyrosine kinase inhibitors that selectively target lung cancer-related epidermal growth factor receptor mutations, management of advanced nonsmall cell lung cancer has been greatly transformed. Improvements in progression-free survival and life quality of the patients were observed in numerous clinical studies. However, overall survival is not prolonged because of later-acquired drug resistance. Recent studies reveal a heterogeneous subclonal architecture of lung cancer, so it is speculated that the tumor may rapidly adapt to environmental changes via a Darwinian selection mechanism. In this review, we aim to provide an overview of both spatial and temporal tumor heterogeneity as potential mechanisms underlying epidermal growth factor receptor tyrosine kinase inhibitor resistance in nonsmall cell lung cancer and summarize the possible origins of tumor heterogeneity covering theories of cancer stem cells and clonal evolution, as well as genomic instability and epigenetic aberrations in lung cancer. Moreover, investigational measures that overcome heterogeneity-associated drug resistance and new assays to improve tumor assessment are also discussed.
Keywords: NSCLC, EGFR, TKIs, drug resistance, tumor heterogeneity
Introduction
Despite many novel cancer treatments developed in the past decades, advanced lung cancer, mostly nonsmall cell lung cancer (NSCLC), is still the leading cause of cancer death and poses a great threat to public health. So far, early resection is still the only way to cure the disease. However, more than two-thirds of the patients are beyond the curable stage at diagnosis.1 The estimated 5-year survival rate of advanced NSCLC is less than 10%, and the median life expectancy is only 4 months if left untreated.1,2 Chemotherapy may slightly prolong survival, but the adverse effects are sometimes unbearable, and the tumor eventually becomes resistant to the drug.3
Based on the “oncogene addiction” theory,4 the development of new compounds targeting tumor-driving pathways (eg, the epidermal growth factor receptor [EGFR] signaling pathway) brings new hope to cancer patients.5 Identification of mutations in these pathways, followed by targeted therapy, gives rise to personalized therapy in several types of cancer.6 In advanced NSCLC, tyrosine kinase inhibitors (TKIs; eg, gefitinib and erlotinib) competitively block EGFR and suppress tumor growth, conferring survival benefits with acceptable adverse effects on patients who have failed chemotherapy.7 In patients with NSCLC and sensitive EGFR mutations, TKIs are recommended as first-line treatments because of their significantly increased response rates and prolonged progression-free survival in this group of patients.8,9
Although early results are encouraging, EGFR TKIs only show effects in a small number of patients (∼10%).7,8 Moreover, even tumors that initially respond to the TKIs eventually become refractory to the therapy,9,10 and the tumors rapidly regrow.8,11 Mechanisms underlying this later-acquired drug resistance are still unknown. However, recent reports reveal a critical role of tumor heterogeneity in the development of drug resistance.12–15 It is now believed that cancer is actually a process of clonal evolution and that every single tumor is a complex hierarchy of tumor subclones resulting from distinctive microenvironmental adaptation.13 These heterogeneous tumor cells provide the material that Darwinian selection can work on; so the fittest subclones can survive through the therapeutic intervention and then dominate regrowth of the tumor.13,16 In this review, we provide an overview of currently recognized causes of tumor heterogeneity, including cancer stem cells (CSCs) and genomic instability, as well as epigenetic aberrations, and discuss their roles in NSCLC. Spatial and temporal tumor heterogeneity as a mechanism of the primary and acquired EGFR TKI resistance in advanced NSCLC is also elucidated. Finally, new approaches to tackling the challenge of tumor heterogeneity are summarized, which may bring new hope to future targeted therapy.
Origins of tumor heterogeneity in NSCLC
CSC and acquired drug resistance in NSCLC
According to the CSC theory, individual tumors are organized into a hierarchy composed of subsets of tumorigenic stems cells and their nontumorigenic progeny cells.16–19 Heterogeneous subclonal lineages in solid tumor are branched from distinctive CSCs and are dynamically maintained by these regenerating cells.15,16 Thus, identification of the CSCs followed by specific treatment targeting developmental signaling pathways (eg, Notch, hedgehog, and transforming growth factor-beta pathways) may be more effective in suppressing tumor growth and preventing drug resistance.15,20
The existence of CSCs in lung cancer is supported by the fact that only a small population of tumor cells (<1.5%) from adenocarcinoma samples possess clonal forming and tumorigenic ability.21 These lung CSCs are thought to be derived from the self-renewing epithelial and bronchioalveolar cells19,22–24 as a result of oncogenic KRAS and EGFR activation23,25 and may exhibit increased aldehyde dehydrogenase activity.20 It has been demonstrated that CSC-like cell population, characterized by elevated expression of cell cycle genes and increased aldehyde dehydrogenase activity, is increased in the EGFR mutant lung cancer cell line with acquired resistance to erlotinib,26,27 indicating a potential role of CSCs in EGFR TKI resistance. However, some nontumorigenic progeny subclones may regain tumorigenic capacity and transform back to stem-like cells.16 Without specific biomarkers, it is very difficult to distinguish CSCs from nontumorigenic cells.19,20 Therefore, CSCs targeting therapy are still quite premature, and research in this field is complicated.16,18
Genomic and chromosomal instability of NSCLC
In addition to CSCs, genomic and chromosomal instability may also contribute to heterogeneity in solid tumors. In normal lung cells, the genome is replicated with high fidelity, and mutations are poorly tolerated, which is attributed to stringent intrinsic checking mechanisms such as base and nucleotide excision repair, mismatch repair, telomere maintenance, and double-strand break repair.28 Malfunctioning checking mechanisms greatly increase the mutation rate and significantly accelerate the process of clonal evolution, leading to carcinogenesis and tumor heterogeneity.28 Chromosomal instability has been shown to correlate with shorter survival in patients with NSCLC,29 possibly because these unstable cancer cells display a higher multidrug-resistant capacity compared with stable cells.30 In mice, lung adenocarcinoma with chromosome instability induced by overexpression of a mitotic checkpoint gene Mad2 is highly aneuploid, correlating with a higher tumor recurrent rate after anticancer treatment.31 Moreover, next-generation sequencing techniques have identified a number of hallmark genomic mutations that are involved in DNA maintenance and mitotic progression, which may predict the prognosis of NSCLC.32–34 Some of these genomic mutations may be used for targets able to be drugged, highlighting the importance of maintaining genomic stability for tumor control.15
Epigenetic aberrations of NSCLC
Apart from genomic mutations, gene expression and phenotypic changes of tumor cells can be affected by epigenetic aberrations including abnormal DNA methylation at CpG islands, dysregulated histone modification, and changes in pathways regulating these epigenetic mechanisms.15,35–37 Compared with genomic mutations, epigenetic changes are reversible and more plastic under environmental pressures, adding further complexity to tumor heterogeneity and contributing to the development of drug resistance.35,38 In NSCLC, global DNA hypomethylation is considered a main cause of genomic instability,39 as well as abnormal oncogene expression.40 A recent analysis of cancer-specific differentially DNA-methylated regions by whole-genome bisulfite sequencing reveals drastic stochastic differences in the differentially methylated regions (DMRs) and a significant loss of sharply delimited methylation boundaries at CpG islands of the DNA in samples from several malignant tumors, including lung cancer.41 Loss of the epigenetic marks in these cancer cells perhaps indicates the epigenome has been “reset”; thus, these cells have greater potential to reshape themselves under selective environmental pressures.41 Consistently, status of global histone modification is strongly associated with prognosis of patients with NSCLC, and it can predict tumor recurrence in early-stage patients.42,43 Notably, epigenetic mechanisms may function in a different dimension from genomic heterogeneity during tumor evolution, through which the tumor cells are not only passively selected by the environments but also may actively change themselves to adapt external stress.35,41
Microenvironmental adaptation and tumor heterogeneity
The extrinsic compartments of tumor cells are composed of disorganized stromal cells including fibroblasts and vasculature and immune cells.44 Distinct microenvironments characterized by varied degrees of selective pressures such as oxygen, acidity, and tumor growth factors may select for mutations that engender subclonal survival and expansion, thus exerting great influence on tumor heterogeneity.45 Moreover, the extrinsic tumor microenvironment may also participate in the development of drug resistance by forming an adaptive, reciprocal signaling loop with tumor cells, thus providing a protective compartment in response to anticancer treatments.44,46 Hepatocyte growth factor (HGF), for instance, is secreted by stromal fibroblasts under the stimulation of tumor-derived factors and strongly propagates tumor expansion by activating MET signaling pathway in NSCLC.47–49 High levels of tumor HGF are associated with EGFR TKI resistance because the activation of MET induces the common downstream prosurvival signaling, bypassing EGFR inhibition.49,50 Moreover, the tumor vascular network featured by dysregulated angiogenesis and reorganization of existing vessels also contributes to tumor heterogeneity. Poorly developed tumor vasculature results in variations in oxygen and nutrient supply within the tumor, as well as affecting drug delivery.44 Microvessel density as an index of angiogenesis may predict survival of the patients with lung cancer,51 and elevated expression proangiogenic ligand vascular endothelial growth factor (VEGF) is associated with poorer prognosis in NSCLC.52 It is therefore conceived that coinhibition of VEGF signaling may confer a benefit on tumor suppression. However, coadministration of VEGF targeted therapy has been shown to reduce the delivery of radiolabeled chemotherapy in patients with NSCLC, as determined by positron emission tomography.53 Nevertheless, a combinational strategy targeting tumor stromal cells remains one of the most important research areas in personalized therapy44 and will be further discussed later.
Spatial tumor heterogeneity and TKI resistance in NSCLC
Intertumor heterogeneity and TKI resistance
As previously mentioned, individual tumors are driven by distinct prosurvival signaling pathways as a result of heterogeneous genomic mutations.5,6 Optimal outcomes of personalized therapy can be achieved by matching the specific lesions with the corresponding treatments, whereas tumors without sensitive mutations are unsusceptible to treatment.6 So far, a number of mutations with the tumor driving capacity have been identified54–57 (Table 1). Frequencies of these driver mutations in NSCLC are associated with histologic subtypes, sex, ethics, age, and past smoking history.58–60 The patients with wild-type EGFR are insensitive to gefitinib9 or erlotinib treatment.8,61 KRAS and BRAF mutants, being mutually exclusively expressed with EGFR in ∼25% and 3% of patients with NSCLC, respectively,60,62 predict poorer prognosis in patients after EGFR-targeted treatment.63,64 Similarly, NSCLC patients harboring EML4-ALK fusion oncogene (∼7%)65 are sensitive to TKI crizotinib but resistant to EGFR TKIs.66,67 Hence, intertumor heterogeneity of different mutations may confer primary resistance to any targeted therapy.68,69
Table 1.
Distribution of known tumor-driving mutations and chromosomal fusions in advanced NSCLC
| Mutation/fusion | Estimated frequency, % |
|---|---|
| Gene mutations | |
| EGFR | 10–30 |
| TP53 | 20–25 |
| KRAS | 15–25 |
| BRAF | 1–3 |
| PIK3CA | 2–3 |
| AKT1 | 1 |
| MAP2K1 | 1 |
| HER2 | 2 |
| MET | <1 |
| Chromosomal fusions | |
| EML4-ALK | 3–7 |
| RET | 1–2 |
| ROS1 | 1–2 |
| Unknown |
Abbreviations: NSCLC, nonsmall cell lung cancer; EGFR, epidermal growth factor receptor; TP53, tumor protein p53; KRAS, Kirsten rat sarcoma viral oncogene homolog; BRAF, v-raf murine sarcoma viral oncogene homolog B; ALK, anaplastic lymphoma kinase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; AKT1, v-akt murine thymoma viral oncogene homolog 1; MAP2K1, mitogen-activated protein kinase 1; HER2, human epidermal growth factor receptor 2.
The sensitivity of different EGFR mutations to TKIs also varied between individuals. The EGFR gene is located on chromosome 7p12–7p13,74 whereas the NSCLC-relevant mutations occur in exons 18–21, encoding the kinase domain of the receptor.54 Although more than 188 EGFR mutations have been identified,75 85% of TKI-sensitive clinical cases harbor only two major mutations:76 in-frame deletions of exon 19 (45%–50%) and a point mutation L858R in exon 21 (40%–45%; Figure 1). Other uncommon TKI-sensitive mutations include amino acid substitution mutations of G719X in exon 18 and L861X in exon 21.76,77 These mutations cause profound activation of both prosurvival and antiapoptotic signaling cascades but also enhance the affinity of the receptor to gefitinib and erlotinib.78,79 In contrast, an insertion mutation in exon 20 (∼4% of EGFR mutations) has never been shown to confer any preferential binding or clinical responses to TKIs77,80,81 but, similarly, elevates the activity of EGFR kinase.82 In addition, deletion mutation in exon 19 seems to have a better response to gefitinib and erlotinib than mutations at other sites.79,83
Figure 1.
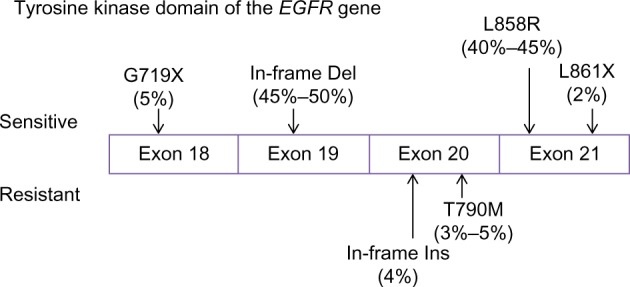
Tumor-driving mutations in the tyrosine kinase domain of EGFR (epidermal growth factor receptor).
Notes: The most common EGFR mutations are in-frame deletions (Del) in exon 19 (45%–50%), which remove four high conserved amino acid residues (the LREA motif) of EGFR, and a point mutation in exon 21 (40%–45%), causing substitution of an arginine by a leucine (L858R). Both of them are sensitive to tyrosine kinase inhibitors (TKIs). Other TKI-sensitive mutations include point mutations such as G719X in exon 18 and L861X in exon 21. In contrast, patients harboring a T790M point mutation or in-frame insertions (Ins) in exon 20 are less sensitive to TKIs. T790M mutation is also an important mechanism in acquired TKI resistance. Data were derived from previous reports.54,59,60,76,77
Variations in the expression of enzymes associated with EGFR signaling may also account for diverse responses to EGFR inhibition. BIM, a BCL2 proapoptotic family member regulated by ERK signaling, has recently been identified as a key mediator of TKI-induced apoptosis.84–86 Levels of pretreatment BIM expression in tumors specimens of NSCLC are different,87 possibly as a result of an intronic deletion polymorphism in the coding region of BIM.88 Low BIM RNA levels are associated with poor outcomes for EGFR inhibition.87 In contrast, elevated tumoral expression of MCL1, an oncogenic member of the BCL2 family,89,90 may predispose NSCLC patients with EGFR-sensitizing mutants to native resistance.91 In addition, mutations in PIK3CA, a p110α catalytic subunit of PI3K, may diminish gefitinib-induced apoptosis by activating bypass Akt signaling, thus conferring resistance to the TKIs.92 The PIK3CA mutant is found in 4% of lung cancers and may cooccur with the EGFR mutation.93
Intratumor heterogeneity and TKI resistance
Apart from differences between tumors, intratumor heterogeneity also presents at histologic, cellular, molecular, genetic, and epigenetic levels.94 Early histological analysis of dissected NSCLC samples showed varied levels of extracellular receptors and stress-responsive genes in different regions of the tumor, indicating distinct subclonal interplay with external stress.95,96 Using a next-generation whole-genome sequencing technique, Gerlinger et al reveal that heterogeneous somatic mutations, divergent allelic profiles and ploidy heterogeneity vastly present in different regions of microdissected samples from the same renal tumor.94 About 63%–69% of all somatic mutations are not detectable across every tumor region, and genes associated with good or poor prognosis can be detected at different regions of the same tumor, both of which reflect drastic subclonal diversity within the tumor.94 The presence of intratumor heterogeneity largely diminishes the reliability of single-tumor biopsy prevalently used in hospital and poses a challenge to the current protocol of personalized therapy.94
In lung cancer, whole-genome analysis of samples from different regions of the tumor is still lacking. By conducting deep digital sequencing analysis of 17 treatment-naïve lung adenocarcinomas, Govindan et al15 found a multiclonal signature indicated by distinct variant allele frequency features in more than half of the samples. Some of these subclones were believed to be involved in progression and migration of the tumor.15 It has been reported that some TKIs desensitizing mutations such as EGFR T790M mutation97,98 and amplification of MET98,99 (both discussed later) may concomitantly present with TKIs sensitizing EGFR mutations in a minor group of patients before the TKI treatment. Considering heterogeneity within the tumor, it is not hard to imagine some less-prevalent subclones may be missed by the single-region biopsy, so that the actual coexistence rate of TKI-sensitive and TKI-resistant subclones may even be higher,94 which may be further examined by genotyping microdissected samples from surgically resected lung tumors.
Notably, some researchers claim coexistence of both wild-type and mutant EGFR within the same tumor,100,101 as well as discordant EGFR mutational status between the primary tumor and the metastatic sites.102,103 These results are challenged by the reports showing highly homogeneous distribution of EGFR mutations across the primary tumor104 and at the metastatic lesions.104,105 The high wild-type EGFR detection rate in the former studies was thought to be explained by normal tissue contamination, as well as technical variations (eg, the minor presence of EGFR mutations that may be neglected by less-sensitive methods).104,106,107 Moreover, although some groups reported a conversion from EGFR mutant to wild-type after TKI treatment in occasional cases,108 most of the studies show highly consistent EGFR mutations in tumors before and after the treatment.57,104,109 It is hard to imagine why the conversion rate of mutant to wild-type after TKI treatment is so low if intratumor heterogeneity of EGFR mutation is not rare.100,110 In addition, Govindan et al applied computational analysis to the deep sequencing data of NSCLC samples and found that EGFR mutation exists in all subclones as a founder mutation of the tumor, suggesting this mutation may be acquired at the very initial phase of tumorigenesis.15
Temporal tumor heterogeneity and acquired TKI resistance in NSCLC
TKI resistance after chemotherapy
Temporal heterogeneity reflects the dynamic tumor evolution in response to environmental changes including therapeutic interventions. Adaptive tumor subclones after this natural selection may then dominate the tumor clonal architecture, leading to disease progression.14,94,111 Therefore, temporal tumor heterogeneity is perhaps more related to acquired resistance to chemotherapy or EGFR-targeted therapy in NSCLC.108,112
It has been noticed that the predictive value of pretreatment EGFR mutation status seen in the first-line TKI regimen is diminished in cohorts of TKI treatment after chemotherapy, and the tumor seems to be less sensitive to EGFR-targeted therapy after chemotherapy.7,9,113 Bai et al compared EGFR mutation status before and after neoadjuvant chemotherapy in both lung tumor tissue and blood samples and found that EGFR mutant-positive NSCLC significantly decreased after chemotherapy.114 However, these results may be misinterpreted because the reduction of EGFR mutant in the blood can also be affected by the amount of tumor cells being exposed to circulation, and chemotherapy may increase fibrotic content in the tumor, thus affecting the detection of mutant EGFR in the tumor.115 De Pas et al compared EGFR expression on mediastinal lymph nodes at the primary biopsy with its expression on both the primary tumor and the residual mediastinal nodes after three cycles of chemotherapy in 47 patients with NSCLC and found reduced EGFR immunoreaction in 12% of the patients.116 Platinum-based agents may reduce the sensitivity of lung cancer cells to EGFR TKIs as a result of PTEN loss and EGFR-independent AKT activation.117 Although the precise mechanism of this chemotherapy-induced resistance to TKIs is still unclear, this issue merits further investigation because it may necessitate rebiopsy after chemotherapy for better guidance in targeted therapy.109,114
Secondary mutations and acquired TKIs resistance in NSCLC
In relapsed NSCLC after TKI treatment, secondary mutations that desensitize EGFR to its inhibitors are frequently detected (Table 2). T790M, which represents threonine-to-methionine substitution at position 790 in the gatekeeper residue of EGFR kinase domain because of a point mutation within exon 20, presents in about 50% of EGFR mutant NSCLCs that become resistant to TKIs after the treatment.109,112,118,119 EGFR of lung cancer cells with both TKI-sensitive mutations and T790M mutation regain affinity to ATP at the hydrophobic pocket, hence abrogating competitive inhibition of the receptor by gefitinib and erlotinib.120 Other secondary mutations in the EGFR kinase domain that are associated with acquired resistance include L747S,121 D761Y,122 and T854A.123 L747S resulting from deletion in exon 19 is located at the head of the loop between strand β3 and helix αC of the receptor and is believed to attenuate BIM upregulation and mitochondrial apoptosis induced by TKIs.124 D761Y from exon 19 mutation occurs in the middle of the helix αC and forms a salt bridge that interacts with the α- and β-phosphates when ATP is present, reducing sensitivity of the EGFR to TKIs.122 T854A mutation in exon 21 interferes the contact of erlotinib to the ATP pocket of the receptor, hence abrogating the inhibition of tyrosine phosphorylation by erlotinib.123 These secondary mutations are rarer and cause less-potent resistance to TKIs than T790M.54
Table 2.
Temporal tumor heterogeneity and acquired TKI resistance in advanced NSCLC
| Mechanisms | Estimated frequency, %
|
References | |
|---|---|---|---|
| Before TKIs | After TKIs | ||
| Resistant EGFR mutations | |||
| T790M | 1–2 | 50 | 57,97,109,139 |
| D761Y, L747S, and T854A | <1 | <5 | 12,76 |
| Bypass signaling | |||
| MET amplification | <5 | 5–20 | 12,130,139 |
| HGF overexpression | 30 | 60 | 140,141 |
| HER2 amplification | 2 | 12 | 142 |
| PIK3CA mutations | 2–3 | 5 | 112,143 |
| CRKL amplification | 3 | 9 | 144,145 |
| Phenotypic changes | |||
| Small-cell transformation | <1 | 3–14 | 57,112 |
| EMT | <1 | 20–44 | 26,133 |
Abbreviations: TKI, tyrosine kinase inhibitor; NSCLC, nonsmall cell lung cancer; EGFR, epidermal growth factor receptor; HGF, hepatocyte growth factor; HER2, human epidermal growth factor receptor 2; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; CRKL, CRK-like protein gene; EMT, epithelial-mesenchymal transition.
Notably, it remains debatable whether T790M mutation truly developed after TKI exposure or whether the mutation preexists before the treatment and become more prevalent in the tumor architecture under selective pressure of the TKIs. There have been some reports showing T790M coexists with sensitive EGFR mutations before TKI treatments in a minor group of patients.97,125 Moreover, T790M mutation is significantly less prevalent in brain metastases, possibly because routine doses of TKIs hardly penetrate the blood–brain barrier and thus exert less selective pressure on the brain lesion.54,126,127 In fact, the presence of secondary mutations in resistant NSCLC may not be an “all or none” phenomenon but, rather, a dynamic process in association with environmental pressure. Discontinuation of EGFR targeted therapy in resistant patients often leads to accelerated tumor growth and symptom progression,128 suggesting at least some tumor cells are still under suppression by the EGFR inhibitors.54,128 Notably, in the absence of EGFR inhibition, the expression of mutations associated with acquired resistance may reversibly disappear, as revealed by serial biopsies.112,128 Consistently, reinitiation of EGFR-targeted therapy after a short period of TKI suspension may slow disease progression again in some cases.112,128,129 Hence, larger cohorts are needed to evaluate whether dynamic mutation monitoring may benefit the patients who acquired resistance to targeted therapy.
Bypass signaling of EGFR inhibition
Compensatory activations of the “oncogene addiction” pathways via bypass signaling after EGFR inhibition represent another mechanism of acquired TKI resistance (Table 2). Focal amplification of MET oncogene, for instance, is observed in about 5%–20% of NSCLCs after treatment with EGFR inhibitors.109,112,130 MET encodes a transmembrane tyrosine kinase receptor for HGF. Activation of MET phosphorylates the downstream ERBB3, activating Akt signaling. Amplification of MET oncogene causes hyperactivation of PI3K and Akt, bypassing the blockade of this pathway by EGFR inhibitors and promoting tumor survival.130 In addition, amplification of HER2 and PI3KCA mutations, both of which activate the common downstream pathways, are detected in about 12% and 5% of EGFR mutant NSCLC patients, respectively.57,112,131 Similar to T790M mutation, MET amplification may also exist before the treatment.99 Turke et al reported MET amplification in rare tumor cells (<1%) from treatment-naïve NSCLC samples (5 of 27 patients).99 Four of these five patients later developed TKI resistance as a result of MET amplification, suggesting acquired resistance may emerge as a result of the survival advantage of pre-existing resistant tumor subclones.12,99,112
Phenotypic changes and epigenetic alterations in acquired TKI resistance
Apart from genetic aberrations, phenotypic changes of the tumor cells such as epithelial to mesenchymal transition132–134 and transformation to a small-cell-like carcinoma77,81 are frequently observed in NSCLC after TKI treatments, possibly as a result of altered epigenetic modifications.135 For instance, gefitinib-induced DNA hypermethylation is associated with decreased expression of micro-RNA (miRNA)-200c, resulting in overexpression of aldehyde dehydrogenase isoform 1, which contributes to the epithelial-to-mesenchymal transition and the presence of stem cell-like properties.26 Moreover, DNA hypermethylation at the promoter region of death-associated protein kinase may silence this gene in the NSCLC cell line,136 leading to erlotinib resistance, whereas transient induction of death-associated protein kinase by gene transfection resensitizes the cells to erlotinib.136 Other hypermethylated tumor DNA such as Wnt antagonist SFRP5137 and mitotic stress checkpoint gene CHFR138 have also been reported to affect TKI sensitivity. Although it is still unclear to what extent epigenetic mechanisms are involved in acquired TKI resistance, considering its importance in clonal evolution and tumor heterogeneity, more efforts should be invested on research and new drug development in this area.38
Advances in molecular diagnosis and tumor monitoring in NSCLC
Next-generation sequencing for the search of new molecular targets
Next-generation high-throughput sequencing techniques now allow us to map the genomic landscape of lung cancer at lower cost, but at orders of magnitude higher speed, compared with the traditional Sanger technique.146,147 These new platforms exponentially enhance our capacity to search for new oncogenes that can be made into drugs.15,148 Imielinski et al149 conducted whole-exome and whole-genome sequencing on 183 paired samples of lung adenocarcinoma and normal lung tissues from treatment-naïve patients, from which 25 hallmark carcinogenic genes including 19 previously reported genes and 6 novel mutants (NKX2-1, TERT, PTEN, MDM2, CCND1, and MYC) were identified. They also detected frequent somatic mutations in epigenetic and splicing factor genes including U2AF1, ARID1A, RBM10, SETD2, and BRD3, suggesting epigenetic regulation as a new hallmark of lung carcinogenesis.149 In 2012, a comprehensive genomic analysis of squamous cell lung cancer was reported by the Cancer Genome Atlas Research Network, showing statistically recurrent mutations in 11 genes, including TP53 mutation in almost all specimens and significantly altered signaling pathways such as NRF2/KEAP1, PI3K/AKT, and CKKN2A/RB1. By matching these genomic aberrations with the currently available US Food and Drug Administration-approved targeted therapeutic agent library, the researchers found 64% of patients in this study possess at least one gene that can potentially be a drug.150 Moreover, the emerging single-cell profiling technique based on whole-genome amplification enables us to process sparse clinical biopsy samples from fine-needle aspirates or core biopsy specimens in which only hundreds of tumor cells are available.151,152 With the development of these advanced techniques, we should be able to obtain a better view of tumor pathology, and new drug development may be greatly accelerated in the foreseeable future.146
Dynamic tumor monitoring by circulating biomarkers
Because cancer cells continue to evolve, dynamically monitoring molecular changes in the tumor should be involved in the personalized therapy. Rebiopsy of the tumor is a straightforward measure but usually confronts ethical issues.153 Instead, dynamic monitoring circulating tumor biomarkers, also called liquid biopsy, may become a more realistic option.154
Circulating tumor cells and DNA
Malignant tumor cells are known to present in the blood of patients.155 However, isolation of these cancer cells from blood samples is rather difficult.156 Maheswaran and coworkers developed a microfluidic-based device in which blood flows through highly condensed micropores coated with epithelial-cell adhesion molecule antibody. The device is able to capture and quantify circulating tumor cells (Ct-cells) at relatively higher efficiency.156 By combining this isolating technique with the ultrasensitive Scorpion Amplification Refractory Mutation System, the researchers claimed 100% specificity and 92% sensitivity in detecting EGFR mutations and secondary T790M mutation from Ct-cells compared with direct tumor biopsy. Using this technique, they also found an increase of T790M mutant allele in Ct-cells after TKI treatment as a sign of drug resistance.156 Moreover, using the whole-genome amplification technique, Ni et al recently reported whole-exome sequencing profiles of single Ct-cells obtained from patients with lung adenocarcinoma.157 These cells exhibit distinct patterns of single-nucleotide variation, but highly reproducible copy number variations. Genes relevant to drug resistance and phenotypic transitions were found to be enriched after chemotherapy.157 In addition, the detection of mutated tumor DNA in cell-free plasma has also been reported, and the cost is relatively lower, as cell isolation is not required.158 However, the sensitivity of direct genotyping for DNA mutation in the plasma is markedly lower (43%–73% compared with for direct tumor biopsy),110,114,159–161 possibly because circulating tumor DNA (Ct-DNA) is more fragmented and it is difficult to extract fragmented tumor DNA from the blood for subsequent amplification.160 Nevertheless, it has been reported that sensitive EGFR mutants are reduced in the Ct-DNA from the patients with NSCLC after TKI treatment, accompanied by the emergence of resistant DNA mutants.114,161 Although larger cohort studies are required before the spread of plasma genotyping, preliminary results have shown a promising future of both Ct-Cells and Ct-DNA in tumor monitoring.
Other circulating nucleotide biomarkers
In addition to Ct-Cells and Ct-DNA, specific miRNAs and methylated tumor DNA in the plasma may also be used as biomarkers of tumor evolution in patients with NSCLC.162–164 Compared with Ct-DNA and Ct-Cells, detection of methylated DNA in the plasma is less expensive and more time-saving.35,165 It also has been reported that methylated RARB2 and RASSF1A are increased in the plasma of patients with NSCLC.166 Plasma levels of these methylated genes decrease after tumor resection and chemotherapy, whereas a rebound rise of these plasma biomarkers manifests tumor relapse.166 So far, very few methylated biomarkers have been identified in the plasma with a predictive value of TKI resistance. Salazar et al showed that unmethylated CHFR may predict prolonged survival in patients receiving EGFR TKIs.138 Other hypermethylated tumor DNA associated with TKI resistance, such as death-associated protein kinase,136 SFRP5,137 may also be worth further validation as plasma biomarkers for tumor monitoring. Moreover, by introducing microelectronic technology, multiple methylated tumor DNAs can be detected and quantified simultaneously, which may further improve efficiency of the detection.167
miRNA is a class of short (18–25 nucleotides), noncoding RNA that binds to complementary mRNA for selective degradation, thus causing a cascade of effects.168 Circulating miRNAs, being more stable than other plasma nucleotides, may emerge as another group of biomarkers for cancer monitoring.168–170 In female nonsmokers with lung adenocarcinoma, levels of plasma miR-195 and miR-122 can differentiate the patients who may benefit from EGFR TKI treatment.171 Circulating miR-21, an important regulator of cell apoptosis and proliferation,172,173 was found to be elevated in the patients with NSCLC after TKI treatment, indicating drug resistance.172 With the development of nanotechnology, highly sensitive probes in plasma are able to directly detect miRNA without labeling or amplification, which may significantly reduce the time and cost of cancer monitoring.174
Proteomic biomarkers and VeriStrat
In addition to nucleotide biomarkers, proteins in the blood may also provide valuable information for tumor evaluation before and after EGFR-targeted therapy. VeriStrat, a mass spectrometry-based high-throughput platform, can analyze proteomic profiles using only 5 μL serum from the patients and subsequently classify the patients for “good” or “poor” response to TKIs.175 To our knowledge, VeriStrat is the only liquid biopsy tool for NSCLC patient stratification before TKI treatment that has been validated in a Phase III clinical trial,176,177 which revealed its prognostic value in predicting overall survival and time to progression.177–180 However, the VeriStrat test seems to be more powerful in selecting patients who may have worse outcomes on TKIs than on chemotherapy, rather than finding patients who may benefit more from TKIs because the overall survival of “good” patients on either treatment is similar.177 In addition, results of VeriStrat may also indicate temporal changes of the tumor. Lazzari et al processed sequentially collected serum samples from 111 NSCLC patients receiving gefitinib and found that one third of these patients converted from good to poor after the treatment, in relevance to drug resistance.181 Further proteomic analysis identified overexpressed serum amyloid A protein 1 (SAA1) in plasma from patients with poor VeriStrat results, which is responsible for the generation of four mass signals in the test.182 Dynamic change of this protein in the plasma during the course of TKI treatment may merit further investigations.182
Advances in the next generation of targeted therapy
Next-generation irreversible TKIs
Apart from the advances in tumor diagnostics, new inhibitors of EGFR have also been developed and examined in clinical trials (Table 3). First-generation TKIs such as erlotinib and gefitinib inhibit EGF receptors by reversibly competing with ATP, but the inhibition can be abrogated by secondary EGFR mutations (eg, T790M) because of the increased affinity of EGFR to ATP.5,183 Second-generation TKIs (eg, afatinib [BIBW2992], dacomitinib [PF00299804], and neratinib [HKI-272]) form irreversible covalent bonds with the EGF receptor and also inhibit other EGFR family members (ERBB2 and ERBB4), and thus may be more potent in tumor suppression.183–185 Afatinib, an irreversible EGFR/ ERBB2 dual inhibitor, has been extensively studied in a serial of clinical trials of NSCLC.186–190 In patients who had failed both chemotherapy and TKI treatment, afatinib significantly prolonged progression-free survival and increased tumor response rate compared with placebo, although no benefit was seen in overall survival.187 Dacomitinib, another pan-ERB inhibitor, also exhibits superiority over erlotinib as the second-line therapy after chemotherapy in unselected patients with NSCLC.191 The main adverse events of the second-generation TKIs include diarrhea, dysphagia, and sore mouth,192 possibly as a result of pan-ERB inhibition.193 Severe gastrointestinal reactions of the second-generation TKIs may reduce bioavailability of the drugs.193 To specifically target T790M mutation, several third-generation TKIs have been developed and are currently undergoing clinical or preclinical investigations. Of note, these highly specific inhibitors may cause much less damage to EGFR in the gastrointestinal tract and skin hair bulbs compared with their predecessors,194,195 and hence intestinal absorption of these drugs may be increased. It is hoped these new drugs may resolve the current clinical dilemma and improve outcomes of targeted therapy.12
Table 3.
Clinical trials of next-generation TKIs in advanced NSCLC
| Targets and trial code | Trial Phase | Lines of treatment | EGFR mutations | Trial design | Outcomes | References |
|---|---|---|---|---|---|---|
| Second-generation irreversible TKIs | ||||||
| Afatinib (BIBW2992) | ||||||
| EGFR/HER2 | ||||||
| LUX-Lung 1 | IIb/III | Third/fourth | Unselected | Afatinib versus placebo in Asian patients | Improved PFS (3.3 versus 1.1 months) and ORR (7% versus 0.5%), no benefit in term of OS | 187 |
| LUX-Lung 2 | II | First/second | Positive | Afatinib after chemotherapy | ORR, 61% | 190 |
| LUX-Lung 3 | III | First | Positive | Afatinib versus pemetrexed + cisplatin | Improved PFS (11.1 versus 6.9 months) | 188 |
| LUX-Lung 4 | II | Second | Positive in 72.6% of patients | Afatinib after TKIs | ORR, 8.2%; median PFS: 4.4 months; median OS, 19 months | 186 |
| LUX-Lung 5 | III | Second | Unselected | Afatinib + chemotherapy versus chemotherapy after afatinib | Ongoing | |
| LUX-Lung 6 | III | First | Positive | Afatinib versus gemcitabine + cisplatin in Asian patients | Improved PFS (11.0 versus 5.6 months) | 189 |
| Dacomitinib (PF00299804) | ||||||
| EGFR/HER2/HER4 | ||||||
| NCT00769067 | II | Second/third | Unselected | Dacomitinib versus erlotinib after chemotherapy | Improved median PFS (2.86 versus 1.91 months) and median OS (9.53 versus 7.44 months) | 191 |
| NCT00548093 | II | Second/third | Positive | Dacomitinib | ORR, 8%; median PFS, 18 weeks | 196 |
| Neratinib (HKI-272) | ||||||
| EGFR, HER2 | ||||||
| Canertinib (CI-1033) | ||||||
| EGFR/HER2/HER4 | ||||||
| NCT00266877 | II | Second | Positive | Neratinib after TKIs | ORR, 3% (all in patients with G719X mutation) | 193 |
| NCT00050830 | II | Second | Unselected | CI-1033 after chemotherapy | 1-year survival rate, 26%–29%; ORR, 2%–4% | 197 |
| N/A | I/II | First | Unselected | CI-1033 + paclitaxel + carboplatin | ORR, 26%; median PFS, 5.1 months; median OS, 12.4 months | 198 |
| Third-generation irreversible TKIs | ||||||
| CO-1686/ | ||||||
| EGFR (T790M mutation) | ||||||
| NCT01526928 | I/II | Second | Positive | CO-1686 after TKIs | Ongoing | |
| AZD9291/ | ||||||
| EGFR (T790M mutation) | ||||||
| NCT01802632 | I/II | Second | Positive | AZD9291 after TKIs | Ongoing | |
| NCT02094261 | II | Second | Positive | AZD9291 in patients with T790M mutation after TKIs | Ongoing | |
| WZ4002/ | ||||||
| EGFR (T790M mutation) | Preclinical | |||||
Abbreviations: TKI, tyrosine kinase inhibitor; NSCLC, nonsmall cell lung cancer; EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; PFS, progression free survival; ORR, overall response rate; OS, overall survival; HER4, human epidermal growth factor receptor 4.
Combinational therapy
As previously discussed, activation of pathways bypassing EGFR is another adaptive mechanism against TKIs. In murine lung tumors cotransfected with mutant EGFR and inducible MET oncogenes, treatment with EGFR inhibitor alone failed to inhibit the tumor growth, but addition of the MET inhibitor crizotinib significantly shrinks the tumor.195,199 In HGF overexpressing lung cancer cell lines, crizotinib and gefitinib together suppress TKI-resistant tumor cell growth.200 Clinical evaluation of the effectiveness of dual MET/EGFR inhibition is currently undergoing.201,202 Moreover, HSP90, a protein-folding chaperone participating in EGFR stabilization in the tumor, may also emerge as a target for tumor control.203–205 Inhibition of HSP90 in lung cancer cell lines and animal models increases the sensitivity to TKIs and enhances tumor suppression 203,206,207 In an early clinical trial, HSP90 inhibitor ganetespib showed a notable disease control rate in patients refractory to chemotherapy and EGFR TKIs, and the adverse effects were acceptable.208 Further clinical cohorts are needed to evaluate the efficacy and safety of HSP90 inhibitors alone or in combination with other targeted treatments.209
Abnormal activation of angiogenesis manifested by overexpression of VEGF is another hallmark of progressing NSCLC.210,211 In patients with advanced NSCLC, chemotherapy plus VEGF targeting monoclonal antibody bevacizumab has been shown to prolong progression-free survival (6.2 versus 4.5 months) and overall survival (12.3 versus 10.3 months) compared with chemotherapy alone.212 Close crosstalk between VEGF and EGFR signaling cascades provides a rationale to concurrently inhibit both pathways. The benefit of combining bevacizumab with erlotinib for the treatment of recurrent NSCLC after chemotherapy has been observed in Phase I/II studies, and the adverse effects are tolerable.213,214 However, subsequent Phase III trials comparing combined treatment of erlotinib and bevacizumab with erlotinib alone as the second-line treatment showed no superiority of the combination in terms of progression-free survival and overall survival.215,216 Notably, these trials are all conducted in unselected patients.210,211 VeriStrat-based proteomic analysis may effectively distinguish NSCLC patients who are likely to benefit from the dual inhibition. Patients with a good VeriStrat result had significantly longer progression-free and overall survival (18.9 versus 6.3 weeks and 71.4 versus 19.9 weeks, respectively) than the patients with a poor result.217 It is hoped that outcomes of the dual EGFR/VEGF inhibition may be improved in patients with activating EGFR mutations.218
Summary and perspectives
Despite tremendous efforts made to improve outcomes of cancer care in the past decades, the death rate of patients with advanced NSCLC is still very high. Personalized cancer treatments targeting specific tumor-driving mutations provide new options for patients who failed chemotherapy, greatly transforming cancer care.219 However, despite an initial response, the patients eventually die from disease progression as a result of drug resistance. With a deeper understanding of tumor biology, we now realize this later-developed drug resistance may turn up as a result of tumor heterogeneity and clonal adaptation. In fact, complexity of the adaptive process is probably much beyond our imagination because CSCs, abnormalities in mitotic-maintaining genes, and epigenetic mechanisms may all contribute to the process. In terms of EGFR-targeted therapy, resistant mechanisms (eg, EGFR T790M mutation and MET amplification) emerge as a result of the tumor evolution after EGFR inhibition by gefitinib or erlotinib. Therefore, biomarkers that are sensitive to the resistant tumor clones and a new compound with specific effects on these resistant mechanisms have become the focus of research in this field (Figure 2). Moreover, instead of biopsy at a single time, next-generation personalization may involve multiple rounds of tumor biopsies, followed by changes of the treatment to overcome the dynamic tumor evolution and drug resistance.6,220,221 At present, laboratory findings are rapidly translated to clinical applications. Novel molecular targeted compounds and less invasive diagnostic tests are currently undergoing clinical evaluation, with some showing encouraging results. Cost/effectiveness evaluation is needed before these new tests and drugs can be spread for clinical use. Future studies should further elucidate the mechanism underlying tumor evolution, and measures to retard its process merit more investigation. With the advances seen in this highly active field, improvements in the clinical outcome of the patients with advanced NSCLC can be expected in the foreseeable future.
Figure 2.
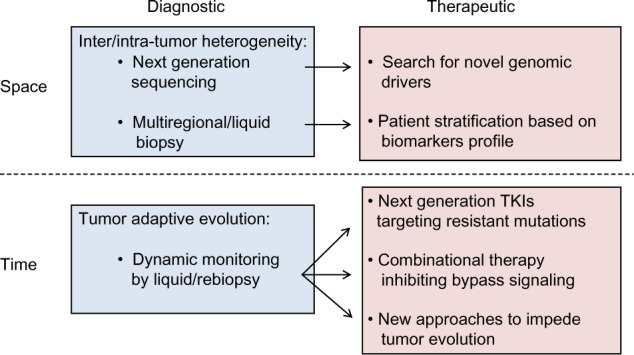
Schematic summary of measures to overcome tumor heterogeneity and drug resistance in lung cancer.
Notes: Spatial heterogeneity indicates inter-/intratumor differences at the genomic, epigenetic, and proteomic levels, whereas temporal heterogeneity reflects dynamic tumor evolution over time. Some diagnostic and therapeutic approaches have been validated in patients for clinical translation.
Abbreviation: TKI, tyrosine kinase inhibitors.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Rapp E, Pater JL, Willan A, et al. Chemotherapy can prolong survival in patients with advanced non-small-cell lung cancer – report of a Canadian multicenter randomized trial. J Clin Oncol. 1988;6(4):633–641. doi: 10.1200/JCO.1988.6.4.633. [DOI] [PubMed] [Google Scholar]
- 3.Schiller JH, Harrington D, Belani CP, et al. Eastern Cooperative Oncology Group Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346(2):92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 4.Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction – a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3(8):448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- 5.Goffin JR, Zbuk K. Epidermal growth factor receptor: pathway, therapies, and pipeline. Clin Ther. 2013;35(9):1282–1303. doi: 10.1016/j.clinthera.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 6.McDermott U, Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol. 2009;27(33):5650–5659. doi: 10.1200/JCO.2009.22.9054. [DOI] [PubMed] [Google Scholar]
- 7.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. National Cancer Institute of Canada Clinical Trials Group Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Carcereny E, Gervais R, et al. Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 9.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin- paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 10.Han JY, Park K, Kim SW, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;30(10):1122–1128. doi: 10.1200/JCO.2011.36.8456. [DOI] [PubMed] [Google Scholar]
- 11.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 12.Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31(31):3987–3996. doi: 10.1200/JCO.2012.45.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marte B. Tumour heterogeneity. Nature. 2013;501(7467):327. doi: 10.1038/501327a. [DOI] [PubMed] [Google Scholar]
- 14.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 15.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150(6):1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 18.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138(5):822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Singh S, Chellappan S. Lung cancer stem cells: Molecular features and therapeutic targets. Mol Aspects Med. 2013 doi: 10.1016/j.mam.2013.08.003. http://dx.doi.org/10.1016/j.mam.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peacock CD, Watkins DN. Cancer stem cells and the ontogeny of lung cancer. J Clin Oncol. 2008;26(17):2883–2889. doi: 10.1200/JCO.2007.15.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carney DN, Gazdar AF, Bunn PA, Jr, Guccion JG. Demonstration of the stem cell nature of clonogenic tumor cells from lung cancer patients. Stem Cells. 1982;1(3):149–164. [PubMed] [Google Scholar]
- 22.Rawlins EL, Hogan BL. Epithelial stem cells of the lung: privileged few or opportunities for many? Development. 2006;133(13):2455–2465. doi: 10.1242/dev.02407. [DOI] [PubMed] [Google Scholar]
- 23.Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 24.Giangreco A, Arwert EN, Rosewell IR, Snyder J, Watt FM, Stripp BR. Stem cells are dispensable for lung homeostasis but restore airways after injury. Proc Natl Acad Sci USA. 2009;106(23):9286–9291. doi: 10.1073/pnas.0900668106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin C, Song H, Huang C, et al. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE. 2012;7(12):e53817. doi: 10.1371/journal.pone.0053817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73(10):3051–3061. doi: 10.1158/0008-5472.CAN-12-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, et al. Stem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: a novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle. 2013;12(21):3390–3404. doi: 10.4161/cc.26417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 29.Birkbak NJ, Eklund AC, Li Q, et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011;71(10):3447–3452. doi: 10.1158/0008-5472.CAN-10-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee AJ, Endesfelder D, Rowan AJ, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71(5):1858–1870. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464(7287):436–440. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SC, Jung Y, Park J, et al. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS ONE. 2013;8(2):e55596. doi: 10.1371/journal.pone.0055596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stead LF, Egan P, Devery A, et al. An integrated inspection of the somatic mutations in a lung squamous cell carcinoma using next-generation sequencing. PLoS ONE. 2013;8(11):e78823. doi: 10.1371/journal.pone.0078823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balgkouranidou I, Liloglou T, Lianidou ES. Lung cancer epigenetics: emerging biomarkers. Biomarkers Med. 2013;7(1):49–58. doi: 10.2217/bmm.12.111. [DOI] [PubMed] [Google Scholar]
- 36.Liu SV, Fabbri M, Gitlitz BJ, Laird-Offringa IA. Epigenetic therapy in lung cancer. Front Oncol. 2013;3:135. doi: 10.3389/fonc.2013.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothschild SI. Epigenetic Therapy in Lung Cancer – Role of microRNAs. Front Oncol. 2013;3:158. doi: 10.3389/fonc.2013.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18(19):2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 39.Daskalos A, Nikolaidis G, Xinarianos G, et al. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer. 2009;124(1):81–87. doi: 10.1002/ijc.23849. [DOI] [PubMed] [Google Scholar]
- 40.Daskalos A, Logotheti S, Markopoulou S, et al. Global DNA hypomethylation-induced ΔNp73 transcriptional activation in non-small cell lung cancer. Cancer Lett. 2011;300(1):79–86. doi: 10.1016/j.canlet.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barlési F, Giaccone G, Gallegos-Ruiz MI, et al. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol. 2007;25(28):4358–4364. doi: 10.1200/JCO.2007.11.2599. [DOI] [PubMed] [Google Scholar]
- 43.Song JS, Kim YS, Kim DK, Park SI, Jang SJ. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Int. 2012;62(3):182–190. doi: 10.1111/j.1440-1827.2011.02776.x. [DOI] [PubMed] [Google Scholar]
- 44.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 45.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 46.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9(9):665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura T, Matsumoto K, Kiritoshi A, Tano Y, Nakamura T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactions. Cancer Res. 1997;57(15):3305–3313. [PubMed] [Google Scholar]
- 48.Yamada T, Matsumoto K, Wang W, et al. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin Cancer Res. 2010;16(1):174–183. doi: 10.1158/1078-0432.CCR-09-1204. [DOI] [PubMed] [Google Scholar]
- 49.Yamada T, Takeuchi S, Kita K, et al. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J Thorac Oncol. 2012;7(2):272–280. doi: 10.1097/JTO.0b013e3182398e69. [DOI] [PubMed] [Google Scholar]
- 50.Siegfried JM, Weissfeld LA, Singh-Kaw P, Weyant RJ, Testa JR, Landreneau RJ. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. 1997;57(3):433–439. [PubMed] [Google Scholar]
- 51.Meert AP, Paesmans M, Martin B, et al. The role of microvessel density on the survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2002;87(7):694–701. doi: 10.1038/sj.bjc.6600551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hegde PS, Jubb AM, Chen D, et al. Predictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumab. Clin Cancer Res. 2013;19(4):929–937. doi: 10.1158/1078-0432.CCR-12-2535. [DOI] [PubMed] [Google Scholar]
- 53.Van der Veldt AA, Lubberink M, Bahce I, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21(1):82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 54.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 56.Stella GM, Luisetti M, Pozzi E, Comoglio PM. Oncogenes in non-small-cell lung cancer: emerging connections and novel therapeutic dynamics. The lancet. Respir Med. 2013;1(3):251–261. doi: 10.1016/S2213-2600(13)70009-2. [DOI] [PubMed] [Google Scholar]
- 57.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jänne PA, Engelman JA, Johnson BE. Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology. J Clin Oncol. 2005;23(14):3227–3234. doi: 10.1200/JCO.2005.09.985. [DOI] [PubMed] [Google Scholar]
- 59.Marchetti A, Martella C, Felicioni L, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol. 2005;23(4):857–865. doi: 10.1200/JCO.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 60.Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64(24):8919–8923. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 61.Garassino MC, Martelli O, Broggini M, et al. TAILOR trialists Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. Lancet Oncol. 2013;14(10):981–988. doi: 10.1016/S1470-2045(13)70310-3. [DOI] [PubMed] [Google Scholar]
- 62.Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62(23):6997–7000. [PubMed] [Google Scholar]
- 63.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23(25):5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 64.Pratilas CA, Hanrahan AJ, Halilovic E, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68(22):9375–9383. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 66.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12(11):1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer – is it becoming a reality? Nat Rev Clin Oncol. 2010;7(7):401–414. doi: 10.1038/nrclinonc.2010.64. [DOI] [PubMed] [Google Scholar]
- 69.Lorusso V, Silvestris N, Marech I. TORCH study: how much longer should we continue to use erlotinib in unselected patients with non-small-cell lung cancer? J Clin Oncol. 2013;31(2):288–289. doi: 10.1200/JCO.2012.46.2648. [DOI] [PubMed] [Google Scholar]
- 70.Li T, Kung HJ, Mack PC, Gandara DR. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. J Clin Oncol. 2013;31(8):1039–1049. doi: 10.1200/JCO.2012.45.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mogi A, Kuwano H. TP53 mutations in nonsmall cell lung cancer. J Biomed Biotechnol. 2011;2011:583929. doi: 10.1155/2011/583929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan Y, Zhang Y, Li Y, et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer. 2014;84(2):121–126. doi: 10.1016/j.lungcan.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 73.Sequist LV, Heist RS, Shaw AT, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22(12):2616–2624. doi: 10.1093/annonc/mdr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010;277(2):301–308. doi: 10.1111/j.1742-4658.2009.07448.x. [DOI] [PubMed] [Google Scholar]
- 75.Yeh P, Chen H, Andrews J, Naser R, Pao W, Horn L. DNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clin Cancer Res. 2013;19(7):1894–1901. doi: 10.1158/1078-0432.CCR-12-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 77.Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res. 2011;17(11):3812–3821. doi: 10.1158/1078-0432.CCR-10-3408. [DOI] [PubMed] [Google Scholar]
- 78.Mulloy R, Ferrand A, Kim Y, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67(5):2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 79.Carey KD, Garton AJ, Romero MS, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66(16):8163–8171. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- 80.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13(1):e23–e31. doi: 10.1016/S1470-2045(11)70129-2. [DOI] [PubMed] [Google Scholar]
- 81.Wu JY, Wu SG, Yang CH, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res. 2008;14(15):4877–4882. doi: 10.1158/1078-0432.CCR-07-5123. [DOI] [PubMed] [Google Scholar]
- 82.Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2(11):e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Riely GJ, Pao W, Pham D, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(3 Pt 1):839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 84.Deng J, Shimamura T, Perera S, et al. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67(24):11867–11875. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 85.Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4(10):e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4(10):1681–1689. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Faber AC, Corcoran RB, Ebi H, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1(4):352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18(4):521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 89.Faber AC, Li D, Song Y, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA. 2009;106(46):19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005;4(3):267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 91.Cetin Z, Ozbilim G, Erdogan A, Luleci G, Karauzum SB. Evaluation of PTEN and Mcl-1 expressions in NSCLC expressing wild-type or mutated EGFR. Med Oncol. 2010;27(3):853–860. doi: 10.1007/s12032-009-9296-7. [DOI] [PubMed] [Google Scholar]
- 92.Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116(10):2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kawano O, Sasaki H, Endo K, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54(2):209–215. doi: 10.1016/j.lungcan.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 94.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crouch EC, Stone KR, Bloch M, McDivitt RW. Heterogeneity in the production of collagens and fibronectin by morphologically distinct clones of a human tumor cell line: evidence for intratumoral diversity in matrix protein biosynthesis. Cancer Res. 1987;47(22):6086–6092. [PubMed] [Google Scholar]
- 96.Blackhall FH, Pintilie M, Wigle DA, et al. Stability and heterogeneity of expression profiles in lung cancer specimens harvested following surgical resection. Neoplasia. 2004;6(6):761–767. doi: 10.1593/neo.04301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66(16):7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 98.Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 99.Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bai H, Wang Z, Wang Y, et al. Detection and clinical significance of intratumoral EGFR mutational heterogeneity in Chinese patients with advanced non-small cell lung cancer. PLoS ONE. 2013;8(2):e54170. doi: 10.1371/journal.pone.0054170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008;99(5):929–935. doi: 10.1111/j.1349-7006.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gow CH, Chang YL, Hsu YC, et al. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol. 2009;20(4):696–702. doi: 10.1093/annonc/mdn679. [DOI] [PubMed] [Google Scholar]
- 103.Kalikaki A, Koutsopoulos A, Trypaki M, et al. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer. 2008;99(6):923–929. doi: 10.1038/sj.bjc.6604629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yatabe Y, Matsuo K, Mitsudomi T. Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J Clin Oncol. 2011;29(22):2972–2977. doi: 10.1200/JCO.2010.33.3906. [DOI] [PubMed] [Google Scholar]
- 105.Matsumoto S, Takahashi K, Iwakawa R, et al. Frequent EGFR mutations in brain metastases of lung adenocarcinoma. Int J Cancer. 2006;119(6):1491–1494. doi: 10.1002/ijc.21940. [DOI] [PubMed] [Google Scholar]
- 106.Nagai Y, Miyazawa H, Huqun, et al. Genetic heterogeneity of the epidermal growth factor receptor in non-small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clamp. Cancer Res. 2005;65(16):7276–7282. doi: 10.1158/0008-5472.CAN-05-0331. [DOI] [PubMed] [Google Scholar]
- 107.Kamila WK, Michał S, Paweł K, et al. EGFR activating mutations detected by different PCR techniques in Caucasian NSCLC patients with CNS metastases: short report. Clin Exp Metastasis. 2013;30(8):1063–1071. doi: 10.1007/s10585-013-9603-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jiang SX, Yamashita K, Yamamoto M, et al. EGFR genetic heterogeneity of nonsmall cell lung cancers contributing to acquired gefitinib resistance. Int J Cancer. 2008;123(11):2480–2486. doi: 10.1002/ijc.23868. [DOI] [PubMed] [Google Scholar]
- 109.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17(5):1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011;17(24):7808–7815. doi: 10.1158/1078-0432.CCR-11-1712. [DOI] [PubMed] [Google Scholar]
- 111.Klein CA. Selection and adaptation during metastatic cancer progression. Nature. 2013;501(7467):365–372. doi: 10.1038/nature12628. [DOI] [PubMed] [Google Scholar]
- 112.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366(9496):1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 114.Bai H, Wang Z, Chen K, et al. Influence of chemotherapy on EGFR mutation status among patients with non-small-cell lung cancer. J Clin Oncol. 2012;30(25):3077–3083. doi: 10.1200/JCO.2011.39.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jair Bar DU. Roni Borshtein, Hovav Nechushtan, Amir Onn. EGFR mutation in lung cancer: tumor heterogeneity and the impact of chemotherapy. Chin Clin Oncol. 2013;2(1):1–3. doi: 10.3978/j.issn.2304-3865.2012.12.03. [DOI] [PubMed] [Google Scholar]
- 116.De Pas T, Pelosi G, de Braud F, et al. Modulation of epidermal growth factor receptor status by chemotherapy in patients with locally advanced non-small-cell lung cancer is rare. J Clin Oncol. 2004;22(24):4966–4970. doi: 10.1200/JCO.2004.01.195. [DOI] [PubMed] [Google Scholar]
- 117.Chin TM, Quinlan MP, Singh A, et al. Reduced Erlotinib sensitivity of epidermal growth factor receptor-mutant non-small cell lung cancer following cisplatin exposure: a cell culture model of second-line erlotinib treatment. Clin Cancer Res. 2008;14(21):6867–6876. doi: 10.1158/1078-0432.CCR-08-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 120.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Costa DB, Schumer ST, Tenen DG, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol. 2008;26(7):1182–1184. doi: 10.1200/JCO.2007.14.9039. [DOI] [PubMed] [Google Scholar]
- 122.Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12(21):6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 123.Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14(22):7519–7525. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4(10):1669–1679. doi: 10.1371/journal.pmed.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tokumo M, Toyooka S, Ichihara S, et al. Double mutation and gene copy number of EGFR in gefitinib refractory non-small-cell lung cancer. Lung Cancer. 2006;53(1):117–121. doi: 10.1016/j.lungcan.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 126.Jackman DM, Holmes AJ, Lindeman N, et al. Response and resistance in a non-small-cell lung cancer patient with an epidermal growth factor receptor mutation and leptomeningeal metastases treated with high-dose gefitinib. J Clin Oncol. 2006;24(27):4517–4520. doi: 10.1200/JCO.2006.06.6126. [DOI] [PubMed] [Google Scholar]
- 127.Clarke JL, Pao W, Wu N, Miller VA, Lassman AB. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. J Neurooncol. 2010;99(2):283–286. doi: 10.1007/s11060-010-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Riely GJ, Kris MG, Zhao B, et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res. 2007;13(17):5150–5155. doi: 10.1158/1078-0432.CCR-07-0560. [DOI] [PubMed] [Google Scholar]
- 129.Kurata T, Tamura K, Kaneda H, et al. Effect of re-treatment with gefitinib (‘Iressa’, ZD1839) after acquisition of resistance. Ann Oncol. 2004;15(1):173–174. doi: 10.1093/annonc/mdh006. [DOI] [PubMed] [Google Scholar]
- 130.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 131.Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Barr S, Thomson S, Buck E, et al. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin Exp Metastasis. 2008;25(6):685–693. doi: 10.1007/s10585-007-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010;30(7):2513–2517. [PubMed] [Google Scholar]
- 134.Chung JH, Rho JK, Xu X, et al. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer. 2011;73(2):176–182. doi: 10.1016/j.lungcan.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 135.Walter K, Holcomb T, Januario T, et al. DNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancer. Clin Cancer Res. 2012;18(8):2360–2373. doi: 10.1158/1078-0432.CCR-11-2635-T. [DOI] [PubMed] [Google Scholar]
- 136.Ogawa T, Liggett TE, Melnikov AA, et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle. 2012;11(8):1656–1663. doi: 10.4161/cc.20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhu J, Wang Y, Duan J, et al. DNA Methylation status of Wnt antagonist SFRP5 can predict the response to the EGFR-tyrosine kinase inhibitor therapy in non-small cell lung cancer. J Exp Clin Cancer Res. 2012;31(1):80. doi: 10.1186/1756-9966-31-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Salazar F, Molina MA, Sanchez-Ronco M, et al. First-line therapy and methylation status of CHFR in serum influence outcome to chemotherapy versus EGFR tyrosine kinase inhibitors as second-line therapy in stage IV non-small-cell lung cancer patients. Lung Cancer. 2011;72(1):84–91. doi: 10.1016/j.lungcan.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 139.Li H, Hu H, Wang R, et al. Primary concomitant EGFR T790M mutation predicted worse prognosis in non-small cell lung cancer patients. Onco Targets Ther. 2014;3;7:513–524. doi: 10.2147/OTT.S60122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68(22):9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 141.Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6(12):2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 142.Mazières J, Peters S, Lepage B, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31(16):1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 143.Wang L, Hu H, Pan Y, et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE. 2014;9(2):e88291. doi: 10.1371/journal.pone.0088291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cheung HW, Du J, Boehm JS, et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1(7):608–625. doi: 10.1158/2159-8290.CD-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kim YH, Kwei KA, Girard L, et al. Genomic and functional analysis identifies CRKL as an oncogene amplified in lung cancer. Oncogene. 2010;29(10):1421–1430. doi: 10.1038/onc.2009.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Daniels MG, Bowman RV, Yang IA, Govindan R, Fong KM. An emerging place for lung cancer genomics in 2013. J Thorac Dis. 2013;5(Suppl 5):S491–S497. doi: 10.3978/j.issn.2072-1439.2013.10.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Korf BR, Rehm HL. New approaches to molecular diagnosis. JAMA. 2013;309(14):1511–1521. doi: 10.1001/jama.2013.3239. [DOI] [PubMed] [Google Scholar]
- 149.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Network CGAR. Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Potter NE, Ermini L, Papaemmanuil E, et al. Single-cell mutational profiling and clonal phylogeny in cancer. Genome Res. 2013;23(12):2115–2125. doi: 10.1101/gr.159913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Navin N, Hicks J. Future medical applications of single-cell sequencing in cancer. Genome Med. 2011;3(5):31. doi: 10.1186/gm247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jakobsen JN, Sørensen JB. Intratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;69(2):289–299. doi: 10.1007/s00280-011-1791-9. [DOI] [PubMed] [Google Scholar]
- 154.Murtaza M, Dawson SJ, Tsui DW, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 155.Allard WJ, Matera J, Miller MC, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10(20):6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 156.Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359(4):366–377. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ni X, Zhuo M, Su Z, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci USA. 2013;110(52):21083–21088. doi: 10.1073/pnas.1320659110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kimura H, Kasahara K, Kawaishi M, et al. Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer. Clin Cancer Res. 2006;12(13):3915–3921. doi: 10.1158/1078-0432.CCR-05-2324. [DOI] [PubMed] [Google Scholar]
- 159.Goto K, Ichinose Y, Ohe Y, et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol. 2012;7(1):115–121. doi: 10.1097/JTO.0b013e3182307f98. [DOI] [PubMed] [Google Scholar]
- 160.Liu X, Lu Y, Zhu G, et al. The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J Clin Pathol. 2013;66(12):1065–1069. doi: 10.1136/jclinpath-2013-201728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kuang Y, Rogers A, Yeap BY, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15(8):2630–2636. doi: 10.1158/1078-0432.CCR-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bearzatto A, Conte D, Frattini M, et al. p16(INK4A) Hypermethylation detected by fluorescent methylation-specific PCR in plasmas from non-small cell lung cancer. Clin Cancer Res. 2002;8(12):3782–3787. [PubMed] [Google Scholar]
- 163.Hsu HS, Chen TP, Hung CH, et al. Characterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasma. Cancer. 2007;110(9):2019–2026. doi: 10.1002/cncr.23001. [DOI] [PubMed] [Google Scholar]
- 164.An Q, Liu Y, Gao Y, et al. Detection of p16 hypermethylation in circulating plasma DNA of non-small cell lung cancer patients. Cancer Lett. 2002;188(1–2):109–114. doi: 10.1016/s0304-3835(02)00496-2. [DOI] [PubMed] [Google Scholar]
- 165.Porter JR, Stains CI, Segal DJ, Ghosh I. Split beta-lactamase sensor for the sequence-specific detection of DNA methylation. Anal Chem. 2007;79(17):6702–6708. doi: 10.1021/ac071163+. [DOI] [PubMed] [Google Scholar]
- 166.Ponomaryova AA, Rykova EY, Cherdyntseva NV, et al. Potentialities of aberrantly methylated circulating DNA for diagnostics and post-treatment follow-up of lung cancer patients. Lung Cancer. 2013;81(3):397–403. doi: 10.1016/j.lungcan.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 167.Shin Y. Label-free methylation specific sensor based on silicon microring resonators for detection and quantification of DNA methylation biomarkers in bladder cancer. Sensors Actuators B: Chem. 2013;177:404–411. [Google Scholar]
- 168.Redova M, Sana J, Slaby O. Circulating miRNAs as new blood-based biomarkers for solid cancers. Future Oncol. 2013;9(3):387–402. doi: 10.2217/fon.12.192. [DOI] [PubMed] [Google Scholar]
- 169.Boeri M, Verri C, Conte D, et al. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci USA. 2011;108(9):3713–3718. doi: 10.1073/pnas.1100048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hu Z, Chen X, Zhao Y, et al. Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small-cell lung cancer. J Clin Oncol. 2010;28(10):1721–1726. doi: 10.1200/JCO.2009.24.9342. [DOI] [PubMed] [Google Scholar]
- 171.Zhang H, Su Y, Xu F, Kong J, Yu H, Qian B. Circulating microRNAs in relation to EGFR status and survival of lung adenocarcinoma in female non-smokers. PLoS ONE. 2013;8(11):e81408. doi: 10.1371/journal.pone.0081408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li B, Ren S, Li X, et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer. 2013 doi: 10.1016/j.lungcan.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 173.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang Y, Zheng D, Tan Q, Wang MX, Gu LQ. Nanopore-based detection of circulating microRNAs in lung cancer patients. Nat Nanotechnol. 2011;6(10):668–674. doi: 10.1038/nnano.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Taguchi F, Solomon B, Gregorc V, et al. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multicohort cross-institutional study. J Natl Cancer Inst. 2007;99(11):838–846. doi: 10.1093/jnci/djk195. [DOI] [PubMed] [Google Scholar]
- 176.Butts CA. VeriStrat validated in patients with non-small-cell lung cancer. Lancet Oncol. 2014;15(7):671–672. doi: 10.1016/S1470-2045(14)70203-7. [DOI] [PubMed] [Google Scholar]
- 177.Gregorc V, Novello S, Lazzari C, et al. Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): a biomarker-stratified, randomised phase 3 trial. Lancet Oncol. 2014;15(7):713–721. doi: 10.1016/S1470-2045(14)70162-7. [DOI] [PubMed] [Google Scholar]
- 178.Amann JM, Lee JW, Roder H, et al. Genetic and proteomic features associated with survival after treatment with erlotinib in first-line therapy of non-small cell lung cancer in Eastern Cooperative Oncology Group 3503. J Thorac Oncol. 2010;5(2):169–178. doi: 10.1097/JTO.0b013e3181c8cbd9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Carbone DP, Ding K, Roder H, et al. Prognostic and predictive role of the VeriStrat plasma test in patients with advanced non-small-cell lung cancer treated with erlotinib or placebo in the NCIC Clinical Trials Group BR.21 trial. J Thorac Oncol. 2012;7(11):1653–1660. doi: 10.1097/JTO.0b013e31826c1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stinchcombe TE, Roder J, Peterman AH, et al. A retrospective analysis of VeriStrat status on outcome of a randomized phase II trial of first-line therapy with gemcitabine, erlotinib, or the combination in elderly patients (age 70 years or older) with stage IIIB/IV non-small-cell lung cancer. J Thorac Oncol. 2013;8(4):443–451. doi: 10.1097/JTO.0b013e3182835577. [DOI] [PubMed] [Google Scholar]
- 181.Lazzari C, Spreafico A, Bachi A, et al. Changes in plasma mass-spectral profile in course of treatment of non-small cell lung cancer patients with epidermal growth factor receptor tyrosine kinase inhibitors. J Thorac Oncol. 2012;7(1):40–48. doi: 10.1097/JTO.0b013e3182307f17. [DOI] [PubMed] [Google Scholar]
- 182.Milan E, Lazzari C, Anand S, et al. SAA1 is over-expressed in plasma of non small cell lung cancer patients with poor outcome after treatment with epidermal growth factor receptor tyrosine-kinase inhibitors. J Proteomics. 2012;76(Spec):91–101. doi: 10.1016/j.jprot.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 183.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102(21):7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ji H, Zhao X, Yuza Y, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci USA. 2006;103(20):7817–7822. doi: 10.1073/pnas.0510284103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Katakami N, Atagi S, Goto K, et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol. 2013;31(27):3335–3341. doi: 10.1200/JCO.2012.45.0981. [DOI] [PubMed] [Google Scholar]
- 187.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 188.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 189.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 190.Yang JC, Shih JY, Su WC, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13(5):539–548. doi: 10.1016/S1470-2045(12)70086-4. [DOI] [PubMed] [Google Scholar]
- 191.Ramalingam SS, Blackhall F, Krzakowski M, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30(27):3337–3344. doi: 10.1200/JCO.2011.40.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yang JC, Hirsh V, Schuler M, et al. Symptom control and quality of life in LUX-Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3342–3350. doi: 10.1200/JCO.2012.46.1764. [DOI] [PubMed] [Google Scholar]
- 193.Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(18):3076–3083. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 194.Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nanjo S, Yamada T, Nishihara H, et al. Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors. PLoS ONE. 2013;8(12):e84700. doi: 10.1371/journal.pone.0084700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Reckamp KL, Giaccone G, Camidge DR, et al. A phase 2 trial of dacomitinib (PF-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non-small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer. 2014;120(8):1145–1154. doi: 10.1002/cncr.28561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jänne PA, von Pawel J, Cohen RB, et al. Multicenter, randomized, phase II trial of CI-1033, an irreversible pan-ERBB inhibitor, for previously treated advanced non small-cell lung cancer. J Clin Oncol. 2007;25(25):3936–3944. doi: 10.1200/JCO.2007.11.1336. [DOI] [PubMed] [Google Scholar]
- 198.Chiappori AA, Ellis PM, Hamm JT, et al. A phase I evaluation of oral CI-1033 in combination with paclitaxel and carboplatin as first-line chemotherapy in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2006;1(9):1010–1019. [PubMed] [Google Scholar]
- 199.Xu L, Kikuchi E, Xu C, et al. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res. 2012;72(13):3302–3311. doi: 10.1158/0008-5472.CAN-11-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chen X, Zhou JY, Zhao J, Chen JJ, Ma SN, Zhou JY. Crizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small-cell lung cancer cells. Anticancer Drugs. 2013;24(10):1039–1046. doi: 10.1097/CAD.0000000000000011. [DOI] [PubMed] [Google Scholar]
- 201.Nakagawa T, Takeuchi S, Yamada T, et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther. 2012;11(10):2149–2157. doi: 10.1158/1535-7163.MCT-12-0195. [DOI] [PubMed] [Google Scholar]
- 202.Goldman JW, Laux I, Chai F, et al. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer. 2012;118(23):5903–5911. doi: 10.1002/cncr.27575. [DOI] [PubMed] [Google Scholar]
- 203.Rice JW, Veal JM, Barabasz A, et al. Targeting of multiple signaling pathways by the Hsp90 inhibitor SNX-2112 in EGFR resistance models as a single agent or in combination with erlotinib. Oncol Res. 2009;18(5–6):229–242. doi: 10.3727/096504009x12596189659240. [DOI] [PubMed] [Google Scholar]
- 204.Wang X, Song X, Zhuo W, et al. The regulatory mechanism of Hsp90alpha secretion and its function in tumor malignancy. Proc Natl Acad Sci USA. 2009;106(50):21288–21293. doi: 10.1073/pnas.0908151106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65(14):6401–6408. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- 206.Ueno T, Tsukuda K, Toyooka S, et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer. 2012;76(1):26–31. doi: 10.1016/j.lungcan.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 207.Shimamura T, Li D, Ji H, et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008;68(14):5827–5838. doi: 10.1158/0008-5472.CAN-07-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Socinski MA, Goldman J, El-Hariry I, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. 2013;19(11):3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Garon EB, Finn RS, Hamidi H, et al. The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol Cancer Ther. 2013;12(6):890–900. doi: 10.1158/1535-7163.MCT-12-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ferrara N. The role of vascular endothelial growth factor in pathological angiogenesis. Breast Cancer Res Treat. 1995;36(2):127–137. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 211.Seto T, Higashiyama M, Funai H, et al. Prognostic value of expression of vascular endothelial growth factor and its flt-1 and KDR receptors in stage I non-small-cell lung cancer. Lung Cancer. 2006;53(1):91–96. doi: 10.1016/j.lungcan.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 212.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 213.RS, O’Neill VJ, Fehrenbacher L, et al. Phase II study of efficacy and safety of bevacizumab in combination with chemotherapy or erlotinib compared with chemotherapy alone for treatment of recurrent or refractory non small-cell lung cancer. J Clin Oncol. 2007;25(30):4743–4750. doi: 10.1200/JCO.2007.12.3026. [DOI] [PubMed] [Google Scholar]
- 214.Herbst RS, Johnson DH, Mininberg E, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23(11):2544–2555. doi: 10.1200/JCO.2005.02.477. [DOI] [PubMed] [Google Scholar]
- 215.Herbst RS, Ansari R, Bustin F, et al. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet. 2011;377(9780):1846–1854. doi: 10.1016/S0140-6736(11)60545-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Johnson BE, Kabbinavar F, Fehrenbacher L, et al. ATLAS: randomized, double-blind, placebo-controlled, phase IIIB trial comparing bevacizumab therapy with or without erlotinib, after completion of chemotherapy, with bevacizumab for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(31):3926–3934. doi: 10.1200/JCO.2012.47.3983. [DOI] [PubMed] [Google Scholar]
- 217.Akerley W, Boucher K, Rich N, et al. A phase II study of bevacizumab and erlotinib as initial treatment for metastatic non-squamous, non-small cell lung cancer with serum proteomic evaluation. Lung Cancer. 2013;79(3):307–311. doi: 10.1016/j.lungcan.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 218.Spigel DR, Burris HA, III, Greco FA, et al. Randomized, double-blind, placebo-controlled, phase II trial of sorafenib and erlotinib or erlotinib alone in previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(18):2582–2589. doi: 10.1200/JCO.2010.30.7678. [DOI] [PubMed] [Google Scholar]
- 219.Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013;382(9893):720–731. doi: 10.1016/S0140-6736(13)61715-8. [DOI] [PubMed] [Google Scholar]
- 220.Gasparini G, Longo R. The paradigm of personalized therapy in oncology. Expert Opin Ther Targets. 2012;16(Suppl 1):S7–S16. doi: 10.1517/14728222.2011.637921. [DOI] [PubMed] [Google Scholar]
- 221.Ma PC. Personalized targeted therapy in advanced non-small cell lung cancer. Cleve Clin J Med. 2012;79(Electronic Suppl 1):eS56–eS60. doi: 10.3949/ccjm.79.s2.12. [DOI] [PubMed] [Google Scholar]