

Abstract
A 29 year-old healthy Saudi female presented with a 1 week history of headache followed by decreased vision in both eyes. Biomicroscopy revealed anterior uveitis without hypopyon, posterior synechia or keratic precipitate. Fundus examinations were remarkable for serous retinal detachment and hyperemic discs. Fundus fluorescein angiogram showed a hot disc with multiple pinpoint leakage in both eyes. CT scan and MRI were normal, all uveitis workups were negative.
Five months later, the patient presented with complete criteria of Vogt–Koyanagi–Harada disease including a 2 weeks history of tinnitus, alopecia, poliosis and vitiligo. Headache alone followed by decreased vision before the onset of neurological and auditory symptoms can be an initial presentation of VKH disease. VKH should be considered in the differential diagnosis of atypical presentation of symptoms.
Keywords: Vogt–Koyanagi–Harada disease, Headache, Panuveitis
Introduction
Vogt–Koyanagi–Harada disease (VKH) is a bilateral intraocular inflammation usually associated with systemic manifestations such as meningismus, dysacusis, poliosis, and vitiligo.1–3 It is an autoimmune disease that targets melanocyte rich organs, such as the eye, inner ear, meninges, and skin.4
For unknown reasons, there is a greater preponderance in, females compared to males, in patients between the second and fifth decades and in heavily pigmented races.1
The diagnostic criteria of VKH disease are categorised into complete, incomplete and probable.5 Headache alone does not fulfil the diagnostic criteria and is insufficient for the diagnosis.
The Vogt–Koyanagi–Harada disease is clinically divided into four phases, prodromal, acute uveitic, convalescent, and chronic recurrent phase.1 The longer the duration of the disease, the greater the number of ocular complications associated with worse visual outcomes.6
Here we report a case of patient with headache as an initial presentation of VKH disease with typical complete clinical features manifesting later over the course of 1 year follow up.
Case report
A 29 year old Saudi female not known to have any medical illnesses presented with a 1 week history of headache followed by decreased vision in both eyes. There was no history of previous surgery or ocular trauma. There was no history of tinnitus, weakness, alopecia, vitiligo, poliosis, vomiting, back pain, numbness or joint pain. At presentation, visual acuity was 20/50 and 20/100 in the right eye and left eye, respectively. Anterior segment examination demonstrated +2 cells in anterior chamber bilaterally. Both pupils had normal reaction to direct light. The automated perimetry, colour vision and the intraocular pressure in each eye were within normal limits. Fundus examination showed multiple serous retinal detachments, deep yellow lesions consistent with choroiditis and hyperemic disc in both eyes without vitritis (Fig. 1).
Figure 1.
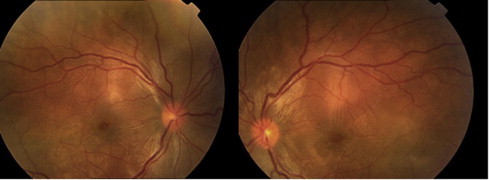
Bilateral multiple serous retinal detachment and disc hyperemia.
Fundus fluorescein angiogram (FFA) showed hot disc with the classic findings of multiple areas with pinpoint hyperfluorescent spots at the level of retinal pigment epithelium with leakage and pooling of dye in the subretinal space of both eyes (Fig. 2). Computed tomography (CT) scan and magnetic resonance imagining (MRI) were normal. All appropriate uveitis investigations including tuberculin skin tests, antinuclear antibody, angiotensin converting enzyme, venereal diseases research laboratory, blood titre for toxoplasma, rubella, cytomegalovirus, and herpes were negative.
Figure 2.
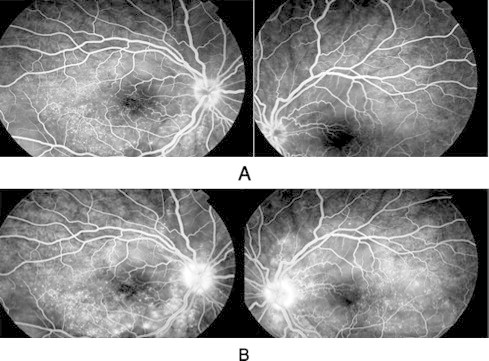
Early (A) and late (B) phase of fluorescein angiogram showing hot disc with multiple pinpoint hyperfluorescent area with leakage of dye in both eyes.
The patient was diagnosed as a probable case of VKH disease and started on topical prednisolone acetate 1% six times a day and oral prednisone 80 mg/day for 1 week then a weekly taper of 5 mg and cycloplegic agent (homatropine 2% two times a day). There was a marked improvement in intraocular inflammation after 2 weeks of treatment and the vision improved to 20/25 in each eye.
One month later, the patient was placed on tapering oral steroid when she started to complain of numbness of fingers and around the mouth. Magnetic resonance imaging (MRI) of the brain with and without contrast was performed to rule out multiple sclerosis. Both fundi showed normal retina and optic discs with resolving serous retinal detachment and disc hyperemia (Fig. 3).
Figure 3.
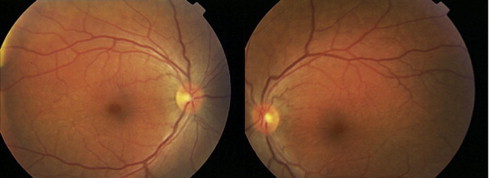
Resolved serous retinal detachment and optic disc hyperemia in both eyes, in comparison to Fig. 1.
Two months later, the patient elected to discontinue her medications without consultation and presented with severe anterior chamber reaction in both eyes and posterior synechia in the right eye. Topical and systemic steroids were resumed with cycloplegic agent.
Five months later, the patient presented with the complete diagnostic criteria for Vogt–Koyanagi–Harada disease including a 2 weeks history of tinnitus, alopecia, poliosis and vitiligo. She could not tolerate the oral steroid anymore, hence we prescribed systemic cyclosporine 75 mg/day and the oral steroid was reduced to 25 mg/day.
At one year follow up, the patient was asymptomatic but clinically she developed a sunset glow fundus bilaterally (Fig. 4). The previous finding of hyperfluorescent areas and leakage on fluorescein angiogram was resolved (Fig. 5).
Figure 4.
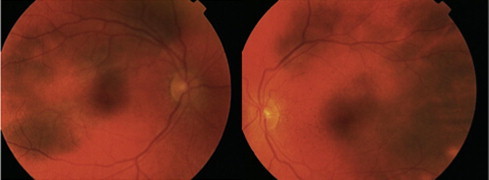
Bilateral sunset glow fundus.
Figure 5.
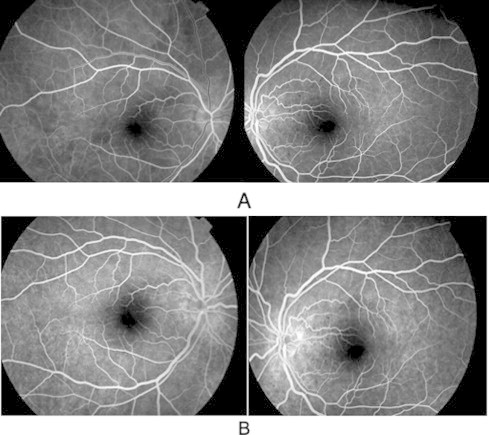
After treatment (A) early and (B) late phase of fluorescein angiogram showing bilateral resolving of hyperfluorescent area with no leakage. Compare with Fig. 2.
The plan was to continue the oral cyclosporine and low dose oral steroid for one more year to control the VKH disease.
Discussion
Our patient presented with rare and unusual initial findings of VKH disease manifesting as headache only without other features of meningisms (malaise, fever, nausea, abdominal pain, stiffness of the neck and back or combination of these factors) as described by revised diagnostic criteria of VKH,5 followed by decreased vision bilaterally. Most commonly, VKH patients present with auditory and neurological findings followed by decreased vision7 and headache alone is insufficient for the diagnosis of VKH disease.
On the literature review to the best of my knowledge very few cases had been reported with headache as an initial manifestation of VKH disease (Cho et al.,7 Tavsanli et al.8 and Kosma et al.9).
Although headache is a common symptom in VKH cases it is not usually the only initial symptom and is most commonly accompanied by disturbances in visual acuity. Beniz et al.10 studied 48 patients, most presented with an initial complaint of diminished visual acuity, although 32 patients (67%) had headache but it was not the initial presentation. Chee et al.11 reviewed 178 eyes of 89 patients, the most common presenting symptom was blurring of vision (98.88%) and it was accompanied by headaches in 51.69% of patients. Tugal-Tutkun et al12 analysed 45 patients, and found headache was the most frequent symptom which was present in 31 patients (69%) but the decreased visual acuity also occurred at presentation. For example they found 0.1 visual acuity in 23 eyes (60.5%), 0.1–0.5 visual acuity in ten eyes (26.3%), and better than 0.5 in five eyes (13.2%).
Differing features of headache have been described in VKH. For example, Kosma et al.9 described the headache as continuous, diffuse and throbbing in nature, while Cho et al.7 described it as a thunderclap headache which is different from our patient as she had diffuse headache, moderate in intensity without any other specific features of headache.
Early in the course of the disease our patient fulfilled the criteria of probable VKH disease as she had bilateral ocular involvement with no previous history of trauma or ocular surgery, no evidence of other ocular disease, no neurological (headache only is insufficient) and auditory findings. Five months later patient fulfils the criteria of complete VKH disease as she developed auditory (tinnitus) and integumentary findings (alopecia, poliosis and vitiligo).
The management for this condition is high dose systemic corticosteroid early in the disease, which is associated with decreased dermatological feature, ocular inflammation, recurrence, and better visual outcome.13–15 Systemic corticosteroid in acute VKH disease whether oral therapy only or a combination of intravenous and oral, do not appear to matter significantly in terms of change in visual acuity or development of visually significant complications.16
In conclusion headache alone followed by decreased vision before the onset of neurological and auditory symptoms can be an initial presentation of VKH disease, which should be considered in the differential diagnosis of atypical cases of VKH disease.
Conflict of interest
The authors declared that there is no conflict of interest.
References
- 1.Moorthy R.S., Inomata H., Rao N.A. Vogt–Koyanagi–Harada syndrome. Surv Ophthalmol. 1995;39:265–292. doi: 10.1016/s0039-6257(05)80105-5. [DOI] [PubMed] [Google Scholar]
- 2.Kitamura M., Takami K., Kitaichi N. Comparative study of two sets of criteria for the diagnosis of Vogt–Koyanagi–Harada’s disease. Am J Ophthalmol. 2005;139:1080–1085. doi: 10.1016/j.ajo.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 3.Yamaki K., Hara K., Sakuragi S. Application of revised diagnostic criteria for Vogt–Koyanagi–Harada disease in Japanese patients. Jpn J Ophthalmol. 2005;49:143–148. doi: 10.1007/s10384-004-0165-9. [DOI] [PubMed] [Google Scholar]
- 4.da Silva F.T., Hirata C.E., Olivalves E., Oyamada M.K., Yamamoto J.H. Fundus-based and electroretinographic strategies for stratification of late-stage Vogt–Koyanagi–Harada disease patients. Am J Ophthalmol. 2009;148(6):939–945.e3. doi: 10.1016/j.ajo.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 5.Read R.W., Holland G.N., Rao N.A. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131:647–652. doi: 10.1016/s0002-9394(01)00925-4. [DOI] [PubMed] [Google Scholar]
- 6.Read R.W., Rechodouni A., Butani N. Complications and prognostic factors in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2001;131(5):599–606. doi: 10.1016/s0002-9394(01)00937-0. [DOI] [PubMed] [Google Scholar]
- 7.Cho J.H., Ahn J.Y., Byeon S.H. Thunderclap headache as initial manifestation of Vogt–Koyanagi–Harada disease. Headache. 2008;48:153–155. doi: 10.1111/j.1526-4610.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 8.Tavsanli M., Uluduz D., Saip S. Vogt- Koyanagi-Harada disease: headache as an initial manifestation. J Headache Pain. 2008;9(4):255–256. doi: 10.1007/s10194-008-0045-7. Epub 2008 May 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosma K.K., Kararizou E., Markou I. Headache as a first manifestation of Vogt–Koyanagi–Harada disease. Med Princ Pract. 2008;17(3):253–254. doi: 10.1159/000117802. Epub 2008. [DOI] [PubMed] [Google Scholar]
- 10.Beniz J., Forster D.J., Lean J.S., Smith R.E., Rao N.A. Variations in clinical features of the Vogt–Koyanagi–Harada syndrome. Retinal. 1991;11:275–280. doi: 10.1097/00006982-199111030-00001. [DOI] [PubMed] [Google Scholar]
- 11.Chee S.P., Jap A., Bacsal K. Spectrum of Vogt–Koyanagi–Harada disease in Singapore. Int Ophthalmol. 2007;27:137–142. doi: 10.1007/s10792-006-9009-6. [DOI] [PubMed] [Google Scholar]
- 12.Tugal-Tutkun I., Ozyazgan Y., Akoya Y.A. The spectrum of Vogt–Koyanagi–Harada disease in Turkey. Int Ophthalmol. 2007;27:117–123. doi: 10.1007/s10792-006-9001-1. [DOI] [PubMed] [Google Scholar]
- 13.Read R.W. Vogt–Koyanagi–Harada disease. Ophthalmol Clin North Am. 2002;15:333–341. doi: 10.1016/s0896-1549(02)00025-1. [DOI] [PubMed] [Google Scholar]
- 14.Miyanaga M., Kawaguchi T., Shimizu K. Influence of early cerebrospinal fluid-guided diagnosis and early high-dose corticosteroid therapy on ocular outcomes of Vogt–Koyanagi–Harada disease. Int Ophthalmol. 2007;27:183–188. doi: 10.1007/s10792-007-9076-3. [DOI] [PubMed] [Google Scholar]
- 15.Chee S.P., Jap A., Bacsal K. Prognostic factors of Vogt–Koyanagi–Harada disease in Singapore. Am J Ophthalmol. 2009;147:154–161. doi: 10.1016/j.ajo.2008.07.044. [DOI] [PubMed] [Google Scholar]
- 16.Read R.W., Yu F., Accorinti M. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2006;142(1):119–124. doi: 10.1016/j.ajo.2006.02.049. [DOI] [PubMed] [Google Scholar]