

Abstract
Background
Clenbuterol (Cl), a β2 agonist, is associated with enhanced myocardial recovery during left ventricular assist device (LVAD) support, and exerts beneficial remodelling effects during mechanical unloading (MU) in rodent heart failure (HF). However, the specific effects of combined Cl+β1 blockade during MU are unknown.
Methods and Results
We studied the chronic effects (4 weeks) of β2-adrenoceptor (AR) stimulation via Cl (2 mg/kg/day) alone, and in combination with β1-AR blockade using metoprolol ((Met), 250 mg/kg/day), on whole heart/cell structure, function and excitation-contraction (EC) coupling in failing (induced by left coronary artery (LCA) ligation), and unloaded (induced by heterotopic abdominal heart transplantation (HATx)) failing rat hearts. Combined Cl+Met therapy displayed favourable effects in HF: Met enhanced Cl's improvement in ejection fraction (EF) whilst preventing Cl-induced hypertrophy and tachycardia. During MU combined therapy was less beneficial than either mono-therapy. Met, not Cl, prevented MU-induced myocardial atrophy, with increased atrophy occurring during combined therapy. MU-induced recovery of Ca2+ transient amplitude, speed of Ca2+ release and sarcoplasmic reticulum Ca2+ content was enhanced equally by Cl or Met mono-therapy, but these benefits, together with Cl's enhancement of sarcomeric contraction speed, and MU-induced recovery of Ca2+ spark frequency, disappeared during combined therapy.
Conclusions
Combined Cl+Met therapy shows superior functional effects to mono-therapy in rodent HF, but appears inferior to either mono-therapy in enhancing MU-induced recovery of EC coupling. These results suggest that combined β2-AR simulation +β1-AR blockade therapy is likely to be a safe and beneficial therapeutic HF strategy, but is not as effective as mono-therapy in enhancing myocardial recovery during LVAD support.
Introduction
The use of LVADs in end stage HF continues to expand [1]. LVADs are used primarily as “bridge to transplantation”. In a minority of patients they have been used as a “bridge to recovery” (BTR), where sufficient functional recovery of the failing heart is achieved, allowing device removal. However, explantation rates remain universally low (≤10%) [2], [3].
HF-associated remodelling is a complex series of whole heart, cellular, molecular, structural and functional changes affecting various pathways, including EC coupling, the extracellular matrix (ECM), apoptosis and adrenergic receptor signalling [4]. Previously thought to be uni-directional, such adverse remodelling is now considered reversible, a process termed reverse remodelling. Pharmacological intervention induces mild reversal of HF remodelling, demonstrating functional and prognostic benefit [5]–[7], however, a more substantial degree of reverse remodelling is achieved via LVAD [8]. Despite profound LVAD-induced reverse remodelling, functional myocardial recovery is rare. This disparity is proposed to arise due to negative MU-induced changes occurring during prolonged unloading [9], [10]. Indeed, in animals and humans, initial contractile recovery of failing hearts is seen to subsequently worsen during prolonged MU [3], [11]. Time dependent worsening of EC coupling [12], Ca2+ handling [11] and myocardial atrophy [13] are proposed negative drivers of contractile performance.
“Combination therapy” describes a strategy in which pharmacotherapy is combined with LVAD support, to enhance LVAD–induced positive remodelling and/or combat LVAD-induced “negative” remodelling [10]. In our institute this strategy - the “Harefield Protocol” - successfully enhanced rates of functional recovery (>63%) in patients with dilated cardiomyopathy [14], [15]. In this unique protocol, the β2-adrenoceptor (AR) agonist Cl was employed alongside conventional HF pharmacotherapy, primarily as an agent to prevent myocardial atrophy. Cl's ability to induce physiological cardiac hypertrophy [16] mandated its inclusion in this protocol, but whether anti-atrophic properties contributed to enhancing functional recovery is unclear [17]. Hence, Cl's precise role in enhancing functional recovery remains poorly defined and controversial. Beyond hypertrophic effects, Cl demonstrates other wide-ranging favourable whole heart/cellular functional, structural, metabolic, cell-survival, EC coupling and gene expression effects in non-failing [16], [18], failing [19], [20] and unloaded [19], [21] hearts.
BTR remains controversial, but the clinical success of combination therapy (14, 15) justifies continued scrutiny of this strategy. Therefore, the search for other novel pharmacological strategies aimed at enhancing LVAD-induced functional recovery continues, and we have recently shown the pacemaker current inhibitor ivabradine and Met to show positive effects on myocardial size, EC coupling and the ECM during mechanical unloading in rodent HF [22].
The efficacy of β-AR blockade in HF therapy is established [4], [6], [23] and is predominantly secondary to blockade of detrimental β1-AR signalling [4]. Greater understanding of the roles played by β1-/β2-AR signalling indicates distinct and often opposing physiological and pathological actions on cardiac structure and function [24], with chronic β1-AR signalling associated with pro-apoptotic and maladaptive remodelling [25] and chronic β2-AR signalling with cardio-protective actions, improved function and myocyte viability [26], [27]. Supporting this, combined β1-AR blockade (Met) and β2-AR stimulation (fenoterol) has shown synergistic beneficial remodelling effects in rodent HF [24], although lack of synergy (Cl+Met) has also been demonstrated [20]. These studies instituted pharmacotherapy early after LCA ligation (<3 weeks) and combined drug effects in an established rodent HF model remain unknown. β2-AR stimulation (Cl) demonstrates positive effects during MU in rodent studies [19], [28], however, the specific effects of combined β2-AR stimulation and β1-AR blockade remain unstudied within this setting.
In this study, we report for the first time the chronic effects of combined β2-AR stimulation (Cl) and β1-AR blockade (Met) on reverse remodelling during MU in a rodent HF model. In particular, we have examined the drug effects on whole heart/cell size, function, EC coupling and T-tubule structure in a rat model of established HF, 12 weeks post LCA ligation, and during MU using HATx.
Methods
Ethics Statement
The investigation conformed to the Guide for the care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No 85–23, revised 1996). Experiments were approved by the Harefield Heart Science Centre ethics review panel.
(For extended methods please refer to Methods S1).
Animal models
Induction of heart failure
HF was induced via permanent LCA ligation as previously described [11], [19]–[22], [24]. Animals were anaesthetised with isoflurane (5% induction, 1.5% maintenance) mixed with O2 (2–3 L/min), intubated and mechanically ventilated via rodent ventilator, and a left thoracotomy performed. The pericardium was opened and heart visualised, the proximal LCA was identified and a suture (6–0 prolene) placed underneath the artery and lightly tied. Heamostasis was achieved and the chest closed. Sham operated animals underwent the identical protocol but the suture was not tied. Acute 24 hour mortality was 25% in the LCA ligation group and 0% in the sham group. Male syngeneic Lewis rats (10 weeks old ∼200 g, Harlan, UK) were used for all experiments to avoid the need for immunosuppression following subsequent heterotopic abdominal transplant (HATx) in the mechanical unloading (MU) groups. 1 week after surgery LV dimensions and function (ejection fraction (EF)) were assessed by transthoracic echocardiography (TTE) (Acuson Sequoia™ 256; Acuson, USA), as previously described [18]–[21], [24]. LAD ligation animals with an EF less than 40% were included in the study, as in our experience animals with baseline function greater than this were unlikely to develop severe dysfunction 12 weeks following surgery. 12 weeks following LCA ligation the presence of severe cardiac dysfunction was confirmed via repeat TTE, and animals were randomised to groups (n = 8). All groups had similar average EF and variability. 3 month mortality was 1% following LCA ligation and 0% in sham group. TTE was performed under general anaesthesia with isoflurane 1%, mixed with O2 (2–3 L/min) to provide adequate sedation but minimal cardiac depression. Experiments were not performed in non-failing hearts as these show an exaggerated atrophic response during unloading compared with failing hearts [29].
In order to test the effects of pharmacotherapy (Cl and/or Met) in combination with MU, failing hearts (12 weeks after LCA ligation) were transplanted into the abdomen of syngeneic male Lewis rats (weight ∼250 g) as previously described [11]–[13], [19], for a period of 4 weeks. In brief, both animals (recipient and donor) were anaesthetised with isoflurane (5% induction, 1% maintenance) mixed with O2 (2–3 L/min), and self-ventilated throughout the procedure. The failing heart was removed from the donor animal under cardioplegic arrest, and the donor aorta anastomosed to the recipient abdominal aorta and the donor pulmonary artery to the recipient inferior vena cava. Total ischaemic time was <40 minutes and operative mortality <4%, with no deaths occurring in any of the treatment groups during the 4 week treatment period.
Recipients were then assigned to 4 treatment groups: (1) Cl (Sigma, England) 2 mg/kg/day via subcutaneous infusion using osmotic minipump (Model 2002, Alzet), (2) Met (Sigma, England) 250 mg/kg/day in drinking water, (3) Met 250 mg/kg/day +Cl 2 mg/kg/day via drinking water and osmotic minipump respectively, (4) no drug treatment (MUHF+Cl, MUHF+Met, MUHF+MetCl and MUHF, respectively). Animals in Met-treated and MUHF group also received an osmotic minipump containing normal saline. All groups contained 8 animals and treatment duration was 4 weeks. At end of treatment Cl levels in blood were measured as previously described [18], with no difference between the different groups treated (in µM: HF+Cl = 0.17+/−0.01, n = 8; MUHF+Cl = 0.17+/−0.01, n = 8; HF+MetCl = 0.16+/−0.01; n = 8; MUHF+MetCl = 0.16+/−0.02, n = 8; p = 0.93).
In order to test the effects of Cl and Met in non-transplanted failing hearts, HF animals were randomly allocated to 4 treatment groups: (1) Cl 2 mg/kg/day, (2) Met 250 mg/kg/day, (3) combined Met+Cl treatment, (4) no drug treatment (HF+Cl, HF+Met, HF+MetCl and HF, respectively). Drug administration was via drinking water and osmotic minipump, exactly as described above. At the end of the treatment period in vivo heart function was measured via repeat TTE.
We have compared the effects of Met during mechanical unloading with those of ivabradine in a previously published study [22]. Therefore, we have included this data here for comparison with the effects of Cl and combined MetCl therapy. The data acquired from the Met group are contemporary and obtained in otherwise identical conditions to the data for the Cl and MetCl groups.
In-vivo assessment of heart rate
4 animals per treatment group received implantable ECG telemetry transmitters (CA-F40 Data Sciences International, Minneapolis, MN) on day 0 of the 4 week treatment period, for in vivo HR and arrhythmia studies. Animals underwent continuous 24 hour ECG recording at weekly intervals during the treatment period. In vivo ECG recordings were acquired using Dataquest ART 3.1 software (Data Sciences International, Minneapolis, MN), and offline HR/arrhythmia analysis performed using ECG-Auto 2.4 software (EMKA, France).
Animal anaesthesia, analgesia and euthanasia
Adequacy of anaesthesia during all procedures was monitored by loss of reflexes, degree of muscle relaxation and respiration rate. Respiration rate and body temperature were monitored throughout all procedures and body temperature maintained at 37°C, via heating mat. Analgesia was provided by subcutaneous (SC) injection of vetergesic (buprenorphine), just prior to skin incision. For LCA ligation and implantation of telemetry device a single dose of 0.03 mg/kg was utilised, and for heterotopic abdominal heart transplant both the donor and recipient received a single dose of 0.05 mg/kg. Post-operative condition of the rats was monitored, and repeat SC injection administered as required (0.01–0.05 mg/kg) every 12 hours during the first week post-operative week. At the end of the 4 week treatment period animals were sacrificed via schedule 1 cervical dislocation. In each group, 4 hearts were utilised for acute cellular experiments, and 4 for histological analysis.
Cardiomyocyte studies
LV cardiomyocytes were isolated by standard enzymatic digestion for 8–10 minutes using collagenase (1 mg/ml, Worthington, USA) and hyaluronidase (0.6 mg/ml, Sigma, England) [19] and used within 6 h.
Assessment of sarcomere shortening and calcium handling
Simultaneous assessment of sarcomere shortening and Ca2+ handling in field stimulated cardiomyocytes was performed using an inverted microscope (TE 200), Ionoptix system (Ionoptix Corporation, USA) and the Ca2+ sensitive, single-excitation dual emission fluorescent dye Indo-1 AM (Molecular Probes, USA). In addition, sarcoplasmic reticulum (SR) Ca2+ uptake (SERCA) and sodium-calcium exchanger (NCX) contribution to Ca2+ extrusion were assessed using rapid caffeine application as previously described [19].
Assessment of electrophysiological parameters
Cells were studied using a MultiClamp 700A (Axon Instruments) in whole cell patch configuration. Current-voltage relationships for L-type Ca2+ current were studied and normalized to cell capacitance, as previously described [19].
Assessment of cell volume, t tubules and calcium sparks with confocal microscopy
Cell volume and t-tubule organisation were studied using the membrane binding dye di-8-Anepps (Molecular Probes, Eugene, OR, USA) and the Zeiss Axiovert microscope (Carl Zeiss, Oberkochen, Germany) with an LSM 510 confocal attachment. In addition, local changes in Ca2+ levels were assessed using the fluorescent dye Fluo-4 AM (Molecular Probes, USA) and the same confocal microscope as previously described [29].
Statistical analysis
Statistical comparison of data was performed using one-way analysis of variance followed by Bonferroni post-hoc test for individual significant differences, or Student's t-test where appropriate. All statistical analyses were performed using Prism 4 software (Graph-Pad Software, Inc.) and P<0.05 was considered significant. Data are expressed as mean ±SEM [n], where n is the number of cells unless otherwise specified. All of the experiments were performed using a minimum of four animals, unless otherwise stated.
Results
Heart rate
There was no difference in average HR between sham-operated and HF group at any time point (mean HR (bpm) at 4 weeks: sham 332±4.8 vs. HF 329±5.5; P>0.05). HR of transplanted failing hearts (MUHF) was significantly lower (∼20%) than that of non-transplanted failing hearts at all time-points ( Figure 1A ). Cl caused a consistent increase in HR (∼12%) at all time-points during MU, with Met either alone, or in combination with Cl reducing HR by approximately 15% ( Figure 1A ). Assessment of HR of non-transplanted failing hearts revealed similar drug effects: Cl caused a significant increase in HR (∼10%) compared to HF group during first 3 weeks of therapy, but this effect was not present at week 4. Met caused significant and equivalent HR reduction (∼20%) either alone, or in combination with Cl, compared to HF group at all-time points ( Figure 1B ).
Figure 1. Average HR of mechanically unloaded failing hearts (MUHF) (A) and non-transplanted failing hearts (B) measured using telemetry devices (n = 4 per group).
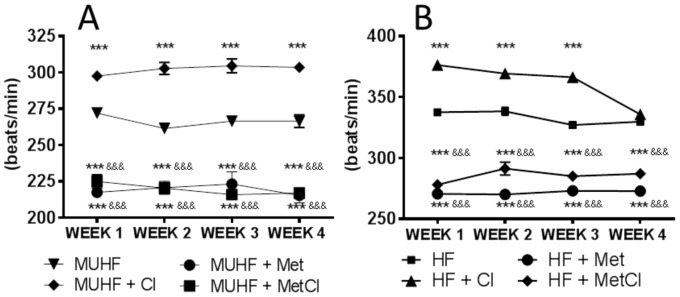
Cl increased HR significantly at all time-points during MU, whereas Met caused equal HR reduction either alone, or in combination with Cl at all time-points during MU. ***P<0.001 vs. MUHF and &&&P<0.001 vs. MUHF+Cl. Cl increased HR in-non-transplanted failing hearts significantly but this effect was lost at week 4. Met caused equal HR reduction in-non-transplanted failing hearts either alone, or in combination with Cl, at all time-points. ***P<0.001 vs. HF and &&&P<0.001 vs. HF+Cl.
LV function
LCA ligation induced LV dysfunction after 12 weeks (time of randomisation), characterised by reduced LVEF (%): sham 83.3±0.8 vs. HF 37.6±0.7; reduced LVFS (%): sham 48.3±0.7 vs. HF 16.6±0.5; and LV chamber dilatation (diastolic diameter (cm)): sham 0.60±0.03 vs. HF 0.98±0.04, P<0.001 for all parameters.
No difference in these parameters was detected between treatment groups prior to commencing therapy ( Figure 2A and B ). In-vivo function of transplanted failing hearts was not assessed, but that of non-transplanted hearts was assessed via repeat TTE at the end of the 4 week treatment period. Cl improved contractile function of non-transplanted failing hearts, and improvement in LVEF was enhanced further by combination with Met ( Figure 2A and B ). Met-induced increase in EF and FS was not statistically significant.
Figure 2. Effect of Cl, Met and combined MetCl therapy on contractile function in non-transplanted failing hearts, EF (A) and FS (B): No difference in baseline values in treatment groups was seen (light grey bars).

Average values at end of treatment period are shown (black bars): Cl-treated group showed improved EF and FS compared to untreated HF group, and improvement in EF was further enhanced in MetCl group. Met-induced improvement in EF and FS was not statistically significant. ***P<0.001 (n = 8 per group).
Cardiac and cardiomyocyte size
LCA ligation induced cardiac and cellular hypertrophy, shown by increased total HW, HW∶BW ratio and cardiomyocyte volumes, compared to sham-operated group ( Table 1 ). MU of failing hearts induced myocardial atrophy at whole heart and cardiomyocyte level, with both HW and cardiomyocyte volumes falling below those of sham values ( Figure 3A and B ). HW of MUHF group was ∼40% smaller than that of HF group and ∼17% smaller than sham, with cell volume being ∼55% lower than in HF group and ∼26% smaller than sham volumes. Cl had no effect on myocardial atrophy. Met prevented both whole heart and cellular atrophy, restoring mean HW and cardiomyocyte volumes to sham values, whilst combined MetCl therapy worsened MU-induced atrophy ( Figure 3A and B ).
Table 1. Anatomical and cell volume data (16 weeks after LCA ligation).
| Sham | HF | |
| N | 8 | 8 |
| Body weight (g) | 386±5.6 | 415±6.2** |
| Heart weight (g) | 1.2±0.05 | 1.6±0.05*** |
| Heart weight/Body weight ×1000 | 3.0±0.1 | 3.6±0.1*** |
| Cell volume (µm3) | 36248±12012 | 59705±13508*** |
**P<0.01,
***P<0.001 vs. sham.
Figure 3. Effect of Cl, Met and combined MetCl treatment on heart weight (HW) (A) and cardiomyocyte volume measured using confocal microscopy (B) during unloading (MUHF).

Prevention of MU-induced cardiac and cardiomyocyte atrophy is achieved by Met and not Cl, with combined MetCl therapy increasing atrophy. *P<0.05, **P<0.01 and ***P<0.001, (HW and cardiomyocyte volume data acquired from 8 and 4 hearts per group, respectively). Effect of Cl, Met and combined MetCl therapy on heart weight∶body weight ratio (HW∶BW) (C) and cardiomyocyte volume (D). HF-induced cardiac hypertrophy was enhanced by Cl therapy but this effect disappeared during combined MetCl therapy. HF-induced myocyte hypertrophy was partially attenuated by Met, but this effect was lost during combination MetCl therapy (HW and cardiomyocyte volume data acquired from 8 and 4 hearts per group, respectively).
In non-transplanted failing hearts Cl augmented cardiac hypertrophy, an effect that disappeared during combined MetCl therapy ( Figure 3C ). At a cellular level Met induced partial regression of cellular hypertrophy, but this effect was also lost during combined MetCl therapy ( Figure 3D ).
Cardiomyocyte EC coupling
MU-induced recovery of deranged EC coupling
LCA ligation caused impaired cardiomyocyte contractility and deranged Ca2+ cycling: speed of sarcomeric contraction, relaxation and Ca2+ release, along with Ca2+ transient amplitude were all decreased, sarcomeric contraction amplitude remaining unchanged ( Figure 4 A–E ). MU normalised speed of sarcomeric contraction and this effect was further enhanced by Cl, with Met and MetCl therapy having no added effect ( Figure 4B ). MU partially improved speed of relaxation, and this effect was antagonised by Met, with no additional observed changes following Cl and MetCl treatment ( Figure 4C ). In addition, MU recovered Ca2+ transient amplitude, speed of Ca2+ release and SR Ca2+ content, and this recovery was further enhanced by both Cl and Met mono-therapy equally, but not by combined MetCl treatment ( Figure 4 D–F).
Figure 4. Effects of Cl, Met and MetCl therapy on cardiomyocyte contractility (A–C) and Ca2+ handling (D–F) during mechanical unloading (MUHF), measured using Indo-1 and Ionoptix system.

Cl's enhancement of MU-induced recovery of speed of sarcomeric contraction (B) and Met's antagonism of MU-induced improvement in speed of relaxation (C) are shown. Cl and Met's enhancement of MU-induced recovery of Ca2+ transient amplitude (D), speed of Ca2+ release (E) and SR Ca2+ content (F), and lack of enhancement during combined MetCl therapy is shown. *P<0.05, **P<0.01 and ***P<0.001. Representative traces of sarcomeric contractions (G) and Ca2+ transients (H) are shown.
An increased SR Ca2+ leak, secondary to altered RyR function, is one mechanism contributing to reduced SR Ca2+ load in HF [30]. Ca2+ sparks are complex entities regulated by multiple factors, and represent a measure of diastolic SR Ca2+ release [29]. Ca2+ spark frequency was increased in HF group compared to sham group suggesting increased SR Ca2+ leak ((sparks/100 µm/sec): sham 0.80±0.17 [37] vs. HF 3.00±0.34 [40]; P<0.001). MU normalised HF-induced increase in Ca2+ spark frequency (sparks/100 µm/sec): MUHF 0.82±0.13 [36] vs. HF 3.00±0.48 [40]; P<0.001. This recovery was unaffected by either Cl or Met mono-therapy, but abolished by combined MetCl therapy (sparks/100 µm/sec): MUHF+Cl 1.67±0.27 [35] vs. MUHF+Met [30] 1.70±0.26; P>0.05, both P>0.05 vs. MUHF, and P<0.001 vs. MUHF+MetCl [30] 3.51±0.36; P<0.001 vs. MUHF and P>0.05 vs. HF. MU also caused recovery of HF-induced depression of L-type Ca2+ current and t-tubule density ( Figure 5 ). However, this recovery was largely antagonised by all three drug treatments; with only maintenance of L-type Ca2+ current recovery occurring in the combined MetCl-treated group ( Figure 5A and B ).
Figure 5. MU-induced recovery of depressed L-type Ca2+ current (A) and t-tubule density (B).

Maintenance of L-type Ca2+ current recovery by combined MetCl therapy, and antagonism of such recovery by Cl or Met mono-therapy (A) is shown, along with antagonism of t-tubule density recovery by all treatments (B). *P<0.05, **P<0.01 and ***P<0.001. Representative di-8-Anepps stained cells from sham (C), HF (D), MUHF (E), MUHF+Cl (F), MUHF+Met (G) and MUHF+MetCl (H) groups are shown. Data for the Met group has been previously published [22] and added here for comparison.
Effects of Cl and Met on deranged EC coupling in non-transplanted failing hearts
Met therapy, either alone or in combination with Cl normalised speed of sarcomeric contraction, relaxation and Ca2+ release, along with Ca2+ transient amplitude, while Cl mono-therapy displayed no effect ( Figure 6B–E ). Met, Cl and combined MetCl therapy all recovered SR Ca2+ content, with the greatest improvement seen following Met therapy ( Figure 6F ). Cl alone caused significant reduction in spark frequency (HF+Cl 1.10±0.17 [30]; P<0.01 vs. HF), whereas spark frequency was no different to HF in the Met or MetCl-treated groups (HF+Met 2.10±0.22 [30] and HF+MetCl 2.00±0.32 [32], both groups P>0.05 vs. HF). Drug therapies had differing effects on L-type Ca2+ current and t-tubule density. HF-induced depression of both these parameters was normalised by Cl; Met therapy alone, or in combination with Cl showed no effect ( Figure 7 ).
Figure 6. Effects of Cl, Met and MetCl therapy on cardiomyocyte contractility (A–C) and Ca2+ handling (D–F) from non-transplanted failing hearts, measured using Indo-1 and Ionoptix system are shown.

Full recovery of speed of sarcomeric contraction (B), relaxation (C), Ca2+ transient amplitude (D) and speed of Ca2+ release (E) caused by Met therapy, either alone or in combination with Cl, and lack of improvement following Cl mono-therapy is shown. Superiority of Met mono-therapy in recovering depressed SR Ca2+ content is also shown (F). *P<0.05, **P<0.01 and ***P<0.001.
Figure 7. Full recovery of HF-induced depression of L-type Ca2+ current (A) and t-tubule density (B) caused by Cl, and lack of improvement in these parameters following Met and MetCl therapy are shown.

***P<0.001. Representative di-8-Anepps stained cells from sham (C), HF (D), HF+Cl (E), HF+Met (F) and HF+MetCl (G) groups. Data for the Met group has been previously published [22] and added here for comparison.
Discussion
In the first part of this study we showed that Met but not Cl prevented MU-induced myocardial atrophy, with combined MetCl therapy worsening atrophic remodelling. MU induced recovery of deranged EC coupling, with either Cl or Met mono-therapy, but not combined MetCl therapy, broadly enhancing this recovery. MU-induced recovery of Ca2+ transient amplitude, speed of Ca2+ release and SR Ca2+ content was enhanced by Cl or Met mono-therapy equally. However, these positive effects, together with Cl's enhancement of speed of sarcomeric contraction, were lost during combined MetCl therapy, along with MU-induced recovery of Ca2+ spark frequency.
The second part of this study showed combined β2-AR stimulation (Cl) and β1-AR blockade (Met) to improve depressed whole heart function in HF, to a greater degree than either mono-therapy. Cl's improvement in LVEF was enhanced by Met, but the mechanisms underlying this superiority appeared independent of cellular EC coupling. Met and Cl mono-therapy displayed distinct EC coupling effects. Met's effects were more favourable than those of Cl, with lack of evidence supporting additive or synergistic benefit, and more suggestive of selected antagonism during combined therapy. Met alone, and in combination with Cl normalised speed of sarcomeric contraction, relaxation, Ca2+ release and Ca2+ transient amplitude, and enhanced SR Ca2+ content; whereas Cl mono-therapy improved SR Ca2+ content to a lesser degree than Met, and normalised deranged Ca2+ spark frequency, L-type Ca2+ current and t-tubule density, positive effects that were lost during combined MetCl therapy. Of note, undesirable Cl-induced hypertrophic/chronotropic effects were prevented during combined therapy.
These results suggest that A) Cl's success in enhancing functional recovery in LVAD-supported patients is unrelated to myocardial size, and reliant on positive EC coupling effects, B) combined β2-AR stimulation +β1-AR blockade is likely to be a safe and beneficial HF therapeutic pharmacological strategy and C) either the strategy of β2-AR stimulation or β1-AR blockade is likely to be superior to that of combined β2-AR stimulation +β1-AR blockade in enhancing functional recovery during LVAD support. This is, however, in contrast with the current clinical experience with β1-AR blockade in patients with LVADs and could be explained by the type and degree of adverse remodelling obtained in our models. It cannot be excluded that subpopulations of LVAD patients with specific, lower degrees of dysfunction may benefit from this pharmacological treatment and more clinical studies are warranted.
Combined β2-AR stimulation and β1-AR blockade during mechanical unloading
Myocardial atrophy
LVAD-mediated unloading of failing ventricles regresses cardiac and cellular hypertrophy [8]. Whether such regression progresses during prolonged clinical LVAD support towards myocardial “atrophy” i.e. decreased cardiac and cell size to sub-normal values, remains controversial [31]. In contrast, convincing experimental data derived mainly from rodent studies employing HATx clearly demonstrate atrophy of normal [13], [28] and failing [11] hearts. It must be emphasised that in this context atrophy relates to decreased size and not function.
A key question surrounding Cl is whether anti-atrophic properties contributed to enhanced recovery rates produced by the Harefield Protocol [18]. Findings of similar myocyte size in both explanted and non-recovered patients treated by this protocol, suggest Cl's pro-recovery effects were unrelated to myocardial size [17]. This present study reinforces this prospect. We show Cl to be ineffective in preventing myocardial atrophy associated with prolonged unloading (4 weeks), and this agrees with previous work [28] in non-failing rat hearts undergoing 2 weeks unloading. In contrast, we previously showed Cl to limit regression of rodent HF myocyte hypertrophy during short-term unloading (1 week) [19]. However, such a brief period of MU, during which atrophic remodelling is regarded as being sub-maximal [11], [13] was considered inadequate, and a poor representation of prolonged clinical LVAD support.
As we have previously shown [22], Met prevented myocardial atrophy, but this effect was lost during combined MetCl therapy with actual worsening of atrophy. The latter observation is unexpected, if compared with the effects of the combined therapy in normally-loaded hearts; this can be due to additional detrimental consequences of mechanical unloading and require to be further studied. Atrophic remodelling is complex and the multiple pathways involved poorly defined [32]. The ubiquitin proteosome, calpain, lysosomal proteolysis and authophagy systems, and mTOR IGF-1/PI3K/AKT and ERK-1 pathways are all altered during MU [32]. These growth regulatory pathways, along with the TGF-β, CAMKII and calcineurin/NFAT hypertrophic signalling pathways, known components under β-AR influence [33], may represent Met's route of action, and augmentation of myocyte number (not assessed in this study) via regenerative mechanisms is another possibility. This finding is particularly important as, to date, no pharmacotherapy has proven effective in attenuating myocardial atrophy, with success via haemodynamic loading strategies alone [34], re-emphasising the critical importance of load in regulation of cardiac mass [13]. Such loading, potentially brought about by HR reduction and subsequent augmentation of LV filling, may have driven Met's anti-atrophic actions; but the lack of effect during combined MetCl therapy, despite an equivalent reduction in HR, makes this mechanism unlikely.
EC coupling during mechanical unloading
LVAD support induces reversal/alteration in expression and function of numerous Ca2+ handling elements adversely remodelled during HF. Recovery of SR Ca2+ content, L-type Ca2+ current fast inactivation and Ca2+ transient amplitude is seen [35], and these parameters show positive correlation with functional recovery in patients treated with the Harefield Protocol [17]. LVAD-induced enhanced SERCA 2A expression [36] and stabilisation of RyR function [37] are potentially involved mechanisms.
In the present study, MU recovered HF-induced deranged EC coupling with normalisation of Ca2+ transient amplitude, speed of Ca2+ release and sarcomeric contraction, SR Ca2+ content, Ca2+ spark frequency, L-type Ca2+ current amplitude and t-tubule density. Drug effects on this recovery were not clear-cut but overall, the positive effects of either mono-therapy disappeared during combined therapy. MU-induced recovery of Ca2+ transient amplitude, speed of Ca2+ release and SR Ca2+ content, proven markers of recovery during LVAD support [17], were equally enhanced by Cl and Met mono-therapy, with Cl also enhancing speed of sarcomeric contraction. These effects were lost during combined therapy along with MU-induced recovery of RyR function (normalised Ca2+ spark frequency).
Cl's effects may originate from improved myofilament sensitivity [19] and SERCA2a expression [28] previously shown during MU. MU is associated with increased β2-AR mRNA expression [21] and adenoviral mediated β2-AR overexpression with improved function following MU [38]. These findings, coupled with results from our study, re-enforce the rationale for the use of Cl/β2-AR agonists during LVAD support. Specific study of β1-AR blockade during MU is lacking. Hence, mechanisms driving Met's effects are speculative. Enhancement of MU-induced recovery of β-AR responsiveness [35] and/or RyR and SERCA/phospholamban function/phosphorylation/expression [36], [37], proven actions of β-blockers in non-unloaded failing myocardium [4] may be involved. Despite mainly beneficial effects during MU, the antagonism of L-type Ca2+ current and t-tubule density recovery highlight potentially unwanted effects of Cl and Met, the significance of which requires further investigation.
Combined β1-AR blockade and β2-AR stimulation in heart failure
β-adrenergic signalling
Deranged β-adrenergic signalling is instrumental in HF pathogenesis [4]. Selective down-regulation/de-sensitisation of β1-ARs promotes relative augmentation of β2: β1-ARs ratio and signalling [4], [39]. Down-regulation of β1-AR signalling is thought protective against heightened sympathetic stimulation, and the clinical efficacy of β1-AR blockade is proven [4], [6], [23].
Augmented β2-AR signalling is also considered cardio-protective [24], and the idea that β2-AR agonism may be beneficial in clinical HF is long-standing [40], but remains controversial and poorly studied in humans. Undesirable β2-AR agonist effects such as elevation of HR and arrhythmia risk shown by certain studies [41], [42], along with fears over increasing LV mass, both independent prognostic risk factors in cardiac disease [43], [44] tempered enthusiasm for using β2-AR agonists in HF therapy. These factors, coupled with expanding evidence supporting β-blocker efficacy [4], [6], [23], and the phenomenon of β2-AR agonist tachyphylaxis, rendered the notion of therapeutic β2-AR agonism illogical and unsound. Hence, little clinical data exists regarding β2-AR agonist administration within this context, with data derived from a few small, non-randomised studies in HF patients [45]–[46] and chronic lung disease patients with HF, receiving β2-AR agonist therapy [42], [47]. Results are unclear, showing both sustained [42], [45] and transient [46] improvements in cardiac function, as well as negative functional effects [47].
This study re-enforces the concept that β2-AR agonism represents a beneficial HF therapeutic strategy, albeit alongside that of clinically proven β1-AR blockade [24]. Cl's undesirable effects of cardiac hypertrophy and tachycardia were inhibited by Met with no evidence of pro-arrhythmic behaviour ( Figure S1 in Methods S1 ). Combined MetCl therapy displayed superior improvement in whole heart systolic function to either mono-therapy, although a cellular mechanism explaining this superiority was not apparent. Met and Cl showed different positive EC coupling effects with evidence of selected antagonism rather than synergy. Met's improvement of cellular contraction, relaxation, Ca2+ release, Ca2+ transient amplitude, and SR Ca2+ was not enhanced by Cl. T-tubule disruption uncouples the normally tight relationship between L-type Ca2+ channels and RyRs, reducing EC coupling efficiency [48]. Cl improved this coupling shown by recovery of L-type Ca2+ current and t-tubule density, along with RyR function (normalised Ca2+ spark frequency). Such improvements appeared not to alter cellular contractility, but were abolished by Met.
Additive anti-apoptotic effects of either Cl [20] or fenoterol [24] in combination with Met, demonstrated by previous rodent HF studies [20], [24] may be responsible for the whole heart functional effects. Such a mechanism is plausible, as opposing actions of the two β-adrenergic signalling pathways on cell survival is recognised; β1-AR signalling broadly promoting apoptosis [25] and β2-AR signalling driving improved myocyte viability [26], [27]. Restoration of Ca2+ handling protein expression e.g SERCA2a or RyR [18], [20], myo-filament sensitivity [19] and myocardial stiffness/diastolic function [49] represent other properties of Cl and Met, that may underpin the augmented function seen during combined therapy; and afterload (blood pressure (BP)) reduction is another. However, at similar doses to those used in this study, chronic Met [20], [50], Cl [20] or combined therapy [20] has not been seen to significantly alter BP. β2-AR agonist tachyphylaxis is recognised following chronic administration [24], [45]. Temporal functional effects were not assessed in this study, and whether tachyphylaxis occurred is unknown. The loss of Cl's chronotropic effect at week 4 suggests it may have done. In a longer rodent HF study (1 year), Met was shown to prevent β2-AR agonist (fenoterol) tachyphylaxis (caused by decreased β2-AR density), producing extension of fenoterol's positive functional effects. Such a process may also explain the findings from our, albeit shorter study.
Conclusions
In conclusion, our study shows that Met but not Cl prevented MU-induced myocardial atrophy, with atrophic worsening during combined therapy. Cl and Met broadly enhanced MU-induced recovery of deranged EC coupling, but this enhancement was lost during combined therapy. These results suggest that A) Cl's success in enhancing functional recovery in LVAD-supported patients is unrelated to myocardial size and reliant on positive EC coupling effects and B) either the strategy of β2-AR stimulation or β1-AR blockade is likely to be superior to that of combined β2-AR stimulation +β1-AR blockade in enhancing functional recovery during LVAD support. Such findings warrant further investigation.
In the second part of this study, we show for the first time in a model of established rodent HF that combined β2-AR simulation (Cl) +β1-AR blockade (Met) displays superior functional effects to either mono-therapy, and undesirable Cl-induced hypertrophic/chronotropic effects are prevented by Met. Mechanisms underlying this superiority appeared independent of cellular EC coupling and require further investigation. These results suggest that this pharmacological strategy is likely to be safe and beneficial in HF therapy.
Limitations
Met was used in this study to facilitate comparison with previous studies using this rodent HF model. Bisoprolol and not Met is used in the Harefield Protocol. As such, findings from this study may not be directly applicable to this specific clinical scenario.
Supporting Information
A detailed description of Material and Methods is provided. The file also contains Figure S1 showing the ventricular ectopic (VE) rate in sham, failing and treated failing hearts.
(DOC)
Funding Statement
The authors are grateful to the Magdi Yacoub Institute (HSC 87/04) for financial support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kirklin JK, Naftel DC, Kormos RL, Stevenson LW, Pagani FD, et al. (2011) Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant 30: 115–123 10.1016/j.healun.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 2. Mancini DM, Beniaminovitz A, Levin H, Catanese K, Flannery M, et al. (1998) Low incidence of myocardial recovery after left ventricular assist device implantation in patients with chronic heart failure. Circulation 98: 2383–2389 10.1161/01.CIR.98.22.2383 [DOI] [PubMed] [Google Scholar]
- 3. Maybaum S, Mancini D, Xydas S, Starling RC, Aaronson K, et al. (2007) Cardiac improvement during mechanical circulatory support: a prospective multicenter study of the LVAD Working Group. Circulation 115: 2497–2505 10.1161/CIRCULATIONAHA.106.633180 [DOI] [PubMed] [Google Scholar]
- 4. Bristow MR (2011) Treatment of chronic heart failure with beta-adrenergic receptor antagonists: a convergence of receptor pharmacology and clinical cardiology. Circ Res 109: 1176–1194 10.1161/CIRCRESAHA.111.245092 [DOI] [PubMed] [Google Scholar]
- 5. Colucci WS, Packer M, Bristow MR, Gilbert EM, Cohn JN, et al. (1996) Carvedilol inhibits clinical progression in patients with mild symptoms of heart failure, US Carvedilol Heart Failure Study Group. Circulation 94: 2800–2806 10.1161/01.CIR.94.11.2800 [DOI] [PubMed] [Google Scholar]
- 6. Lechat P, Escolano S, Golmard JL, Lardoux H, Witchitz S, et al. (1997) Prognostic value of bisoprolol-induced hemodynamic effects in heart failure during the Cardiac Insufficiency BIsoprolol Study (CIBIS). Circulation. 96: 2197–2205 10.1161/01.CIR.96.7.2197 [DOI] [PubMed] [Google Scholar]
- 7. St John SM, Pfeffer MA, Plappert T, Rouleau JL, Moye LA, et al. (1994) Quantitative two-dimensional echocardiographic measurements are major predictors of adverse cardiovascular events after acute myocardial infarction. The protective effects of captopril. Circulation 89: 68–75 10.1161/01.CIR.89.1.68 [DOI] [PubMed] [Google Scholar]
- 8. Margulies KB (2002) Reversal mechanisms of left ventricular remodeling: lessons from left ventricular assist device experiments. J Card Fail 8: S500–S505 10.1054/jcaf.2002.129264 [DOI] [PubMed] [Google Scholar]
- 9. Klotz S, Jan Danser AH, Burkhoff D (2008) Impact of left ventricular assist device (LVAD) support on the cardiac reverse remodeling process. Prog Biophys Mol Biol 97: 479–496 10.1016/j.pbiomolbio.2008.02.002 [DOI] [PubMed] [Google Scholar]
- 10. Yacoub MH, Tansley P, Birks EJ, Banner NR, Khaghani A, et al. (2001) A novel combination therapy to reverse end-stage heart failure. Transplant Proc 33: 2762–2764 doi:2132/jtrans.proc.2001.1321 [DOI] [PubMed] [Google Scholar]
- 11. Oriyanhan W, Tsuneyoshi H, Nishina T, Matsuoka S, Ikeda T, et al. (2007) Determination of optimal duration of mechanical unloading for failing hearts to achieve bridge to recovery in a rat heterotopic heart transplantation model. J Heart Lung Transplant 26: 16–23 10.1016/j.healun.2006.10.016 [DOI] [PubMed] [Google Scholar]
- 12. Ito K, Nakayama M, Hasan F, Yan X, Schneider MD, et al. (2003) Contractile reserve and calcium regulation are depressed in myocytes from chronically unloaded hearts. Circulation 107: 1176–1182 10.1161/01.CIR.0000051463.72137.96 [DOI] [PubMed] [Google Scholar]
- 13. Korecky B, Masika M (1991) Use of heterotopically isotransplanted rat heart to study the effect of load on cardiac mass. J Cardiovasc Pharmacol 17 Suppl 2S105 doi:17.1314/JCardPh.12123 [DOI] [PubMed] [Google Scholar]
- 14. Birks EJ, Tansley PD, Hardy J, George RS, Bowles CT, et al. (2006) Left ventricular assist device and drug therapy for the reversal of heart failure. N Engl J Med 355: 1873–1884 10.1056/NEJMoa053063 [DOI] [PubMed] [Google Scholar]
- 15. Birks EJ, George RS, Hedger M, Bahrami T, Wilton P, et al. (2011) Reversal of severe heart failure with a continuous-flow left ventricular assist device and pharmacological therapy: a prospective study. Circulation 123: 381–390 10.1161/CIRCULATIONAHA.109.933960 [DOI] [PubMed] [Google Scholar]
- 16. Wong K, Boheler KR, Bishop J, Petrou M, Yacoub MH (1998) Clenbuterol induces cardiac hypertrophy with normal functional, morphological and molecular features. Cardiovasc Res 37: 115–122 10.1016/S0008-6363(97)00190-9 [DOI] [PubMed] [Google Scholar]
- 17. Terracciano CM, Hardy J, Birks EJ, Khaghani A, Banner NR, et al. (2004) Clinical recovery from end-stage heart failure using left-ventricular assist device and pharmacological therapy correlates with increased sarcoplasmic reticulum calcium content but not with regression of cellular hypertrophy. Circulation 109: 2263–2265 10.1161/01.CIR.0000129233.51320.92 [DOI] [PubMed] [Google Scholar]
- 18. Soppa GK, Smolenski RT, Latif N, Yuen AH, Malik A, et al. (2005) Effects of chronic administration of clenbuterol on function and metabolism of adult rat cardiac muscle. Am J Physiol Heart Circ Physiol 288: H1468–H1476 10.1152/ajpheart.00624.2004 [DOI] [PubMed] [Google Scholar]
- 19. Soppa GK, Lee J, Stagg MA, Felkin LE, Barton PJ, et al. (2008) Role and possible mechanisms of clenbuterol in enhancing reverse remodelling during mechanical unloading in murine heart failure. Cardiovasc Res 77: 695–706 10.1093/cvr/cvm106.Epub2008Jan4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xydas S, Kherani AR, Chang JS, Klotz S, Hay I, et al. (2006) Beta(2)-Adrenergic stimulation attenuates left ventricular remodeling, decreases apoptosis, and improves calcium homeostasis in a rodent model of ischemic cardiomyopathy. J Pharmacol Exp Ther 317: 553–561 10.1124/jpet.105.099432 [DOI] [PubMed] [Google Scholar]
- 21. Tsuneyoshi H, Oriyanhan W, Kanemitsu H, Shiina R, Nishina T, et al. (2005) Heterotopic transplantation of the failing rat heart as a model of left ventricular mechanical unloading toward recovery. ASAIO J 51: 116–120 10.1097/01.MAT.0000150325.05589.8B [DOI] [PubMed] [Google Scholar]
- 23. Waagstein F, Bristow MR, Swedberg K, Camerini F, Fowler MB, et al. (1993) Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet 342: 1441–1446 doi:11.321.lanc/1993.21 [DOI] [PubMed] [Google Scholar]
- 24. Ahmet I, Krawczyk M, Zhu W, Woo AY, Morrell C, et al. (2008) Cardioprotective and survival benefits of long-term combined therapy with beta2 adrenoreceptor (AR) agonist and beta1 AR blocker in dilated cardiomyopathy postmyocardial infarction. J Pharmacol Exp Ther 325: 491–499 10.1124/jpet.107.135335 [DOI] [PubMed] [Google Scholar]
- 25. Zheng M, Zhu W, Han Q, Xiao RP (2005) Emerging concepts and therapeutic implications of beta-adrenergic receptor subtype signaling. Pharmacol Ther 108: 257–268 10.1016/j.pharmthera.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 26. Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, et al. (2001) Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A 98: 1607–1612 10.1073/pnas.98.4.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, et al. (2003) Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest 111: 617–625 10.1172/JCI16326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsuneyoshi H, Oriyanhan W, Kanemitsu H, Shiina R, Nishina T, et al. (2005) Does the beta2-agonist clenbuterol help to maintain myocardial potential to recover during mechanical unloading? Circulation 112: I51–I56 10.1161/CIRCULATIONAHA.104.525097 [DOI] [PubMed] [Google Scholar]
- 29. Ibrahim M, Al Masri A, Navaratnarajah M, Siedlecka U, Soppa GK, et al. (2010) Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J 24: 3321–3329 10.1096/fj.10-156638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bers DM (2006) Altered cardiac myocyte Ca regulation in heart failure. Physiology. (Bethesda) 21: 380–387 10.1152/physiol.00019.2006 [DOI] [PubMed] [Google Scholar]
- 31. Drakos SG, Kfoury AG, Hammond EH, Reid BB, Revelo MP, et al. (2010) Impact of mechanical unloading on microvasculature and associated central remodeling features of the failing human heart. J Am Coll Cardiol 56: 382–391 10.1016/j.jacc.2010.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baskin KK, Taegtmeyer H (2011) Taking pressure off the heart: the ins and outs of atrophic remodelling. Cardiovasc Res 90: 243–250 10.1093/cvr/cvr06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao M, Fajardo G, Urashima T, Spin JM, Poorfarahani S, et al. (2011) Cardiac pressure overload hypertrophy is differentially regulated by {beta}-adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol 301: H1461–H1470 10.1152/ajpheart.00453.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J, Marui A, Ikeda T, Komeda M (2008) Partial left ventricular unloading reverses contractile dysfunction and helps recover gene expressions in failing rat hearts. Interact Cardiovasc Thorac Surg 7(1): 27–31 10.1510/icvts.2007.165563 [DOI] [PubMed] [Google Scholar]
- 35. Dipla K, Mattiello JA, Jeevanandam V, Houser SR, Margulies KB (1998) Myocyte recovery after mechanical circulatory support in humans with end-stage heart failure. Circulation 97: 2316–2322 10.1161/01.CIR.97.23.2316 [DOI] [PubMed] [Google Scholar]
- 36. Heerdt PM, Holmes JW, Cai B, Barbone A, Madigan JD, et al. (2000) Chronic unloading by left ventricular assist device reverses contractile dysfunction and alters gene expression in end-stage heart failure. Circulation. 2000 102: 2713–2719 10.1161/01.CIR.102.22.2713 [DOI] [PubMed] [Google Scholar]
- 37. Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, et al. (2001) Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol 153: 699–708 10.1083/jcb.153.4.699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tevaearai HT, Eckhart AD, Walton GB, Keys JR, Wilson K, et al. (2002) Myocardial gene transfer and overexpression of beta2-adrenergic receptors potentiates the functional recovery of unloaded failing hearts. Circulation 106: 124–129 10.1161/01.CIR.0000020220.79105.FD [DOI] [PubMed] [Google Scholar]
- 39. Brodde OE (1991) Beta 1- and beta 2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev 43: 203–242 doi:6.43.PhRev/5321 [PubMed] [Google Scholar]
- 40. Irmer M, Wollschlager H, Just H (1981) Treatment of severe congestive heart failure with the beta-agonist fenoterol (author's transl). Klinn Wochenschr 59: 639–645 doi:54.32111.KLW.1 [DOI] [PubMed] [Google Scholar]
- 41. Dawson JR, Bayliss J, Norell MS, Canepa-Anson R, Kuan P, et al. (1982) Clinical studies with beta 2 adrenoceptor agonists in heart failure. Eur Heart J 3: 135–141 doi:32.3121/eurheartj/ehn421 [PubMed] [Google Scholar]
- 42. Matera MG, Martuscelli E, Cazzola M (2010) Pharmacological modulation of beta-adrenoceptor function in patients with coexisting chronic obstructive pulmonary disease and chronic heart failure. Pulm Pharmacol Ther 23: 1–8 10.1016/j.pupt.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 43. Kolloch R, Legler UF, Champion A, Cooper-Dehoff RM, Handberg E, et al. (2008) Impact of resting heart rate on outcomes in hypertensive patients with coronary artery disease: findings from the INternational VErapamil-SR/trandolapril STudy (INVEST). Eur Heart J 29: 1327–1334 10.1093/eurheartj/ehn123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990) Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322: 1561–1566 10.1056/NEJM199005313222203 [DOI] [PubMed] [Google Scholar]
- 45.Awan NA, Needham K, Evenson MK, Hermanovich J, Joye JA, et al. (1981) Therapeutic efficacy or oral pirbuterol in severe chronic congestive heart failure: acute hemodynamic and long-term ambulatory evaluation. Am Heart J 102:555–563. PMID: 7270402. [DOI] [PubMed]
- 46.Colucci WS, Alexander RW, Mudge GH, Rude RE, Holman BL, et al. (1981) Acute and chronic effects of pirbuterol on left ventricular ejection fraction and clinical status in severe congestive heart failure. Am Heart J 102: 564–568. PMID: 6115573. [DOI] [PubMed]
- 47. Au DH, Udris EM, Fan VS, Curtis JR, McDonell MB, et al. (2003) Risk of mortality and heart failure exacerbations associated with inhaled beta-adrenoceptor agonists among patients with known left ventricular systolic dysfunction. Chest 123: 1964–1969 10.1378/chest.123.6.1964 [DOI] [PubMed] [Google Scholar]
- 48. Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, et al. (2006) Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A 103: 4305–4310 10.1073/pnas.0509324103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hon JK, Steendijk P, Petrou M, Wong K, Yacoub MH (2001) Influence of clenbuterol treatment during six weeks of chronic right ventricular pressure overload as studied with pressure-volume analysis. J Thorac Cardiovasc Surg 22: 767–774 10.1067/mtc.2001.114354 [DOI] [PubMed] [Google Scholar]
- 50. Wei S, Chow LT, Sanderson JE (2000) Effect of carvedilol in comparison with metoprolol on myocardial collagen postinfarction. J Am Coll Cardiol 36: 276–281 10.1016/S0735-1097(00)00671-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A detailed description of Material and Methods is provided. The file also contains Figure S1 showing the ventricular ectopic (VE) rate in sham, failing and treated failing hearts.
(DOC)