

Abstract
The vault nanoparticle is one of the largest known ribonucleoprotein complexes in the sub-100 nm range. Highly conserved and almost ubiquitously expressed in eukaryotes, vaults form a large nanocapsule with a barrel-shaped morphology surrounding a large hollow interior. These properties make vaults an ideal candidate for development into a drug delivery vehicle. In this study, we report the first example of using vaults towards this goal. We engineered recombinant vaults to encapsulate the highly insoluble and toxic hydrophobic compound All-trans Retinoic Acid (ATRA) using a vault binding lipoprotein complex that forms a lipid bilayer nanodisk. These recombinant vaults offer protection to the encapsulated ATRA from external elements. Furthermore, a cryo-electron tomography (cryo-ET) reconstruction shows the vault binding lipoprotein complex sequestered within the vault lumen. Finally, these ATRA loaded vaults have enhanced cytotoxicity against the hepatocellular carcinoma cell line HepG2. The ability to package therapeutic compounds into the vault is an important achievement toward their development into a viable and versatile platform for drug delivery.
Keywords: Vaults, Nanoparticles, Nanodisk, All-trans Retinoic Acid, Drug Delivery Systems
1. Introduction
Although chemically produced drugs have a long record of success as therapeutic agents, they are not without serious limitations. The vast majority are small hydrophobic molecules that are limited in use due to their poor pharmacokinetic and pharmacodynamic properties. While much attention has focused on generating new compounds or modifying existing ones for improved efficacy, new strategies have begun to emerge to compliment and/or replace the existing dogma of drug therapy. The development of nanoparticle based platforms have enhanced the delivery of current therapeutic compounds and circumvented the adverse pharmacological properties of these conventional drugs.[1] These new drug delivery systems (DDS) overcome current limitations by offering new environments for improved solubility, thereby eliminating the need for many toxic organic solvents. Common examples include the use of dendrimers, liposomes or conjugation to polymers, such as polyethylene glycol (PEG).[2-4] The latter two have had considerable success and have been approved for clinical use despite existing pitfalls, such as size limitations and lack of tissue targeting. Therefore, new nanoparticles and new strategies are still needed.
Recent studies suggest that vaults may be an ideal nanoparticle for development into a DDS. Discovered over a quarter-century ago, the vault is the largest known cellular ribonucleoprotein complex at 13 MDa with a uniform particle size of 71×42×42 nm.[5,6] Native vaults are highly conserved and are present in most eukaryotes.[7] Despite implication in various cellular roles including multidrug resistance, cytoplasmic-nuclear transport, and the innate immune response, a definitive functional role for endogenous vaults has yet to be established.[6,8] Native vaults have a simple composition consisting of only three proteins: the major vault protein (MVP), vault poly (ADP-ribose) polymerase (VPARP), telomerase associated protein 1 (TEP1) and several copies of one or more un-translated vault associated RNA (vRNAs).[6,8-11] MVP constitutes 75% of the native vault particle mass, while its expression alone is necessary and sufficient for recombinant vault particle formation.[12] Recombinant vaults are non-immunogenic and have undergone significant engineering, including cell surface receptor targeting and encapsulation of a wide variety of proteins.[13-19] Electron microscopy studies have shown both endogenous and recombinant vaults to be an ellipsoidal nanoparticle with a thin protein shell encapsulating a large internal volume of 50×106 nm3, which could potentially hold hundreds to thousands of small molecular weight compounds.[9] Thus, recombinant vaults are an attractive target for engineering as a platform for drug delivery.
With the goal of creating a vault capable of encapsulating therapeutic compounds for drug delivery, we have designed a strategy to package another nanoparticle, known as a Nanodisk (ND), into the vault lumen.[20,21] Nanodisks are small discoidal lipid bilayer fragments derived from a truncated form of Apolipoprotein-AI (Apo-AI, amino acids 44-200) containing a series of amphipathic helices that encircle the disk's circumference in a belt-like manner resulting in a nanoparticle with an average diameter of approximately 10 to 20 nm. As a lipid bilayer, NDs provide a rich lipophillic domain that can absorb hydrophobic compounds. NDs have been previously shown to bind a variety of drugs and improve their effectiveness.[22-25] Although these NDs can be modified for tissue-specific targeting directly, lipid surfaces remain exposed, potentially allowing for exchange with surrounding cell membranes, which could limit their usefulness as a DDS.[26] By packaging drug loaded NDs into the vault lumen, the ND and its contents would be shielded from the external medium. Moreover, given the large vault interior, it is conceivable that multiple NDs could be packaged, which would considerably increase the localized drug concentration. We proposed that packaging of NDs into the vault could be achieved through the use of a vault lumen binding domain derived from the C-terminus of VPARP (amino acids 1563-1724).[13] This MVP binding domain, termed INT for vault interaction domain has been used to shuttle a variety of proteins, enzymes, and immunogens into the vault interior.[13-17] By fusing a truncated form of Apo-AI to INT to form ΔApo-AI-INT, we formed a modified ND, which we designate as Nanodisk-INT (NDI) that should package into recombinant vaults. This strategy provides a promising and versatile method for encapsulating a variety of therapeutic compounds into the vault nanoparticle, as depicted schematically in Figure 1.
Figure 1.
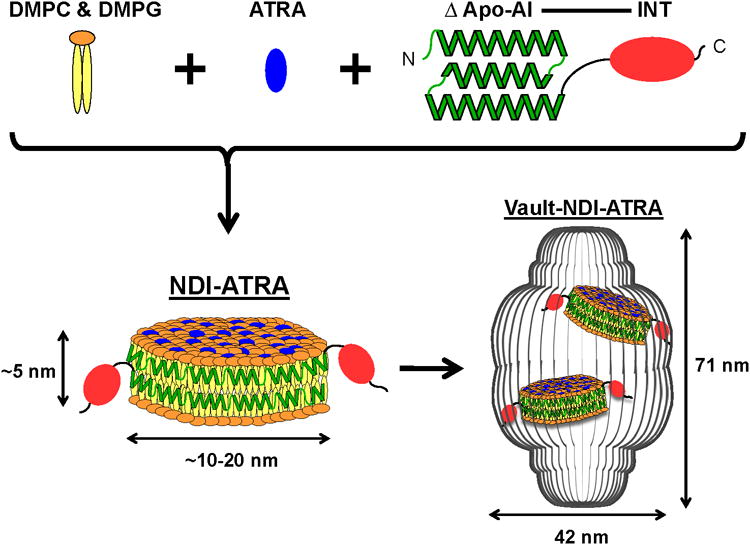
Schematic diagram depicting NDI-ATRA formation and encapsulation into the vault nanoparticle. DMPC & DMPG lipids interact with ΔApo-AI-INT to form a discoidal phospholipid bilayer nanodisk (NDI). The amphipathic helices of ΔApo-AI-INT run perpendicular to the phospholipid acyl chains in a double belt-like manner around the circumference of the nanodisk. Addition of ATRA during formation results in the drugs absorbance into the lipoprotein complex. NDI packaging into the vault nanoparticle is achieved by the vault binding domain INT. Components are not drawn to scale.
To evaluate the vault packaged with drug loaded NDI as a viable nanocapsule DDS, several criteria were analyzed including the ability for NDI to retain drug during nanodisk formation and subsequently upon packaging into the vault, as well as the ability for these drug loaded vaults to retain biological activity. Based on a previous study of nanodisks, we decided to package All-trans Retinoic Acid (ATRA) into recombinant vaults.[24] ATRA is a vitamin A derivative that interacts with the retinoid acid receptor (RAR) and retinoid X receptor (RXR) to function as a gene transcription regulator.[27,28] Binding with its receptors leads to changes in a variety of genes involved in cell proliferation, differentiation, and apoptosis. ATRA has been shown to be useful in the treatment of a variety of illnesses ranging from acne to cancer.[29-31] However, ATRA is not without significant drawbacks including being highly insoluble, teratogenic, and the possibility of leading to Retinoic acid syndrome, properties that make it an ideal candidate for use in a DDS.[32,33]
In the present study we demonstrate for the first time that a small molecule drug can be selectively and stably incorporated into recombinant vaults using NDI nanodisks. Furthermore, these NDI packaged vaults retain the biological activity of the sequestered drug. Incorporation of ATRA into vaults is a promising first step toward developing the vault as a novel nanocapsule DDS. We expect that this NDI strategy will be amendable to a myriad of therapeutic compounds, thereby making the vault a general and versatile vehicle for drug delivery.
2. Results
2.1 NDI-ATRA Formation
The first step towards developing the vault nanoparticle as a DDS was to form NDI using recombinant ΔApo-AI-INT protein with synthetic phospholipids. When purified ΔApo-AI-INT was added to a stock solution of dimyristoylphosphatidylcholine (DMPC) and dimyristoylphosphatidylglycerol (DMPG) (3.5 mg: 1.5 mg) in phosphate buffered saline (PBS), a visible decrease in sample turbidity occurred in a time dependent manner. This decrease in solution turbidity indicated that the lipids were condensed into the smaller, more discrete soluble NDI particles, thereby reducing the excess lipid pellet size following centrifugation (Figure 2a, red circles). When ΔApo-AI-INT protein was co-mixed with DMPC/DMGP and ATRA (2 mg: 5 mg: 0.4 mg) to form NDI-ATRA, the solution did not visibly clear but remained turbid. Following centrifugation and sterile filtration to remove excess lipids and ATRA, NDI-ATRA supernatants retained a visible yellow hue. This solution color is correlated with ATRA, thereby indicating the association of ATRA within the soluble NDI. In contrast, processed supernatants of ATRA alone or DMPC/DMPG-ATRA alone were void of any color. ΔApo-AI-INT protein mixed with ATRA but without DMPC/DMPG also did not retain any color despite recovery of ΔApo-AI-INT within the supernatant (data not shown). Taken together, these results indicate that both ΔApo-AI-INT and DMPC/DMPG are required to form soluble NDI capable of encapsulating ATRA.
Figure 2.

NDI and NDI-ATRA formation. a) In the absence of ΔApo-AI-INT, the DMPC/DMPG lipid mixture remains turbid but pellets upon centrifugation. In the presence of ΔApo-AI-INT, the lipid mix clears and does not pellet upon centrifugation (red circles). A DMPC/DMPG lipid-ATRA mixture behaves similarly in the absence of ΔApo-AI-INT. However, in the presence of ΔApo-AI-INT, the lipid-ATRA mixture does not clear until after centrifugation. Importantly, the supernatant remains yellow, indicating retention of ATRA within the soluble NDI nanoparticles. b) Top: TEM of NDI. Bottom: TEM of NDI-ATRA.
2.2 NDI-ATRA TEM
To further characterize NDI formation in the presence or absence of ATRA, samples were negatively stained and analyzed by transmission electron microscopy (TEM). As expected, ΔApo-AI-INT only samples or ATRA alone showed no stained material (data not shown). Despite a few liposomal vesicles, DMPC/DMPG samples with or without ATRA were also mostly void of stained material (data not shown). These trace liposomes disappeared in the NDI sample as the TEM grid was filled with a mostly homogenous population of small, discrete discoidal particles with an average diameter size of approximately 10 to 20 nm (Figure 2b). NDI prepared in the presence of ATRA appeared morphologically similar to that of NDI.
2.3 NDI-ATRA Spectroscopy
UV spectroscopy verified that ATRA was selectively retained in the NDI-ATRA sample with a detectable maximum absorbance between 341-350 nm, while no signal was detected in the DMPC/DMPG-ATRA or ATRA only samples (Figure 3). Furthermore, NDI-ATRA showed a clear peak regardless of whether diluted into PBS or ethanol. However, when an equal concentration of free drug was scanned, only ATRA diluted into ethanol gave a noticeable absorbance spectrum, whereas, the absorption spectrum of ATRA diluted into PBS was mostly attenuated by the buffer.
Figure 3.
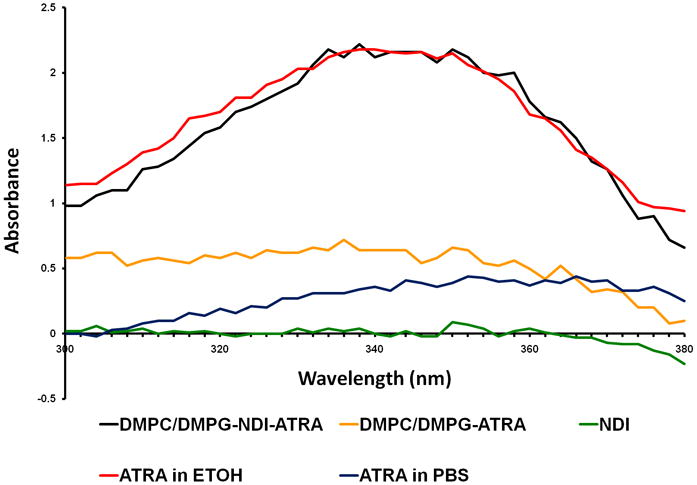
UV spectroscopy of ATRA. NDI-ATRA in PBS (black line) displays a normal ATRA absorption spectrum with peak between 341-350 nm. NDI in PBS alone (green line) has no spectral properties while the DMPC/DMPG-ATRA control in PBS (yellow line) did not retain significant levels of ATRA following excess lipid/drug centrifugation and sterile filtration. An equal concentration of ATRA in ethanol (red line) has a similar spectrum as NDI-ATRA in PBS. However, the spectrum for ATRA in PBS (blue line) is mostly attenuated.
2.4 NDI-ATRA Packaging into CP-MVP Vaults
With the ability of NDI to form morphologically similar nanodisks while retaining ATRA confirmed, we next examined whether they retained the ability to bind inside of the vault lumen. NDI-ATRA packaging into vaults was analyzed by mixing purified recombinant CP-MVP vaults (see methods) with pre-formed NDI-ATRA. NDI-ATRA alone was fractionated over a 20-60% discontinuous sucrose gradient followed by western blot analysis. ΔApo-AI-INT from NDI-ATRA remained in the lighter 20 & 30% sucrose gradient layers (Figure 4a, left panel). Following NDI-ATRA co-incubation with vaults, ΔApo-AI-INT protein showed a shift in its sucrose gradient profile as some of the protein migrated into the denser 40-45% layers which also contained CP-MVP (Figure 4a, right panel). This co-migration of ΔApo-AI-INT into the denser layers of the gradient is only possible by specific association with the larger, denser vault nanoparticle. Numerous INT fusion proteins have been analyzed in a similar manner to confirm vault packaging.[13-19] Meanwhile, a previous cryo-EM reconstruction followed by difference mapping of recombinant vaults containing a luciferase-INT fusion protein provided definitive proof of INT packaging by revealing the presence of additional density in the interior of the vault above and below the particle waist due to INT binding. [13]
Figure 4.
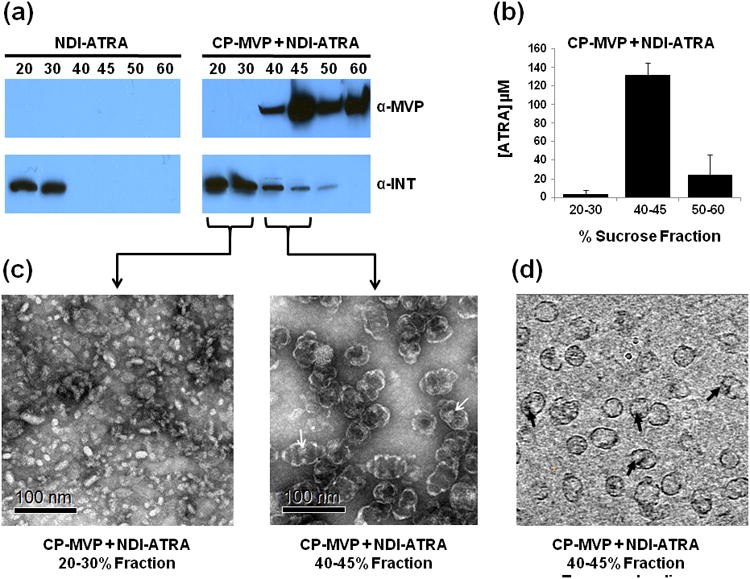
NDI-ATRA vault packaging. a) Western blot analysis of the sucrose gradient fractionation pattern of ΔApo-AI-INT for NDI-ATRA alone or NDI-ATRA co-mixed with CP-MVP vaults. b) ATRA concentration for pooled sucrose gradient fractions of CP-MVP + NDI-ATRA following centrifugation. c) TEM of collected CP-MVP + NDI-ATRA fractions. d) CP-MVP + NDI-ATRA EM tomography slice.
2.5 CP-MVP-NDI-ATRA Spectroscopy
With confirmation that ΔApo-AI-INT protein packaged into the vault, it was important to examine ATRA retention within these nanocapsules. Pooled sucrose gradient fractions were collected and assayed for ATRA concentration. NDI-ATRA by itself had no detectable ATRA in any of the fractions, suggesting that ATRA is not stably associated with the NDI lipoprotein complex during sucrose gradient centrifugation. For the co-mixed vault-NDI-ATRA sample, ATRA was retained in the denser 40-45% and 50-60% fractions with the co-localized vaults, whereas the 20-30% fractions contained relatively little ATRA following sucrose gradient separation despite the majority of ΔApo-AI-INT protein located within that fraction (Figure 4b). The retention of ATRA in the fractions containing vaults suggests that the full NDI-ATRA lipoprotein-drug complex was protected within the vault nanocapsule.
2.6 CP-MVP-NDI-ATRA TEM and Tomography
TEM was conducted on the different sucrose layer fractions to analyze the morphology of NDI-ATRA co-mixed with recombinant CP-MVP vaults (Figure 4c). The 20-30% layer of NDI-ATRA co-mixed with vaults showed intact lipoprotein particles of approximately equal size and shape to those prior to co-mixing, despite the loss of ATRA. The 40-45% layer showed vaults that appeared to have additional interior density (white arrows), likely corresponding with internalized NDI-ATRA. Definitive proof of NDI-ATRA packaging into the vault was obtained from cryo-electron tomography (cryo-ET) studies of the same 40-45% fraction containing CP-MVP co-mixed with NDI-ATRA. Tomography slices showed extra densities representing NDI-ATRA, clearly within the vault lumen (Figure 4d, black arrows). Interestingly, an examination of tomography slices indicated that most vaults appeared to contain only a single area of extra density, suggesting that on average, only a single NDI-ATRA nanoparticle was packaged within the vault.. In addition to the cryo-ET, a single particle reconstruction of a CP-MVP vault containing NDI from a 40-45% sucrose gradient fraction was conducted (Figure 5). A three dimensional volume rendering of the particle clearly showed the location of the large lipoprotein complex of NDI (red) within the vault nanoparticle (green).
Figure 5.
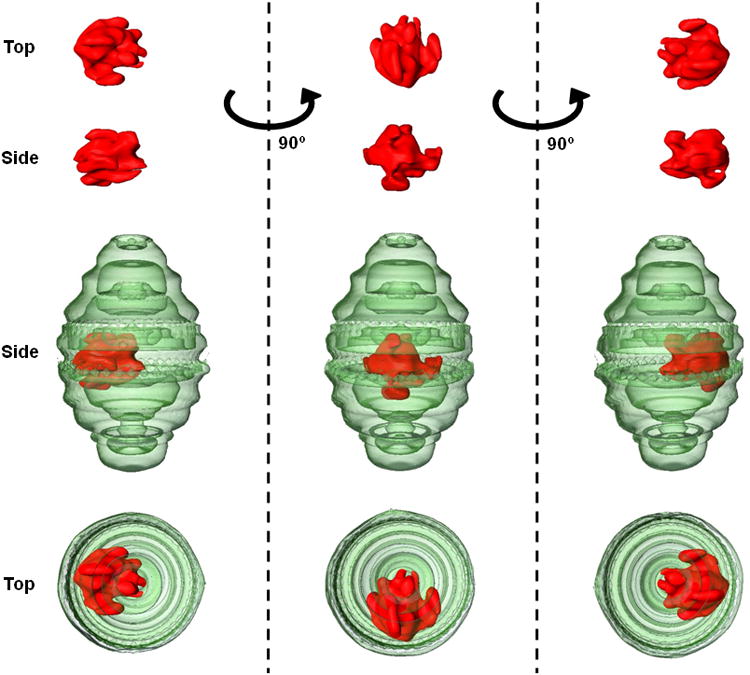
Three dimension volume rendering from a cryo-ET single particle reconstruction of CP-MVP containing NDI. Rotation about the z-axis of the vault particle (green) reveals that the NDI lipoprotein complex (red) is not only located within the vault lumen but that it is associated at a specific position at the vault waist.
2.7 HepG2 Cell Viability
ATRA retention within the vault by NDI was further confirmed by testing these vaults for ATRA biological activity. HepG2 cells, a malignant hepatoma cell line, were plated at 5×104 cells/well and treated with either: free drug, NDI, NDI-ATRA, empty CP-MVP vaults, or CP-MVP vaults containing NDI-ATRA. Cell viability was measured 5 days later using an MTT assay (Figure 6) with all values normalized against untreated cells. Free ATRA caused an 18% drop in cell viability, whereas NDI-ATRA increased the killing efficiency to 37%. Vaults containing NDI-ATRA had a slightly improved efficacy of 42%, while empty vaults caused a 9% drop in cell viability. Interestingly, NDI alone induced a 27% drop in cell viability even though no ATRA was present. These results show that the vaults containing NDI-ATRA were biologically active against HepG2 cells.
Figure 6.
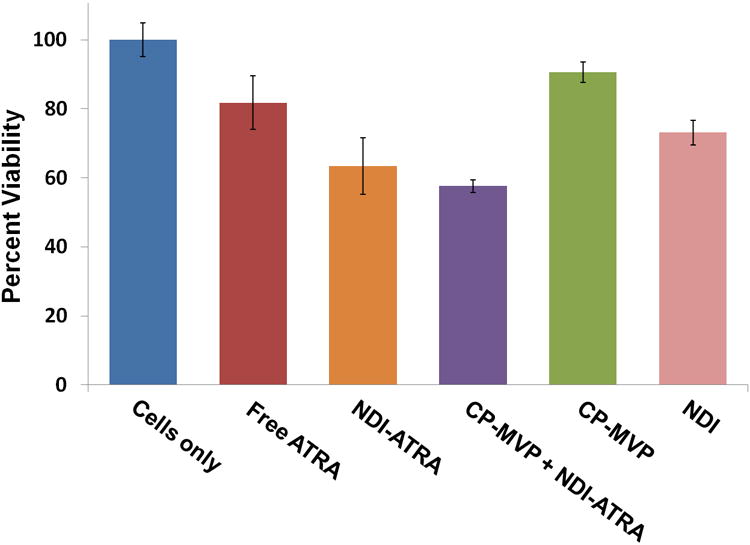
HepG2 cell viability assay. NDI-ATRA and CP-MVP + NDI-ATRA both display increased toxicity than free ATRA over the course of 120 hours.
3. Discussion
Here we show that a recombinant nanodisk containing ATRA, a potent but toxic therapeutic compound, can be packaged inside of the vault nanoparticle. The development of a nanodisk capable of being packaged within vaults was achieved by fusing a truncated form of the lipoprotein Apo-AI to the vault binding domain, INT. ΔApo-AI-INT co-incubated with a synthetic phospholipid dispersion of DMPC/DMPG in aqueous buffer, spontaneously formed NDI nanoparticles. Furthermore, ΔApo-AI-INT co-mixed with DMPC/DMPG in the presence of the extremely hydrophobic ATRA formed nanodisks containing the drug. The adsorption spectrum of NDI-ATRA prepared in PBS indicated that the drug was located within the hydrophobic environment of the nanodisks lipid bilayer. TEM analysis showed NDI-ATRA formed morphologically distinct nanoparticles of proper size. Based on these results, we conclude that NDI-ATRA behaves similarly to previously described nanodisks.[24]
Since NDI-ATRA is a large lipoprotein-drug complex with unique chemical and physical properties, it was uncertain whether preformed NDI-ATRA would package into vaults without denaturing or losing its cargo. Sucrose gradient fractionation indicated that both ΔApo-AI-INT and ATRA were retained in the 40-45% and 50-60% layers containing CP-MVP. These fractions had normal ATRA absorbance spectra despite being in an aqueous buffer, indicating that the hydrophobic lipids DMPC and DMPG likely remained associated with the drug. The 20-30% layer which contained no CP-MVP but only excess ΔApo-AI-INT had little to no associated ATRA despite TEM images showing nanodisks morphologically similar to NDI-ATRA prior to the sucrose gradient centrifugation, further implicating the retention of DMPC and DMPG with ΔApo-AI-INT. It is plausible that ATRA is stripped away from unpackaged NDI-ATRA particles during sucrose gradient centrifugation and subsequently lost. Likewise, CP-MVP vaults mixed with ATRA alone or DMPC/DMPG-ATRA (i.e. lipid-drug mix only) did not retain any detectable amounts of the drug within any fraction following centrifugation (data not shown).
The vault nanoparticle appears to offer internalized NDI-ATRA protection against drug loss during the sucrose gradient centrifugation. This is a promising aspect of packaging NDI-ATRA within vaults for use as a DDS as it demonstrates vault protection of the sequestered drug while also allowing for removal of non-specifically bound ATRA. Interestingly, this contrasts with what was observed when NDI containing the anti-fungal compound amphotericin B (AMB) was packaged (data not shown). Following sucrose gradient separation of NDI-AMB co-mixed with CP-MVP, the AMB remained associated within the 20-30% layers containing ΔApo-AI-INT alone and in the 40-45% and 50-60% layers containing both ΔApo-AI-INT and CP-MVP. This is an interesting paradox given that ATRA is far more insoluble than AMB, with partition constants of 6.3 and 0 respectively and should have an expected tighter association with the NDI lipoprotein complex. It is possible that the larger polyene backbone of AMB provides a greater surface area for binding interactions with the acyl chains of the phospholipids than the smaller ATRA.
TEM of negative stained samples indicated the presence of additional density inside of recombinant vaults packaged with NDI-ATRA. Furthermore, cryo-ET analysis provided additional evidence that the large nanodisk particle was sequestered within the vault lumen. These vaults appeared to primarily package a single NDI-ATRA particle. This is an interesting finding, given that vaults are able to package dozens of copies of other INT fusion proteins with similar molecular weights as that of ΔApo-AI-INT.[13,15] The three dimensional volume rendering for a single particle reconstruction of a vault packaged with NDI alone provided additional proof that the entire lipoprotein complex was sequestered within the vault interior. The particle's large size can only be attributed to the retention of the DMPC/DMPG lipids with ΔApo-AI-INT. However, the resolution limit of this method prohibits a more detailed analysis of the nanodisks morphology. Intriguingly, the nanodisk's large size might explain why, on average, only a single NDI particle was packaged per vault, as it may preclude others from entering the vault due to steric constraints. Furthermore, rotation about the z-axis of the vault revealed a site specific association of NDI within the vault lumen.
Lastly, to ensure that vaults containing NDI-ATRA retained biological activity, they were compared to free ATRA and NDI-ATRA in a cell toxicity assay. Over the course of 5 days, HepG2 cell viability decreased when exposed to samples containing ATRA. Toxicity increased when ATRA was associated within the lipoprotein complex of NDI. Vaults containing NDI-ATRA exhibited a similar marked improvement in efficacy over that of the free drug administered in aqueous buffer. The similar HepG2 cell toxicity induced by NDI-ATRA or recombinant vaults containing NDI-ATRA was seen when normalized concentrations of ATRA were compared between all samples. It may not be possible to detect differences in efficacy between NDI-ATRA or vaults packaged with NDI-ATRA on a 5 day time scale. The toxicity of NDI on cell viability was unexpected and is perhaps attributable to the synthetic lipids DMPC and DMPG altering cell membrane properties. Taken as a whole, these results demonstrate that NDI selectively retains the highly insoluble hydrophobic drug ATRA and can then be packaged into the lumen of the vault nanoparticle where it remains biologically active.
It is important to note that INT exists in an equilibrium state heavily favoring binding into the vault lumen. Moreover, the vault is a dynamic nanoparticle capable of opening and closing or even exchanging entire halves between vaults.[14,34-36] This dynamic nature suggests that NDI-ATRA could escape the vault and be taken up by the cell independently of the vault, though this seems unlikely given the high theoretical binding constant of INT into vaults. Vaults carrying NDI-ATRA are likely taken up by phagocytosis and dissociate within the endosome or lysosome to release ATRA into HepG2 cells. Future experiments utilizing fluorescent labeled vaults and NDI-ATRA will confirm whether or not they are internalized together by the targeted cell. Lastly, additional methods exist to potentially increase the efficacy of NDI-ATRA loaded vaults by selectively targeting specific cell types, increasing entry into the cytoplasm and altering particle dynamics to allow for slow sustained release of the drug into the cell.[17,35]
4. Conclusion
The first steps toward the development of the vault nanoparticle into a versatile and effective DDS are reported in this paper. The ability to encapsulate therapeutic compounds into the vault is a critical and fundamental obstacle in their development for small molecule drug delivery. However, by using another nanoparticle, known as a nanodisk, this obstacle has been overcome. While NDs show significant promise as a DDS by themselves, we believe that their use in conjunction with the vault nanoparticle will have a synergistic effect leading to development of an even more effective DDS. The ability of vaults to provide shelter during delivery to specifically targeted cells will enhance the efficacy of the drug stored inside.
5. Experimental Section
Materials and reagents
All-trans Retinoic Acid (ATRA) was from Sigma-Aldrich Co. Dimyrisotoylphosphatidylcholine (DMPC) and dimyristoylphosphatidylglycerol (DMPG) were from Avanti Polar Lipids Inc. Apolipoprotein-AI cDNA (MGC Clone ID: 3934992) was from Invitrogen. Expression of ΔApo-AI-INT was carried out using the Novagen pET 28 system with BL21 codon plus Escherichia coli cells from Stratagene. NiNTA beads were obtained from Qiagen. Invitrogen's Bac-to-Bac Expression System was used for vault expression in Spodoptera frugiperda (Sf9) cells grown in Sf-900 II media. The hepatocellular carcinoma cell line HepG2 was obtained from ATTC (HB-8065). MTT was from Roche.
NDI construction, expression and purification
Full length human Apolipoprotein A-I cDNA was used to PCR amplify an amine terminal truncated form of the protein consisting of residues leu-44 to glu-200 and designated ΔApo-AI-INT. The 5′ end contains an Nco I restriction site incorporating a start methionine in front of residue 44 resulting in a sequence change of L44V. The 3′ end was engineered to contain an overhang region encoding a TEV protease cleavage site flanked by two small flexible linker regions. The carboxyl-terminus interaction domain (INT) of rat VPARP, residues cys-1563 to gly-1724, was PCR amplified and engineered with a complimentary 5′ end overhang for the TEV linker sequence for subsequent double PCR steps. Additionally, the 3′ end contained an in frame (His)6 tag for Ni+ column purification. Double PCR generated recombinant ΔApo-AI-INT with a predicted theoretical molecular weight of 44 kDa was sub-cloned into the pET28(a)+ vector system then transformed into BL21 Codon Plus E. coli. ΔApo-AI-INT's amino acid sequence is as follows with the linker region underlined and double underline for the TEV protease site.
MVKLLDNWDSVTSTFSKLREQLGPVTQEFWDNLEKETEGLRQEMSKDLEEVKAKVQPYLDDFQKKWQEEMELYRQKVEPLRAELQEGARQKLHELQEKLSPLGEEMRDRARAHVDALRTHLAPYSDELRQRLAARLEALKENGGARLAEYHAKATEHLSTLSEKAKPALEDLRQGLLPVLESFKVSFLSALEEYTKKLNTQTGTENLYFQTGTCTQHWQDAVPWTELLSLQTEDGFWKLTPELGLILNLNTNGLHSFLKQKGIQSLGVKGRECLLDLIATMLVLQFIRTRLEKEGIVFKSLMKMDDPSISRNIPWAFEAIKQASEWVRRTEGQYPSICPRLELGNDWDSATKQLLGLQPISTVSPLHRVLHYSQGHHHHHH.
ΔApo-AI-INT expression and Ni+ column purification was conducted similar to the Novagen manual. Briefly, 500 μl of a saturated glycerol cell stock was used to inoculate 50 ml of LB media containing 50 μg/ml Kanamyacin and 34 μg/ml Chloramphenical overnight at 37 °C and subsequently used to inoculate a 1L final volume culture the following morning. Once O.D.600 nm reached 0.5 - 0.6, isopropyl β-D-1-thiogalactopyranoside was added to a final 1 mM concentration to induce expression at 37 °C with shaking at 250 rpm. After 3 hours, cells were pelleted, decanted and stored at −80 °C. Induced cell pellets were resuspended in binding buffer containing 1% Triton-X100 and sonicated to ensure complete cell lysis. Lysate was centrifuged for 20 minutes at 38,000×g to remove insoluble material. Supernatant was passed through a 0.45 μm filter prior to loading onto an equilibrated 4 ml bed volume NiNTA Ni+ column. The column was washed prior to elution using 250 mM imidazole buffer. Fractions were analyzed by SDS-PAGE. Eluted fractions were dialyzed overnight at 4 °C to remove imidazole. Purified ΔApo-AI-INT samples were collected, flash frozen in liquid nitrogen and stored at −80 °C.
NDI-ATRA formation and spectroscopy
A stock lipid mixture was prepared at a ratio of 3.5 mg DMPC: 1.5 mg DMPG then placed into a glass conical vial and resuspended in a chloroform: methanol (3:1 v/v) solution. In order to coat the vial with an evenly dispersed lipid mixture, mixed lipids were dried under argon (g) to remove chloroform followed by overnight removal of residual methanol in a vacuum chamber. The following day, PBS buffer was added to generate a 1× stock solution with a final mixed lipid concentration of 5 mg/ml. ATRA (0.4 mg) was added from a 15 mg/ml dimethyl sulfoxide (DMSO) stock solution. Lastly, 2 mg of purified ΔApo-AI-INT was added and the final volume adjusted to 4 ml. Samples were placed covered at room temperature and were briefly sonicated to disrupt large liposomes and aggregates during the middle of a 4 hour incubation period. Samples were dialyzed overnight at 4 °C in PBS to remove residual organic solvents, then collected and centrifuged at 16,000×g for 20 minutes at 4 °C to pellet lipid-drug aggregates. Supernatants were collected, 0.22 μm sterile filtered and stored at 4 °C. ATRA concentration was measured from UV absorbance spectra of each sample in a 1:20 dilution into either PBS or 100% ethanol with the long wavelength value being set to baseline. ATRA has a characteristic peak at 341 and 350 nm with known extinction coefficients of 45,300 & 44,300 M−1 cm−1, respectively.[37]
Vault expression, purification and NDI-ATRA packaging
Vaults were formed by expression of baculovirus containing recombinant MVP cDNA in Sf9 insect cells and purified according to previously described methods.[12] The purified vaults, referred to as CP-MVP vaults, were mixed with NDI with or without ATRA at a protein ratio of 2000 μg: 250 μg for 30 minutes at 4 °C and then loaded onto a 20-60% discontinuous sucrose gradient. Samples were centrifuged at 25,000×g for 16 hours. Sucrose fractions were collected, pooled, diluted and centrifuged at 100,000×g for 2 hours at 4 °C to pellet vaults. Vault fractions were resuspended in 20 mM MES buffer pH 6.5, assayed for protein and ATRA then stored at 4 °C.
Transmission electron microscopy, cryo-electron tomography, and reconstruction
Negative staining and TEM was conducted using a JEM 1200EX microscope (JEOL) equipped with a Bioscan 600W digital camera (Gatan Inc.). Cryogenically frozen samples were prepared on Quantifoil grids [38]. An FEI Tecnai F20 transmission cryo-electron microscope was used to acquire tomography tilt series from −70° to +70°. The tilt series were recorded on a 16 megapixel TVIPS CCD camera using a magnification which gives a pixel size of 0.45 nm/pixel at the specimen level. The tomography tilt series were processed with a suite of programs to generate a 3D reconstruction. First, the series were precisely aligned using etomo program from the Imod package.[39] Rough alignment was performed by cross-correlation of image in the tilt series, followed by fine alignment using 10 nm gold beads as fiducial markers. A boundary model was created to adjust the tilt axis before a final, precisely aligned tilt stack was produced for each series. The aligned tilt series was then used to make a 3D reconstruction using GPU-based SIRT (Simultaneous Iterative Reconstruction Technique) reconstruction implemented in the Inspect3D program from FEI. The 3D reconstructions were saved as a stack of X-Y sections stacked along the Z direction. This 3D tomographic reconstruction was further processed by median filtering and pixel binning to enhance structural contrast. Individual vault particles were then segmented from the 3D density maps and displayed by volume renderings of the vault particles with packaged material using Amira (Visage Inc.). For densities corresponding to the vault particle, the known 39-fold symmetry was imposed to improve the signal to noise ratio for volume rendering.[40]
HepG2 cell viability assay
HepG2 cells were grown in Dulbecco/Vogt modified Eagle's minimal essential medium (DMEM) containing penicillin/streptomyocin at 37 °C + 5% CO2. Cells were plated into a 24 well plate at 5×104 cells in 1 ml and grown overnight. After 24 hours, cells were washed and fresh media added. Either media, free ATRA in 20 mM MES buffer pH 6.5, NDI, NDI-ATRA, CP-MVP, or CP-MVP containing NDI-ATRA were added at an equalized ATRA concentration of 1.5 μg/ml. Cells were incubated for 120 hours and then assayed by an MTT assay to determine cell viability.
Acknowledgments
This work was supported by the UC Discovery Grant Program in collaboration with our corporate sponsor, Abraxis Biosciences Inc (Award# BIO07-10671) and by the Mather's Charitable Foundation (Grant# 04095186) and NIH/NIBIB Award R01 EB004553.
The authors would like to thank Drs. Jenny Kim, Jian Yang, Jan Mrazek and Hongrong Liu for their guidance and technical expertise. Additionally, we thank Dr. Zach Tolstyka, Yuwen Chen and Hedi Roseboro for technical assistance.
Contributor Information
Daniel C. Buehler, Department of Biological Chemistry, David Geffen School of Medicine, 615 Charles E. Young Dr. South, University of California, Los Angeles, CA 90095, USA
Daniel B. Toso, Department of Microbiology, Immunology & Molecular Genetics, and Biomedical Engineering Program, 609 Charles E. Young Dr. South, University of California, Los Angeles, California 90095, USA
Dr. Valerie A. Kickhoefer, Department of Biological Chemistry, David Geffen School of Medicine, 615 Charles E. Young Dr. South, University of California, Los Angeles, CA 90095, USA
Prof. Z. Hong Zhou, Department of Microbiology, Immunology & Molecular Genetics, and Biomedical Engineering Program, 609 Charles E. Young Dr. South, University of California, Los Angeles, California 90095, USA; California Nanosystems Institute, University of California, Los Angeles, California 90095
Prof. Leonard H. Rome, Email: lrome@mednet.ucla.edu, Department of Biological Chemistry, David Geffen School of Medicine, 615 Charles E. Young Dr. South, University of California, Los Angeles, CA 90095, USA; California Nanosystems Institute, University of California, Los Angeles, California 90095.
References
- 1.Saha RN, Vasanthakumar S, Bende G, Snehalatha M. Mol Membr Biol. 2010;27:215–231. doi: 10.3109/09687688.2010.510804. [DOI] [PubMed] [Google Scholar]
- 2.Samad A, Alam MI, Saxena K. Curr Pharm Des. 2009;15:2958–2969. doi: 10.2174/138161209789058200. [DOI] [PubMed] [Google Scholar]
- 3.Avgoustakis K. Curr Drug Deliv. 2004;1:321–333. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 4.Charman WN. J Pharm Sci. 2000;89:967–978. doi: 10.1002/1520-6017(200008)89:8<967::aid-jps1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 5.Kedersha NL, Rome LH. J Cell Biol. 1986;103:699–709. doi: 10.1083/jcb.103.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suprenant KA. Biochemistry. 2002;41:14447–14454. doi: 10.1021/bi026747e. [DOI] [PubMed] [Google Scholar]
- 7.Kedersha NL, Miquel MC, Bittner D, Rome LH. J Cell Biol. 1990;110:895–901. doi: 10.1083/jcb.110.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zon A, Mossink MH, Scheper RJ, Sonneveld P, Wiemer EA. Cell Mol Life Sci. 2003;60:1828–1837. doi: 10.1007/s00018-003-3030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikyas Y, Makabi M, Raval-Fernandes S, Harrington L, Kickhoefer VA, Rome LH, Stewart PL. J Mol Biol. 2004;344:91–105. doi: 10.1016/j.jmb.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 10.Kickhoefer VA, Siva AC, Kedersha NL, Inman EM, Ruland C, Streuli M, Rome LH. Journal of Cell Biology. 1999a;146:917–928. doi: 10.1083/jcb.146.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kickhoefer VA, Liu Y, Kong LB, Snow BE, Stewart PL, Harrington L, Rome LH. J Cell Biol. 2001;152:157–164. doi: 10.1083/jcb.152.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephen AG, Raval-Fernandes S, Huynh T, Torres M, Kickhoefer VA, Rome LH. J Biol Chem. 2001;276:23217–23220. doi: 10.1074/jbc.C100226200. [DOI] [PubMed] [Google Scholar]
- 13.Kickhoefer VA, Garcia Y, Mikyas Y, Johansson E, Zhou JC, Raval-Fernandes S, Minoofar P, Zink JI, Dunn B, Stewart PL, Rome LH. Proc Natl Acad Sci U S A. 2005;102:4348–4352. doi: 10.1073/pnas.0500929102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poderycki MJ, Kickhoefer VA, Kaddis CS, Raval-Fernandes S, Johansson E, Zink JI, Loo JA, Rome LH. Biochemistry. 2006;45:12184–12193. doi: 10.1021/bi0610552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Champion CI, Kickhoefer VA, Liu G, Moniz RJ, Freed AS, Bergmann LL, Vaccari D, Raval-Fernandes S, Chan AM, Rome LH, Kelly KA. PLoS One. 2009;4:e5409. doi: 10.1371/journal.pone.0005409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldsmith LE, Pupols M, Kickhoefer VA, Rome LH, Monbouquette HG. ACS Nano. 2009;3:3175–3183. doi: 10.1021/nn900555d. [DOI] [PubMed] [Google Scholar]
- 17.Lai CY, Wiethoff CM, Kickhoefer VA, Rome LH, Nemerow GR. ACS Nano. 2009;3:691–699. doi: 10.1021/nn8008504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng BC, Yu M, Gopal A, Rome LH, Monbouquette HG, Tolbert SH. Nano Lett. 2008;8:3503–3509. doi: 10.1021/nl080537r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kickhoefer VA, Han M, Raval-Fernandes S, Poderycki MJ, Moniz RJ, Vaccari D, Silvestry M, Stewart PL, Kelly KA, Rome LH. ACS Nano. 2009;3:27–36. doi: 10.1021/nn800638x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayburt TH, Grinkova YV, Sligar SG. Nano Lett. 2002;2:853–856. [Google Scholar]
- 21.Ryan RO. Nanobiotechnology. 2010;8:28. doi: 10.1186/1477-3155-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oda MN, Hargreaves PL, Beckstead JA, Redmond KA, van Antwerpen R, Ryan RO. J Lipid Res. 2006;47:260–267. doi: 10.1194/jlr.D500033-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Nelson KG, Bishop JV, Ryan RO, Titus R. Antimicrob Agents Chemother. 2006;50:1238–1244. doi: 10.1128/AAC.50.4.1238-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redmond KA, Nguyen TS, Ryan RO. Int J Pharm. 2007;339:246–250. doi: 10.1016/j.ijpharm.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh AT, Evens AM, Anderson RJ, Beckstead JA, Sankar N, Sassano A, Bhalla S, Yang S, Platanias LC, Forte TM, Ryan RO, Gordon LI. Br J Haematol. 2010;150:158–169. doi: 10.1111/j.1365-2141.2010.08209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iovannisci DM, Beckstead JA, Ryan RO. Biochem Biophys Res Commun. 2009;379:466–469. doi: 10.1016/j.bbrc.2008.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Napoli JL. Clin Immunol Immunopathol. 1996;80:S52–62. doi: 10.1006/clin.1996.0142. [DOI] [PubMed] [Google Scholar]
- 28.Soprano DR, Qin P, Soprano KJ. Annu Rev Nutr. 2004;24:201–221. doi: 10.1146/annurev.nutr.24.012003.132407. [DOI] [PubMed] [Google Scholar]
- 29.Clarke N, Germain P, Altucci L, Gronemeyer H. Expert Rev Mol Med. 2004;6:1–23. doi: 10.1017/S1462399404008488. [DOI] [PubMed] [Google Scholar]
- 30.Okuno M, Kojima S, Matsushima-Nishiwaki R, Tsurumi H, Muto Y, Friedman SL, Moriwaki H. Curr Cancer Drug Targets. 2004;4:285–298. doi: 10.2174/1568009043333023. [DOI] [PubMed] [Google Scholar]
- 31.Liu PT, Krutzik SR, Kim J, Modlin RL. J Immunol. 2005;174:2467–2470. doi: 10.4049/jimmunol.174.5.2467. [DOI] [PubMed] [Google Scholar]
- 32.Patatanian E, Thompson DF. J Clin Pharm Ther. 2008;33:331–338. doi: 10.1111/j.1365-2710.2008.00935.x. [DOI] [PubMed] [Google Scholar]
- 33.Cai W, Zhao H, Guo J, Li Y, Yuan Z, Wang W. Childs Nerv Syst. 2007;23:549–554. doi: 10.1007/s00381-006-0289-y. [DOI] [PubMed] [Google Scholar]
- 34.Goldsmith LE, Yu M, Rome LH, Monbouquette HG. Biochemistry. 2007;46:2865–2875. doi: 10.1021/bi0606243. [DOI] [PubMed] [Google Scholar]
- 35.Yu M, Ng BC, Rome LH, Tolbert SH, Monbouquette HG. Nano Lett. 2008;8:3510–3515. doi: 10.1021/nl080536z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Kickhoefer VA, Ng BC, Gopal A, Bentolila LA, John S, Tolbert SH, Rome LH. ACS Nano. 2010 doi: 10.1021/nn102051r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.H FI, Suzts Ete Z. Archives of Biochemistry and Biophysics. 1991;287:297–304. doi: 10.1016/0003-9861(91)90482-x. [DOI] [PubMed] [Google Scholar]
- 38.Ermantraut E, Wohlfart K, Tichelaar W. Ultramicroscopy. 1998;74:75–81. [Google Scholar]
- 39.Kremer JR, Mastronarde DN, McIntosh JR. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka H, Kato K, Yamashita E, Sumizawa T, Zhou Y, Yao M, Iwasaki K, Yoshimura M, Tsukihara T. Science. 2009;323:384–388. doi: 10.1126/science.1164975. [DOI] [PubMed] [Google Scholar]