

Abstract
Angiotensin-(1-7) [Ang-(1-7)] may have beneficial effects in diabetes mellitus-induced erectile dysfunction (DMIED) but its molecular actions in the diabetic corpus cavernosum (CC) are not known. We characterized the effects of diabetes and/or chronic in vivo administration of Ang-(1-7) on vascular reactivity in the rat corpus cavernosum (CC) and on protein expression levels of potential downstream effectors of the renin-angiotensin-aldosterone system (RAAS) such as angiotensin-converting enzyme (ACE), ACE2, Rho kinases 1 and 2 (ROCK1 and ROCK2), and omega-hydroxylase, the cytochrome-P450 enzyme that metabolizes arachidonic acid to form the vasoconstrictor, 20-hydroxyeicosatetraenoic acid. Streptozotocin-treated rats were chronicically administered Ang-(1-7) with or without A779, a Mas receptor antagonist, during weeks 4 to 6 of diabetes. Ang-(1-7) reversed diabetes-induced abnormal reactivity to vasoactive agents (endothelin-1, phenylepherine, and carbachol) in the CC without correcting hyperglycemia. Six weeks of diabetes led to elevated ACE, ROCK1, ROCK 2, and omega-hydroxylase and a concomitant decrease in ACE2 protein expression levels that were normalized by Ang-(1-7) treatment but not upon coadministration of A779. These data are supportive of the notion that the beneficial effects of Ang-(1-7) in DMIED involve counterregulation of diabetes-induced changes in ACE, ACE2, Rho kinases, and omega-hydroxylase proteins in the diabetic CC via a Mas receptor-dependent mechanism.
1. Introduction
Erectile dysfunction (ED), a measure of sexual dysfunction or impotency in males, is defined as the inability to achieve and/or maintain an erection sufficient to permit satisfactory sexual intercourse. It is commonly associated with diabetes mellitus (DM) with up to 75% of men with diabetes exhibiting some degree of erectile dysfunction (ED) [1–4]. The incidence of ED, in large part also due to the predicted increase in DM [5, 6], will rise to about 300 million sufferers worldwide by 2025 [7, 8] and represents a significant health burden.
DM-induced ED (DMIED) is multifactorial in aetiology comprising both central (neurogenic) and peripheral (vasculogenic) components and appears more severe and more resistant to treatment compared with nondiabetic ED [9, 10]. For example, treatment with phosphodiesterase (PDE) inhibitors such as sildenafil (Viagra) is not always effective in DMIED for reasons that are not entirely clear [3, 7]. Thus, there is a need for newer more effective therapies based on an increased understanding of the underlying mechanisms of DMIED.
The exact molecular mechanisms by which DM induces ED are not fully known but chronic hyperglycemia likely degrades both neural and vascular endothelium penile control systems that eventually leads to a failure in the neuronal response and/or increase in tone and/or contractility of the smooth muscle within the corpus cavernosum (CC) and penile arteries [7, 9]. Experimental evidence suggests that this may occur via hyperglycemia-induced modulation of nitric oxide (NO) signaling and/or proinflammatory cell signaling pathways and/or elevation in oxidative stress via several pathways including increased glycolysis, polyol pathway flux, formation of advanced glycation, and lipoxygenation end-products [4, 11, 12]. Additionally, there is now a growing body of evidence from our laboratory and others [7, 12–14] on the existence of a local renin-angiotensin-aldosterone system (RAAS) in the penis that plays a critical role in erectile function.
Angiotensin II (Ang II), a major effector of the RAAS, is formed from the actions of angiotensin-converting enzyme (ACE) on Angiotensin 1. It is expressed in the corpus cavernosum and via its AT1 receptor activates signaling pathways leading to vasoconstriction, proliferation, fibrosis, and oxidative stress that are thought to play a detrimental role in the progression of DMIED [7, 11–13, 15]. For example, we recently reported in a rat model of type 1 diabetes that Ang II-mediated elevation in oxidative stress, along with a concomitant decrease in antioxidant levels and increased DNA damage, resulted in major cellular degeneration with the diabetic CC that could be blocked either by preventing the formation of Ang II with an ACE inhibitor or by blocking its effects with an AT1 receptor antagonist [12].
In contrast to the detrimental ACE/Ang II/AT1 receptor “branch” of the RAAS, there also now appears to be a counterregulating or opposing beneficial “branch” that comprises the angiotensin-converting enzyme 2 (ACE2) that can form the heptapeptide, angiotensin-(1-7) (Ang-(1-7)) from Ang II, which mediates its effects via the G-protein coupled receptor known as Mas. The ACE2/Ang-(1-7)/Mas receptor pathway is known to oppose the detrimental effects of ACE/Ang II/AT1 receptor in diabetes-induced cardiovascular complications [16–18] and recent evidence suggests it may also be involved in DMIED [7, 12–14, 19]. Several studies have now suggested that Ang-(1-7) has proerectile functions that include enhancing NO-mediated vasodilation, inhibiting penile fibrosis, and attenuating oxidative-stress mediated tissue degeneration [7, 12–14, 20, 21]. For example, we have shown that Ang-(1-7) treatment opposed Ang II-induced oxidative stress and DNA damage that led to penile tissue degeneration in a rat model of type 1 diabetes [12], whereas acute, ex vivo administration of Ang-(1-7) to diabetic rabbit CC segments was effective in attenuating diabetes or Ang II-induced hyperreactivity suggesting a possible role for Ang-(1-7) in the treatment of DMIED [14]. Although these studies suggested that alterations in the fine balance between the ACE/Ang II/AT1 receptor and the ACE2/Ang-(1-7)/Mas receptor pathways will likely be important in the development of DMIED, the exact molecular mechanisms underlying the disease and how Ang-(1-7) treatment may impact on specific effector molecules is not known in detail.
2. Aims
The aim of this study was to characterize the effects of diabetes and/or chronic administration of Ang-(1-7) on vascular reactivity in the rat CC and in the presence or absence of a MAS receptor antagonist on protein expression levels in the rat CC of (a) ACE1 and ACE2, major components of the two counterregulatory branches of the RAAS; (b) Rho kinases (ROCK) 1 and 2, important contractile regulatory proteins known to be important mediators of ED [22, 23]; and (c) the cytochrome P450 4A1 enzyme, omega-hydroxylase, that metabolizes arachidonic acid (AA) to 20-hydroxyeicosatetraenoic acid (20-HETE), a potent vasoconstrictor [24–26] that we previously identified as a potential target in the treatment of DMIED [19]. Both ROCKs and omega-hydroxylase are known downstream effectors of Ang II in the cardiovascular system [24, 25, 27, 28] but the effect of Ang-(1-7) treatment on the expression of these proteins in the diabetic CC is not known and therefore a subject of this study.
3. Methods
3.1. Induction of Diabetes
Experiments were performed on male Wistar rats weighing approximately 300 g. The rats had free access to food and water throughout the study. Diabetes was induced by a single ip injection (55 mg/kg, body weight) of streptozotocin (STZ). Rats with fasting blood glucose levels above 250 mg/dL were considered diabetic and included in the study. The animals were sacrificed six weeks after inducing diabetes. Drug treatment was administered for the last 3 weeks prior to sacrificing the animals at 6 weeks.
3.2. Animal Groups
All studies involving animals were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication number 85-23, Revised in 1985) as approved by Kuwait University Research Administration. Animals included in this study were divided into the following groups (N = 12/group): Group 1: nondiabetic (control) animals, Group 2: STZ-treated diabetic animals, Group 3: STZ + Ang-(1-7), and Group 4: STZ + Ang-(1-7) + A779 (a Mas receptor antagonist). Ang-(1-7) was given at a dose of 576 μg/kg/day intraperitoneally (ip) and A779 at a dose of 744 μg/kg/day ip based on our previous studies in animal models [12, 18, 29–33].
3.3. Vascular Reactivity Experiments
Rats were anesthetized with ketamine and then sacrificed and the penis was removed en bloc. A ventral incision was made and the cavernosal tissue exposed. The corpus cavernosum was cleaned of the adjacent tissue and cut into longitudinal strips of 2 × 10 mm. Strips of the corpus cavernosum were suspended longitudinally in organ-bath chambers containing 25 mL Krebs Henseleit (KH) solution at pH 7.4, for measurement of isometric tension [14, 19]. The composition of KH-solution was as follows (mM): NaCl (118.3), KCl (4.7), CaCl2 (2.5), MgSO4 (1.2), NaHCO3 (25), KH2PO4 (1.2), and glucose (11.2). The tissue bath solution was maintained at 37°C and was aerated with 95% oxygen and 5% carbon dioxide mixture. Reactivity of the isolated strips of the corpus cavernosum was determined by measurement of changes in the isometric tension to vasoactive agonists using computerized automatic organ bath LSI Letica Scientific Instruments (Powerlab/8sp ADInstruments, Panlab, Spain) [14, 19]. The preparations were left for 45 min with changing KH solution at 15 min intervals. A pretension of 1.0 gm was applied and the preparations were allowed to stabilize (45 min) until a stable baseline tone was obtained.
3.4. Effect of Vasoactive Agonists
Cumulative concentration response curves were established to investigate the contractile responses of the corpus cavernosum to the constrictor agonists phenylephrine (PE) and endothelin-1 (ET-1). Ascending concentrations of the agonists PE (10−9–5 × 10−3 M) and ET-1 (10−10–10−6 M) were applied to establish a cumulative dose response curve using corpus cavernosal strips isolated from the different animal groups. The response to any given concentration of the agonists was left to stabilize before adding the next drug concentration. In another set of experiments, the relaxant responses to carbachol were investigated in the cavernosal strips from the different animal groups. The relaxant response to the agonists were determined after precontracting the tissues with submaximal dose of PE (3 × 10−7 M) added to the organ baths. After obtaining a steady level of precontraction, the relaxant effects to carbachol (10−9–10−5 M) were tested on different preparations of the corpus cavernosum. The response to each concentration of the agonists was left to stabilize before adding the next drug concentration. The relaxant responses were expressed as percentage reduction of the tension induced by precontraction with PE.
3.5. Western Blotting Studies
Western blotting for ACE (band detected at molecular weight of approximately 195 kDa), ACE2 (90–100 kDa), ROCK1 and ROCK2 (both at around 160 kDa), and omega-hydroxylase (58 kDa) was performed similar to that described by us previously [32, 34]. Strips of the corpus cavernosum were dissected from the different animal groups, cut into small pieces, and homogenized in appropriate amounts of lysis buffer (pH 7.6) containing 50 mM Tris-base, 5 mM EGTA, 150 mM NaCl, 1% Triton 100, 2 mM Na3VO4, 50 mM NAF, 1 mM PMSF, 20 μM phenylarsine, 10 mM sodium molybdate, 10 μg/mL leupeptin, and 8 μg/mL aprotinin. The tissue was homogenized at half maximum speed for 1 minute and then left to lyse completely by incubation on shaking ice-cold shaker for 1 hour. Lysates were centrifuged at 12000 rpm at 4°C for 15 minutes. Supernatants were then collected and protein concentration was estimated using Lowry's method. Then the samples were mixed with sample loading buffer and lysis buffer to make all the samples of the same protein concentration. The samples were then boiled for 5 min and subjected to SDS polyacrylamide gel electrophoresis. The separated proteins were then transferred to PVDF membrane for 1 hour at 95 V. After blocking, membranes were incubated with the primary antibodies specific for each protein overnight and subsequently with appropriate secondary antibodies conjugated to horseradish peroxidase (Amersham, UK). The following antibodies were used in this study: ACE (H-170): sc-20791, ACE2 (H-175): sc-20998, ROCK1 (H-85): sc-5560, ROCK2 (H-85): sc-5561 (Santa Cruz, USA), and omega-hydroxylase (cytochrome P450 4A1): ab22615 (Abcam, USA). In addition, the anti-β-actin rabbit polyclonal IgG (1 μL/10 mL) Cat. number A-2066 was obtained from Sigma Chemical Co., USA. Immunoreactive bands were detected with SuperSignal Chemiluminescent Substrate (Pierce, UK) using Kodak autoradiography film (G.R.I., Rayne, UK). To ensure equal loading of proteins β-actin levels were detected using primary rabbit anti-human β-actin antibody followed by the secondary anti-rabbit IgG horseradish peroxidase conjugated antibody (Cell Signaling, USA). Images were finally analysed and quantified by densitometry and all data normalized to β-actin levels as described by us previously [32, 34].
3.6. Statistical Analysis
Results were analyzed using GraphPad Prism software. Data are presented as Mean ± SE of “n” number of experiments. Mean values were compared using either 2-way analysis of variance or analysis of variance followed by post hoc test (Bonferroni) as appropriate. The difference was considered to be significant when P value was less than 0.05.
3.6.1. Main Outcome Measures
Vascular responsiveness of isolated CC strips to vasoactive agents (endothelin-1, phenylephrine, and carbachol) and protein levels were determined by western blotting.
4. Results
4.1. Blood Glucose Levels and Body Weights
STZ-induced diabetes led to a significant elevation in blood glucose concentration. Hyperglycemia persisted in the diabetic animals and was 31.2 ± 1.7 mmol·L−1 after 6 weeks of diabetes as compared with 4.7 ± 0.7 mmol·L−1 in the nondiabetic control animals but 3-week chronic treatment with Ang-(1-7) (30.8 ± 1.4 mmol·L−1) did not significantly reduce blood glucose levels. There was a significant reduction in the weights of STZ-diabetic rats (145 ± 8 g) compared with the nondiabetic control animals (219 ± 5 g) after 6 weeks of diabetes, whereas Ang-(1-7) treatment significantly improved the weight of diabetic rats to 188 ± 11 g.
4.2. Chronic Treatment with Ang-(1-7) Reverses Diabetes-Induced Abnormal Vascular Reactivity in the Rat CC
Figures 1 and 2 show that PE and ET-1, respectively, produced a concentration-dependent contraction, while carbachol (Figure 3) produced relaxation in the isolated CC from both nondiabetic control and diabetic rats. In the CC tissue strips from diabetic animals, the constrictor responses to ET-1 and PE were significantly potentiated, whereas the relaxant responses to carbachol were significantly attenuated (P < 0.05). However, diabetes-induced abnormal reactivity to the vasoactive agonists was significantly corrected in cavernosal tissues isolated from diabetic animals chronically treated with Ang-(1-7) (P < 0.05) (Figures 1–3).
Figure 1.
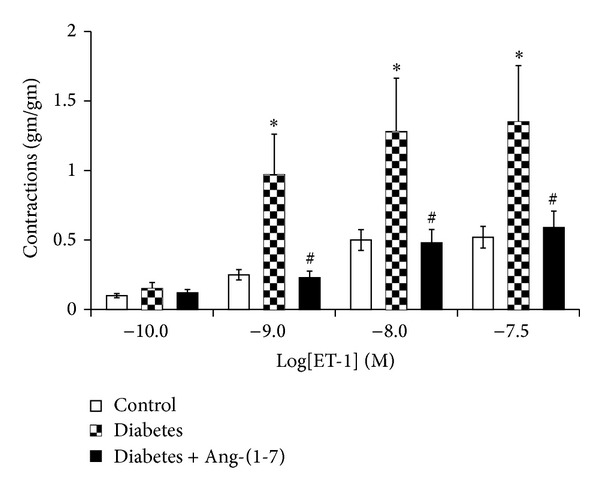
Endothelin-1-induced vasoconstriction in the corpus cavernosum segments from nondiabetic (control), diabetic, and diabetic rats treated with Ang-(1-7) (Mean ± SEM, n = 6–8). Asterisk (*) indicates significantly different mean values as compared to control, and hash (#) indicates significantly different mean values as compared to diabetes. P < 0.05. Tissues isolated from 6-week diabetic rats treated with Ang-(1-7) for the last 3 weeks of the study.
Figure 2.
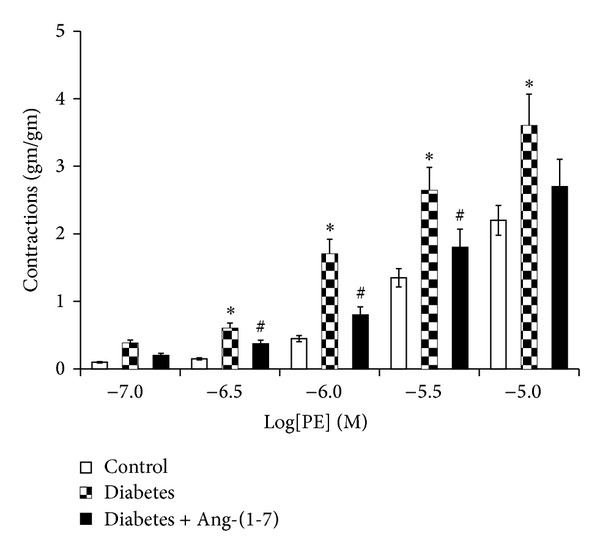
Phenylephrine-induced vasoconstriction in the corpus cavernosum segments from nondiabetic (control), diabetic, and diabetic rats treated with Ang-(1-7) (Mean ± SEM, n = 6–8). Asterisk (*) indicates significantly different mean values as compared to control, and hash (#) indicates significantly different mean values as compared to diabetes. P < 0.05. Tissues isolated from 6-week diabetic rats treated with Ang-(1-7) for the last three weeks of the study.
Figure 3.
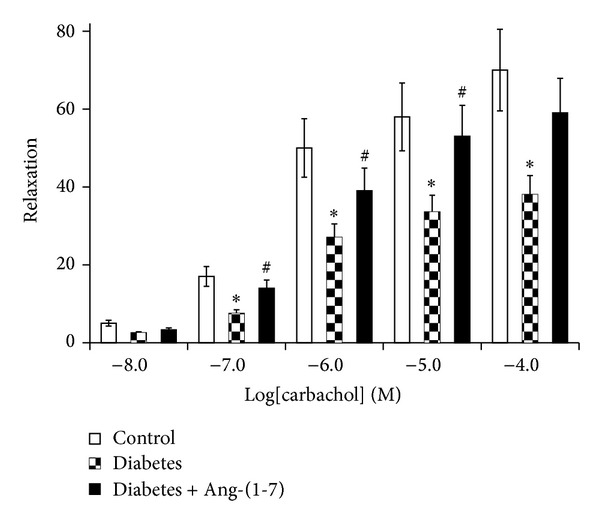
Carbachol-induced relaxation (expressed as %) in the corpus cavernosum segments from nondiabetic (control), diabetic, and diabetic rats treated with Ang-(1-7) (Mean ± SEM, n = 6–8). Asterisk (*) indicates significantly different mean values as compared to control, and hash (#) indicates significantly different mean values as compared to diabetes. P < 0.05. Tissues isolated from 6-week diabetic rats treated with Ang-(1-7) for the last three weeks of the study.
4.3. Chronic Treatment with Ang-(1-7) Attenuates Diabetes-Induced Changes in ACE and ACE2 Protein Expression via Its Mas Receptor
Figure 4 shows that 6 weeks of diabetes led to significantly increased expression of ACE and decreased expression of ACE2 proteins in the rat CC. Chronic treatment with Ang-(1-7) significantly attenuated diabetes-induced changes in both proteins (P < 0.05). Chronic coadministration of the Mas receptor antagonist, A779, with Ang-(1-7) reversed the effects of Ang-(1-7) on ACE and ACE protein expression in the diabetic rat CC (P < 0.05).
Figure 4.
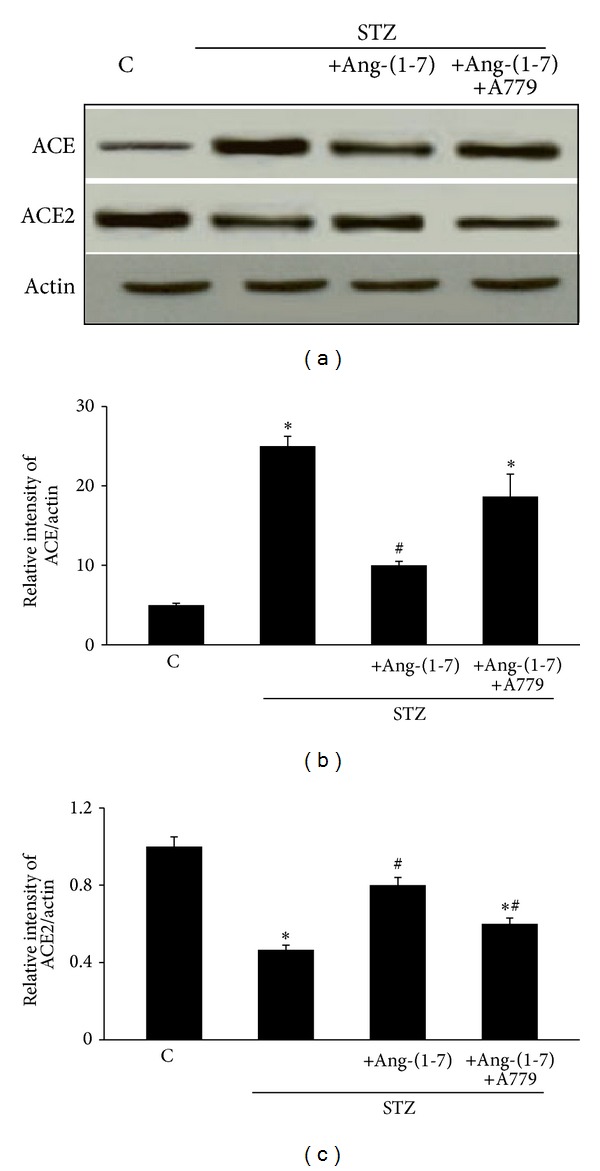
The levels of ACE and ACE2 protein expression in corpus cavernosum of nondiabetic (C), diabetic (STZ), and STZ-rats chronically treated either with Ang-(1-7) alone (STZ + Ang-(1-7)) or in combination with A779 (STZ + Ang-(1-7) + A779). Panel (a) is a representative western blot showing the levels of ACE, ACE2, and the protein loading control, β-actin. Panels (b) and (c) are densitometry histograms showing levels of ACE and ACE2 normalized to actin, respectively. N = 4; Mean ± SD. Asterisk (∗) indicates significantly different (P < 0.05) mean values from normal nondiabetic rats (C), whereas hash (#) indicates significantly different mean values (P < 0.05) from diabetic rats (STZ).
4.4. Chronic Ang-(1-7) Treatment Corrects Diabetes-Induced Elevation in ROCK1 and ROCK2 Protein Expression via Its Mas Receptor
Figure 5 shows that diabetes led to significantly increased protein expression of ROCK1 and ROCK2 in the rat CC (P < 0.05). Chronic treatment with Ang-(1-7) significantly attenuated diabetes-induced changes in both ROCK proteins (P < 0.05). Chronic coadministration of A779 with Ang-(1-7) reversed the effects of Ang-(1-7) on ROCK1 and ROCK2 protein expression in the diabetic rat CC (P < 0.05) to a level that was not significantly different to nondiabetic controls (Figure 5).
Figure 5.
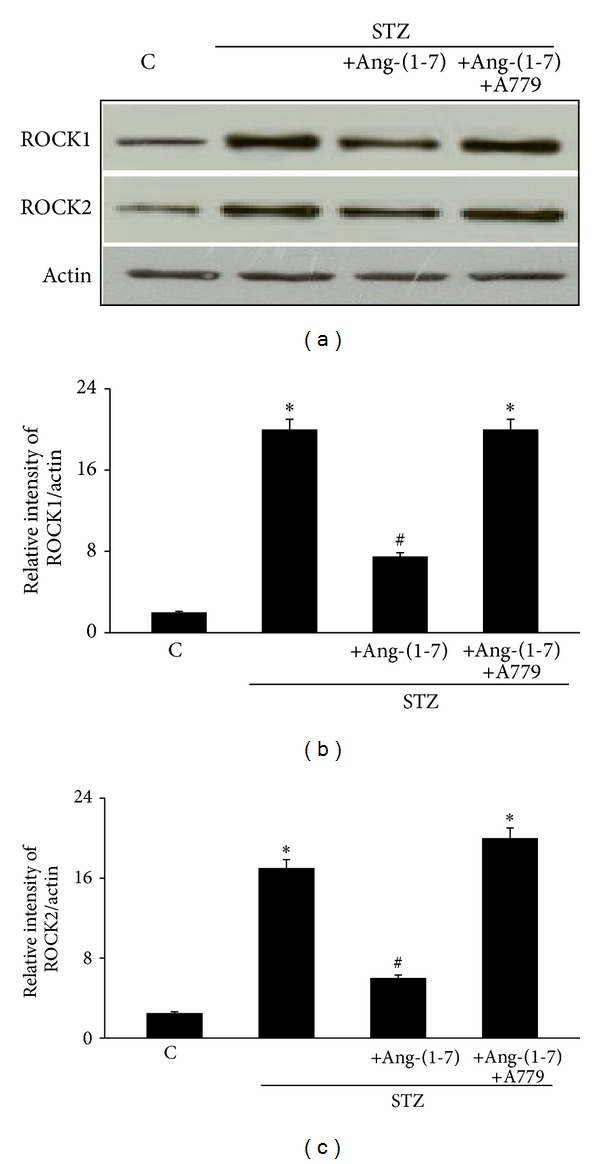
The levels of ROCK1 and ROCK2 protein expression in corpus cavernosum of nondiabetic (C), diabetic (STZ), and STZ-rats chronically treated either with Ang-(1-7) alone (STZ + Ang-(1-7)) or in combination with A779 (STZ + Ang-(1-7) + A779). Panel (a) is a representative western blot showing the levels of ROCK1, ROCK2, and the protein loading control, β-actin. Panels (b) and (c) are densitometry histograms showing levels of ROCK1 and ROCK2 normalized to actin, respectively. N = 4; Mean ± SD. Asterisk (∗) indicates significantly different (P < 0.05) mean values from normal nondiabetic rats (C), whereas hash (#) indicates significantly different mean values (P < 0.05) from diabetic rats (STZ).
4.5. Chronic Treatment with Ang-(1-7) Attenuates Diabetes-Induced Increase in Omega-Hydroxylase Protein Expression via Its Mas Receptor
Figure 6 shows that diabetes led to significantly increased expression of omega-hydroxylase protein in the rat CC (P < 0.05). Chronic treatment with Ang-(1-7) significantly attenuated diabetes-induced changes in omega-hydroxylase protein expression (P < 0.05). Chronic coadministration of A779 with Ang-(1-7) significantly attenuated the effects of Ang-(1-7) on omega-hydroxylase protein expression in the diabetic rat CC (P < 0.05) (Figure 5).
Figure 6.
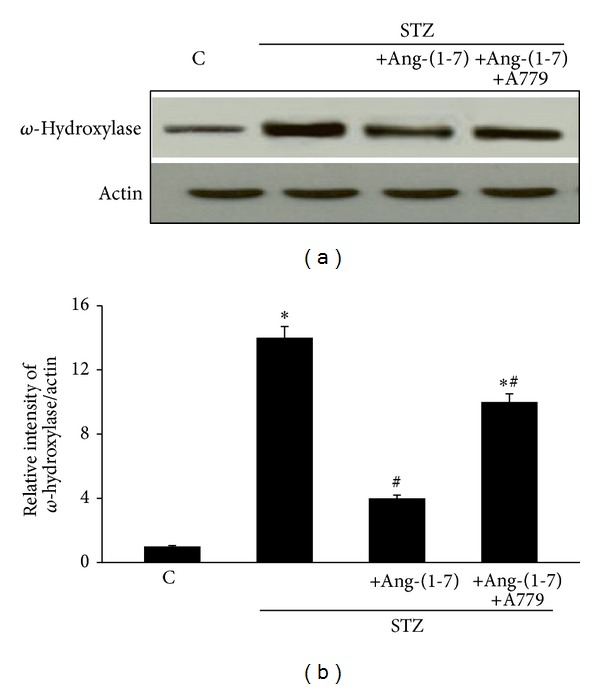
The level of omega-hydroxylase protein expression in the corpus cavernosum of nondiabetic (C), diabetic (STZ), and STZ-rats chronically treated either with Ang-(1-7) alone (STZ + Ang-(1-7)) or in combination with A779 (STZ + Ang-(1-7) + A779). Panel (a) is a representative western blot showing the level of omega(ω)-hydroxylase protein and β-actin as a loading control. Panels (b) and (C) are densitometry histograms showing levels of omega(ω)-hydroxylase protein normalized to actin. N = 4; Mean ± SD. Asterisk (∗) indicates significantly different (P < 0.05) mean values from normal nondiabetic rats (C), whereas hash (#) indicates significantly different mean values (P < 0.05) from diabetic rats (STZ).
5. Discussion
The major findings of this study were that diabetes led to an upregulation in ACE, ROCK1, ROCK2, and omega-hydroxylase proteins and a downregulation in ACE2 protein that was accompanied by abnormal vascular reactivity in the rat CC. Importantly, chronic treatment with exogenous Ang-(1-7) was able to reverse diabetes-induced abnormal CC vascular reactivity without correcting hyperglycemia and via its Mas receptor normalized diabetes-induced changes in ACE, ACE2, ROCK1, ROCK2, and omega-hydroxylase proteins in the rat CC. Our study, therefore, showed for the first time that an imbalance in the ACE-ACE2 enzymes is associated with development of DMIED and further highlights ROCKs and omega-hydroxylase as novel protein effectors by which Ang-(1-7) mediates its beneficial effects in DMIED. Furthermore, since only a relatively short term, 3-week chronic treatment with Ang-(1-7) in animals with preestablished diabetes was effective in reversing DM-induced protein and vascular reactivity changes in the diabetic CC, our study has clinical relevance in suggesting that therapeutic strategies aimed at activating the endogenous ACE2/Ang-(1-7)/Mas receptor signaling pathway for even a short time course may be beneficial in DMIED.
The fact that in our study we were able to measure detectable levels of ACE and ACE2 in normal and diabetic CC provided further evidence for the existence of a local renin-angiotensin system (RAS) within the CC [3, 7, 11]. Moreover, since diabetes led to a significant upregulation of ACE and downregulation of ACE2 in the rat CC (Figure 4), our data implies that disruption in the ACE-ACE2 balance with a resulting hyperactivity of the detrimental ACE/Ang II/AT1 receptor “branch” of the RAAS together with a concomitant attenuation of ACE2/Ang-(1-7)/Mas receptor signaling pathway is associated with the development of DMIED. Our finding in the diabteic rat CC is consistent with the development of other cardiovascular and renal pathologies that are also thought to involve an imbalance in ACE-ACE2 enzymes representing the two opposing “branches” of the RAAS [16, 17, 35].
The observed upregulation of ACE in the diabetic CC (Figure 4), which converts the inactive decapeptide Ang I into the octapeptide Ang II, the major effector of the RAAS, is consistent with previous reports showing higher levels of Ang II in the diabetic CC [36–38] that lead to deleterious effects, such as vasoconstriction, proliferation, fibrosis, and oxidative stress [7, 12, 15]. Pharmacological blockage of Ang II/AT1 actions using ACE inhibitors and/or AT1 receptor blockers (ARBs) has beneficial effects on erectile function in animal models and humans [1, 3, 11–13]. Since both ACE inhibitors and ARBs can increase Ang-(1-7) levels in plasma and tissue [39], it is possible that the beneficial effects of the Ang II blockade on erectile function might, at least in part, be mediated by Ang-(1-7). Indeed, we previously showed that not only modulation of Ang II signaling by captopril and losartan but also administration of Ang-(1-7) restored the diabetes-induced structural changes and oxidative DNA damage in the diabetic CC [12].
Our observation that ACE2 is downregulated in diabetes suggests reduced formation of endogenous Ang-(1-7) in the diabetic rat CC, though its levels were not measured directly in this study. Importantly, our results showed that chronic 3-week daily treatment with exogenous Ang-(1-7) was able to reverse diabetes-induced changes in ACE1 and ACE2 and vascular reactivity in the rat CC presumably by activation of the endogenous ACE2/Ang-(1-7)/Mas receptor pathway and readdressing of the proposed imbalances in the two counterregulatory branches of the RAAS in the diabetic CC.
The fact that administration of Ang-(1-7) also reversed diabetes-induced upregulation of Rho kinases, ROCK1 and ROCK2, suggests a novel mechanism by which Ang-(1-7) may exerts its beneficial effects on DMIED. Upregulation of the Rho/ROCK pathway has been implicated in a variety of cardiovascular complications including ED [13, 15, 22] and may represent a downstream effector of Ang II. ACE/Ang II/AT1 receptor signaling is known to exert its vasoconstrictor effects on smooth muscle via its downstream effects on Rho/ROCK pathway [15, 40, 41]. Thus, ROCK upregulation might be contributing to the observed exaggerated responsiveness of the diabetic rat CC to vasoconstrictor agents (Figures 1 and 2) and attenuated response to relaxant effects of carbachol (Figure 3) in a manner similar to that observed by us in the diabetic mesenteric bed where diabetes-induced altered vascular reactivity was corrected upon ROCK inhibition [34]. Inhibition of ROCKs is also known to improve erectile function by a variety of mechanisms including increased NO signaling and suppressing penile apoptosis and corporal fibrosis [11, 42–44]. As such, ROCK inhibitors are increasingly being considered for the treatment of ED [1, 11]. Rho-kinase inhibitors improve erectile function in hypertensive, diabetic, and aged rats as well as in cavernous nerve injury induced rats [11, 42]. These reports clearly illustrate that direct inhibition of RhoA/Rho-kinase pathway may represent a suitable therapeutic approach in the treatment of ED. However, our data presented here suggests for the first time that chronic treatment with Ang-(1-7) can also reverse upregulation of ROCKs in the diabetic CC and may represent a novel alternative to conventional ROCK inhibitors for the treatment of ED especially in DMIED. Whether attenuation of ROCK protein expression in diabetic CC occurs via a direct effect of Ang-(1-7) or by counterregulation of ACE/Ang II/AT1 receptor pathway is not entirely clear and requires further study.
The fact that diabetes led to an elevation in omega-hydroxylase protein expression implies that DMIED is associated with increased production of 20-HETE-a potential downstream effector of the ACE/Ang II/AT1 receptor “branch” of the RAAS [24] though its levels were not measured here and is a potential limitation of this study. However, we previously showed that Ang II via its AT1 receptors increases 20-hydroxyeicosatetraenoic acid (20-HETE) production in vascular smooth muscle cells [28]. It is now well established that an imbalance in the metabolism of AA and 20-HETE levels contributes to development of cardiovascular dysfunction and end-organ damage [20, 24, 25, 45–48] but this is the first report in the CC showing an imbalance in omega-hydroxylase being associated with DMIED. 20-HETE is a potent vasoconstrictor that has important roles in the regulation of vascular tone in several different tissues [24]. Its upregulation in the diabetic CC implies that it may have a significant role in contributing to the exaggerated vasoconstrictor response to PE and ET-1 observed in the diabetic CC in this study (Figures 1 and 2). Elevated 20-HETE production is also known to occur in other vasculatures as well as the heart, liver, and kidney of diabetic animals [24, 47, 48]. Indeed, we were the first to show that selective inhibition of 20-HETE production with N-hydroxy-N′-(4-butyl-2-methyl-phenyl)-formamidine (HET0016; an inhibitor of CYP450 4A1, omega-hydroxylase) can prevent diabetes-induced hyperreactivity to vasoconstrictors in the rat carotid artery [47] and can further attenuate cardiovascular end-organ damage in animal models of diabetes and/or hypertension [20, 45, 48]. Interestingly, inhibition of 20-HETE also improved vasodilator responses in several vascular beds [20, 45, 48]. Thus, removal of the vasoconstrictor signaling mediated via 20-HETE leads to correction of diabetes-induced abnormal responsiveness to vasoconstrictors and vasodilators. This may also be occurring in our present study in the CC where we observed exaggerated CC vascular reactivity to PE and ET-1 (Figures 1 and 2) and an attenuated responsiveness to the vasorelaxant, carbachol (Figure 3), after 6 weeks of diabetes. Although not directly studied here, we previously showed that acute, ex vivo inhibition of omega-hydroxylase with HET0016 in CC segments resulted in correction of abnormal reactivity to phenylephrine and carbachol in diabetic and aged rats [19]. The fact that in the present study we observed a normalization of the abnormal CC reactivity upon chronic treatment with Ang-(1-7) suggests that Ang-(1-7) can also counterregulate omega-hydroxylase upregulation and the likely increased 20-HETE production in the diabetic CC. Taken together, our data are therefore consistent with the hypothesis that ACE/Ang II/AT1 receptor arm of the RAAS and likely downstream effectors, omega-hydroxylase/20-HETE and Rho/ROCK pathways, are driving the vasoconstriction and other deleterious antierectile actions in DMIED, whereas ACE2/Ang-(1-7)/Mas receptor signaling pathway leads to vasorelaxation and generally beneficial or proerectile actions in DMIED.
The data presented here is also consistent with previous reports on the beneficial actions of Ang-(1-7) in erectile function. Both Ang-(1-7) and its Mas receptor are known to be present in the human CC [11, 21]. Our group previously reported that Ang-(1-7) via Mas receptor can produce nitric oxide-dependent relaxation of the rabbit CC [14]. We further showed that acute, ex vivo preincubation of the diabetic rabbit CC with Ang-(1-7) corrected the diabetes-induced hyperresponsiveness to Ang II [14]. Ang-(1-7) has also been shown to normalize the severely depressed erectile function observed in DOCA-salt hypertensive rats by activation of Mas receptor and subsequent NO release [21] and oral delivery of Ang-(1-7) in a cyclodextrin formulation reduced penile fibrosis and improved cavernosal endothelial function in mice with hypercholesterolemia [7]. Additionally, also the synthetic Mas receptor agonist, AVE 0991, potentiated rat penile erectile response [49]. These studies suggest that proerectile vasculogenic actions of the ACE2/Ang-(1-7)/Mas receptor axis involve increased cavernosal vasodilation by increasing NO production/bioavailability [7, 14, 20, 21, 49], reduced oxidative-stress mediated damage to DNA and penile tissue [12], and blocking development of penile fibrosis [7, 13]. Although the above studies and our data presented here suggest that Ang-(1-7) counteracts the ACE/Ang II/AT1 receptor arm of the RAAS in the CC via its Mas receptor, there is a possibility that it may also block AT1 and/or stimulate AT2 receptors in certain cell types and tissues [7]. However, whether Ang-(1-7), in addition to its effects via Mas receptor, affects AT1 or AT2 receptors in the penis remains to be studied.
In conclusion, in this study we showed that diabetes-induced abnormal cavernosal vascular reactivity, a marker for DMIED, was accompanied by imbalances in ACE-ACE2, ROCK1, ROCK2, and omega-hydroxylase proteins which could be reversed by 3-week chronic treatment with Ang-(1-7) via activation of its Mas receptor. Whether more longer-term treatments with Ang-(1-7) would have any additional benefit remains to be determined. However, despite the caveat that only protein expression and not activity was measured, our study provides a novel insight into the mechanism of action of ACE2/Ang-(1-7)/Mas receptor pathway in the diabetic CC whereby we show for the first time that the beneficial effects of Ang-(1-7) in DMIED are at least partially due to inhibition of omega-hydroxylase, ROCKs, and ACE levels through activation of its Mas receptor. Thus, we further suggest that activating the endogenous ACE2/Ang-(1-7)/Mas receptor pathway of the RAAS system may represent an attractive strategy for the treatment of ED associated with diabetes.
Acknowledgments
This study was funded by a grant from Kuwait University Research Administration Project no. MR04/09. The authors also acknowledge support from the OMICS Research Unit/RCF and the General Facility Grant (SRUL02/13).
Conflict of Interests
The authors report no conflict of interests.
Authors' Contribution
Mariam H.M. Yousif, Ahmed Z. El-Hashim, Saghir Akhtar, and Ibrahim F. Benter were responsible for conception and design of the paper and revising it for intellectual content. Batoul Makki and Mariam H.M. Yousif were responsible foracquisition of data. Mariam H.M. Yousif, Batoul Makki, Saghir Akhtar, and Ibrahim F. Benter drafted the paper. Mariam H.M. Yousif, Batoul Makki, Ahmed Z. El-Hashim, Saghir Akhtar, and Ibrahim F. Benter were responsible for analysis and interpretation of data and the final approval of the completed paper.
References
- 1.Gur S, Peak TC, Kadowitz PJ, Sikka SC, Hellstrom WJG. Review of erectile dysfunction in diabetic animal models. Current Diabetes Reviews. 2014;10(1):61–73. doi: 10.2174/1573399809666131126151024. [DOI] [PubMed] [Google Scholar]
- 2.Zheng H, Liu X, Patel KP. Centrally mediated erectile dysfunction in rats with type 1 diabetes: role of angiotensin II and superoxide. Journal of Sexual Medicine. 2013;10(9):2165–2176. doi: 10.1111/jsm.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson K-E. Mechanisms of penile erection and basis for pharmacological treatment of erectile dysfunction. Pharmacological Reviews. 2011;63(4):811–859. doi: 10.1124/pr.111.004515. [DOI] [PubMed] [Google Scholar]
- 4.Thorve VS, Kshirsagar AD, Vyawahare NS, Joshi VS, Ingale KG, Mohite RJ. Diabetes-induced erectile dysfunction: epidemiology, pathophysiology and management. Journal of Diabetes and its Complications. 2011;25(2):129–136. doi: 10.1016/j.jdiacomp.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Scully T. Diabetes in numbers. Nature. 2012;485:S2–S3. doi: 10.1038/485s2a. [DOI] [PubMed] [Google Scholar]
- 6.Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes Atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Research and Clinical Practice. 2011;94(3):311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 7.Fraga-Silva RA, Montecucco F, Mach F, Santos RAS, Stergiopulos N. Pathophysiological role of the renin-angiotensin system on erectile dysfunction. European Journal of Clinical Investigation. 2013;43(9):978–985. doi: 10.1111/eci.12117. [DOI] [PubMed] [Google Scholar]
- 8.Aytaç IA, McKinlay JB, Krane RJ. The likely worldwide increase in erectile dysfunction between 1995 and 2025 and some possible policy consequences. BJU International. 1999;84(1):50–56. doi: 10.1046/j.1464-410x.1999.00142.x. [DOI] [PubMed] [Google Scholar]
- 9.Malavige LS, Levy JC. Erectile dysfunction in diabetes mellitus. Journal of Sexual Medicine. 2009;6(5):1232–1247. doi: 10.1111/j.1743-6109.2008.01168.x. [DOI] [PubMed] [Google Scholar]
- 10.Price D, Hackett G. Management of erectile dysfunction in diabetes: an update for 2008. Current Diabetes Reports. 2008;8(6):437–443. doi: 10.1007/s11892-008-0076-0. [DOI] [PubMed] [Google Scholar]
- 11.Decaluwé K, Pauwels B, Boydens C, van de Voorde J. Treatment of erectile dysfunction: new targets and strategies from recent research. Pharmacology Biochemistry and Behavior. 2014;121:146–157. doi: 10.1016/j.pbb.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 12.Fraga-Silva RA, Costa-Fraga FP, Savergnini SQ, et al. An oral formulation of Angiotensin-(1-7) reverses corpus cavernosum damages induced by Hypercholesterolemia. Journal of Sexual Medicine. 2013;10(10):2430–2442. doi: 10.1111/jsm.12262. [DOI] [PubMed] [Google Scholar]
- 13.Kilarkaje N, Yousif MHM, El-Hashim AZ, Makki B, Akhtar S, Benter IF. Role of angiotensin II and angiotensin-(1-7) in diabetes-induced oxidative DNA damage in the corpus cavernosum. Fertility and Sterility. 2013;100(1):226–233. doi: 10.1016/j.fertnstert.2013.02.046. [DOI] [PubMed] [Google Scholar]
- 14.Yousif MH, Kehinde EO, Benter IF. Different responses to angiotensin-(1–7) in young, aged and diabetic rabbit corpus cavernosum. Pharmacological Research. 2007;56(3):209–216. doi: 10.1016/j.phrs.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Jin L-M. Angiotensin II signaling and its implication in erectile dysfunction. Journal of Sexual Medicine. 2009;6(3):302–310. doi: 10.1111/j.1743-6109.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- 16.Patel VB, Parajuli N, Oudit GY. Role of angiotensin-converting enzyme 2 (ACE2) in diabetic cardiovascular complications. Clinical Science. 2014;126(7):471–482. doi: 10.1042/CS20130344. [DOI] [PubMed] [Google Scholar]
- 17.Santos RAS, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. Journal of Endocrinology. 2013;216(2):R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 18.Benter IF, Yousif MHM, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. American Journal of Physiology: Heart and Circulatory Physiology. 2007;292(1):H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- 19.Yousif MHM, Benter IF. Role of cytochrome P450 metabolites of arachidonic acid in regulation of corporal smooth muscle tone in diabetic and older rats. Vascular Pharmacology. 2007;47(5-6):281–287. doi: 10.1016/j.vph.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Yousif MHM, Benter IF, Dunn KMJ, Dahly-Vernon AJ, Akhtar S, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in altering vascular reactivity in diabetes. Autonomic and Autacoid Pharmacology. 2009;29(1-2):1–12. doi: 10.1111/j.1474-8673.2009.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Da Costa Gonçalves AC, Leite R, Fraga-Silva RA, et al. Evidence that the vasodilator angiotensin-(1-7)-Mas axis plays an important role in erectile function. The American Journal of Physiology—Heart and Circulatory Physiology. 2007;293(4):H2588–H2596. doi: 10.1152/ajpheart.00173.2007. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Wei L. Rho kinases in cardiovascular physiology and pathophysiology: the effect of fasudil. Journal of Cardiovascular Pharmacology. 2013;62(4):341–354. doi: 10.1097/FJC.0b013e3182a3718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bivalacqua TJ, Champion HC, Usta MF, et al. RhoA/Rho-kinase suppresses endothelial nitric oxide synthase in the penis: a mechanism for diabetes-associated erectile dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(24):9121–9126. doi: 10.1073/pnas.0400520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsaad AM, Zordoky BN, Tse MM, El-Kadi AO. Role of cytochrome P450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug Metabolism Reviews. 2013;45(2):173–195. doi: 10.3109/03602532.2012.754460. [DOI] [PubMed] [Google Scholar]
- 25.Fan F, Sun C-W, Maier KG, et al. 20-Hydroxyeicosatetraenoic acid contributes to the inhibition of K+ channel activity and vasoconstrictor response to angiotensin II in rat renal microvessels. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0082482.e82482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid ω-hydroxylases and 20-HETE formation. Annual Review of Pharmacology and Toxicology. 2005;45:413–438. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- 27.Giani JF, Gironacci MM, Muñoz MC, Turyn D, Dominici FP. Angiotensin-(1-7) has a dual role on growth-promoting signalling pathways in rat heart in vivo by stimulating STAT3 and STAT5a/b phosphorylation and inhibiting angiotensin II-stimulated ERK1/2 and Rho kinase activity. Experimental Physiology. 2008;93(5):570–578. doi: 10.1113/expphysiol.2007.014269. [DOI] [PubMed] [Google Scholar]
- 28.Muthalif MM, Benter IF, Uddin MR, Harper JL, Malik KU. Signal transduction mechanisms involved in angiotensin-(1-7)-stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth muscle cells. Journal of Pharmacology and Experimental Therapeutics. 1998;284(1):388–398. [PubMed] [Google Scholar]
- 29.Benter IF, Yousif MHM, Al-Saleh FM, Raghupathy R, Chappell MC, Diz DI. Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-l-arginine methyl ester. Journal of Cardiovascular Pharmacology. 2011;57(5):559–567. doi: 10.1097/FJC.0b013e31821324b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Hashim AZ, Renno WM, Raghupathy R, Abduo HT, Akhtar S, Benter IF. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways. British Journal of Pharmacology. 2012;166(6):1964–1976. doi: 10.1111/j.1476-5381.2012.01905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renno WM, Al-Banaw AG, George P, Abu-Ghefreh AA, Akhtar S, Benter IF. Angiotensin-(1-7) via the mas receptor alleviates the diabetes-induced decrease in GFAP and GAP-43 immunoreactivity with concomitant reduction in the COX-2 in hippocampal formation: an immunohistochemical study. Cellular and Molecular Neurobiology. 2012;32(8):1323–1336. doi: 10.1007/s10571-012-9858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akhtar S, Yousif MH, Dhaunsi GS, Chandrasekhar B, Al-Farsi O, Benter IF. Angiotensin-(1–7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. British Journal of Pharmacology. 2012;165(5):1390–1400. doi: 10.1111/j.1476-5381.2011.01613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yousif MHM, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF. Characterization of Angiotensin-(1-7) effects on the cardiovascular system in an experimental model of Type-1 diabetes. Pharmacological Research. 2012;66(3):269–275. doi: 10.1016/j.phrs.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Akhtar S, Yousif MHM, Dhaunsi GS, et al. Activation of ErbB2 and downstream signalling via Rho kinases and ERK1/2 contributes to diabetes-induced vascular dysfunction. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0067813.e67813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bader M. ACE2, angiotensin-(1-7), and Mas: The other side of the coin. Pflugers Archiv European Journal of Physiology. 2013;465(1):79–85. doi: 10.1007/s00424-012-1120-0. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y, Li SX, Yao LS, Wang R, Dai YT. Valsartan treatment reverses erectile dysfunction in diabetic rats. International Journal of Impotence Research. 2007;19(4):366–370. doi: 10.1038/sj.ijir.3901534. [DOI] [PubMed] [Google Scholar]
- 37.Abdel Aziz MT, El Asmer MF, Mostafa T, et al. Effects of Losartan, HO-1 Inducers or HO-1 inhibitors on erectile signaling in diabetic rats. Journal of Sexual Medicine. 2009;6(12):3254–3264. doi: 10.1111/j.1743-6109.2009.01517.x. [DOI] [PubMed] [Google Scholar]
- 38.Kifor I, Williams GH, Vickers MA, Sullivan MP, Jodbert P, Dluhy RG. Tissue angiotensin II as a modulator of erectile function. I. Angiotensin peptide content, secretion and effects in the corpus cavernosum. Journal of Urology. 1997;157(5):1920–1925. [PubMed] [Google Scholar]
- 39.Iusuf D, Henning RH, van Gilst WH, Roks AJM. Angiotensin-(1-7): pharmacological properties and pharmacotherapeutic perspectives. European Journal of Pharmacology. 2008;585(2-3):303–312. doi: 10.1016/j.ejphar.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 40.Ying Z, Jin L, Palmer T, Webb RC. Angiotensin II up-regulates the leukemia-associated Rho guanine nucleotide exchange factor (RhoGEF), a regulator of G protein signaling domain-containing RhoGEF, in vascular smooth muscle cells. Molecular Pharmacology. 2006;69(3):932–940. doi: 10.1124/mol.105.017830. [DOI] [PubMed] [Google Scholar]
- 41.Schofield AV, Bernard O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Critical Reviews in Biochemistry and Molecular Biology. 2013;48(4):301–316. doi: 10.3109/10409238.2013.786671. [DOI] [PubMed] [Google Scholar]
- 42.Hannan JL, Albersen M, Kutlu O, et al. Inhibition of Rho-kinase improves erectile function, increases nitric oxide signaling and decreases penile apoptosis in a rat model of cavernous nerve injury. Journal of Urology. 2013;189(3):1155–1161. doi: 10.1016/j.juro.2012.09.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho MC, Park K, Chai JS, Lee SH, Kim SW, Paick JS. Involvement of sphingosine-1-Phosphate/RhoA/Rho-Kinase signaling pathway in corporal fibrosis following cavernous nerve injury in male rats. Journal of Sexual Medicine. 2011;8(3):712–721. doi: 10.1111/j.1743-6109.2010.02147.x. [DOI] [PubMed] [Google Scholar]
- 44.Toque HA, Nunes KP, Yao L, et al. Activated rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: possible involvement of p38 MAPK activation. Journal of Sexual Medicine. 2013;10(6):1502–1515. doi: 10.1111/jsm.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yousif MHM, Benter IF, Roman RJ. Cytochrome P450 metabolites of arachidonic acid play a role in the enhanced cardiac dysfunction in diabetic rats following ischaemic reperfusion injury. Autonomic and Autacoid Pharmacology. 2009;29(1-2):33–41. doi: 10.1111/j.1474-8673.2009.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yousif MHM, Benter IF. Role of 20-hydroxyeicosatetraenoic and epoxyeicosatrienoic acids in the regulation of vascular function in a model of hypertension and endothelial dysfunction. Pharmacology. 2010;86(3):149–156. doi: 10.1159/000317521. [DOI] [PubMed] [Google Scholar]
- 47.Benter IF, Yousif MH, Canatan H, Akhtar S. Inhibition of Ca2+/calmodulin -dependent protein kinase II, RAS- GTPase and 20-hydroxyeicosatetraenoic acid attenuates the development of diabetes-induced vascular dysfunction in the rat carotid artery. Pharmacological Research. 2005;52(3):252–257. doi: 10.1016/j.phrs.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Benter IF, Francis I, Cojocel C, Juggi JS, Yousif MHM, Canatan H. Contribution of cytochrome P450 metabolites of arachidonic acid to hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Autonomic and Autacoid Pharmacology. 2005;25(4):143–154. doi: 10.1111/j.1474-8673.2005.00343.x. [DOI] [PubMed] [Google Scholar]
- 49.da Costa Gonçalves AC, Fraga-Silva RA, Leite R, Santos RAS. AVE 0991, a non-peptide Mas-receptor agonist, facilitates penile erection. Experimental Physiology. 2013;98(3):850–855. doi: 10.1113/expphysiol.2012.068551. [DOI] [PubMed] [Google Scholar]