

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disorder caused by damage of motoneurons leading to paralysis state and long term disability. Riluzole is currently the only FDA-approved drug for the treatment of ALS. The proposed mechanisms of ALS include glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, protein aggregation, SOD1 accumulations, and neuronal death. In this review, we discuss potential biomarkers for the identification of patients with ALS. We further emphasize potential therapy involving the uses of neurotrophic factors such as IGF-I, GDNF, VEGF, ADNF-9, colivelin and angiogenin in the treatment of ALS. Moreover, we described several existing drugs such as talampanel, ceftriaxone, pramipexole, dexpramipexole and arimoclomol potential compounds for the treatment of ALS. Interestingly, the uses of stem cell therapy and immunotherapy are promising for the treatment of ALS.
Keywords: ALS, ADNF-9, biomarkers, ceftriaxone, colivelin, neurotrophic factor, neurodegenerative disease, stem cell
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder caused by loss of cortical, brainstem and spinal motoneurons [reviewed by [1]], which occurs in both forms familial and sporadic ALS. Familial ALS (fALS) is similar to sporadic ALS. It is estimated that 10% of cases are inherited; and 20% caused by mutations in superoxide dismutase 1 gene (SOD1), and the remaining cases present mutations of other known and unknown genes [2]. The disease is linked to a genetic defect on chromosome 21q [3, 4], and chromosome 9q34 [5]. There are other forms of ALS that have been found linked to other chromosomes [reviewed by [6]]. There is also evidence that a failure to degrade ubiquitin-dependent protein can lead to the pathogenesis of ALS [7]. Another cause of ALS includes mutations in valosin-containing protein (VCP) gene [8] and C9ORF72 gene in chromosome 9 [9]. Thus, ALS might be caused or induced through several factors, both genetic and environmental [10-13].
The molecular mechanisms of neuronal death are unknown for ALS. There are several hypotheses suggesting that neuronal death may be caused by oxidative stress, a deficit in trophic factors, chronic inflammation, and possibly glutamate-induced excitoxicity [reviewed by [14, 15]]. Unbalanced glutamate uptake may be critical for ALS disease, as a dramatic loss of glutamate transporters has been found in the ALS motor cortex [16]. Another possible primary mechanism could be related to the toxin itself of the mutant SOD1 [13, 17].
Motoneurons are considered a key factor in ALS; the survival of these neurons depends on their targets, skeletal muscle and myelinated Schwann cells surrounding the peripheral axons [18, 19]. Thus, the spatiotemporal organization of the motoneurons makes it difficult to find efficient procedures for the treatment of this disease. To the best of our knowledge, there are no potential drugs for the treatments of ALS. Riluzole, an inhibitor of glutamate release, is the only compound clinically used in patients suffering from ALS [6]. However, the pool target of different trophic factors might have neuroprotection for the motoneurons [20, 21]. Thus, neurotrophic factors have been the focus in clinical trials to restore the deficits of the motoneurons in ALS. We discussed several outcomes of these potential neurotrophic factors for the treatments of ALS. We also discussed potential biomarkers in ALS, potential compounds for the treatment of ALS, possible uses of stem cell therapy in ALS, and immunotherapy for the treatment of ALS.
2. POTENTIAL BIOMARKERS IDENTIFIED IN PATIENTS WITH ALS
The diagnosis of ALS is a challenging experience. The lack of specific diagnostic tests and of measurable biomarkers (Table 1) delays the diagnosis of this neurodegenerative disorder by an average of 12 months after the onset of symptoms [22]. A diagnostic test, which, if positive, would be characteristic of ALS, would allow early detection of ALS. Thus, earlier disease-modifying treatments would be provided to improve the disease state of the patient. Moreover, the identification of specific biomarkers would be a valuable tool to understand the neuropathophysiology of ALS for the determination of effective drugs for the treatment of ALS.
Table 1.
Summary of Findings Investigated Biomarkers for ALS
| Biomarkers | Mechanism of Action | References |
|---|---|---|
| Neurofilament proteins (Nfs) | NfL levels are increased | [23-26] |
| Cytoplasmic FMR Interacting Protein (CyFIP2) Retino- blastoma Binding Protein 9 (RbBP9) |
Change in the expression levels and RNA editing of CyFIP2 and RbBP9 genes |
[27] |
| Transglutamine enzymes activity (TGases) | TGases is elevated at early stages and decreased at the terminal stages | [26] |
| Transactive response DNA-binding protein 43 (TDP-43) | Increased levels of abnormal TDP-43 at the onset of ALS | [27-29] |
Neurofilament heavy chain proteins might be a promising biomarker in the disease progression of ALS. Neurofilament proteins (Nfs) are target in large myelinated neurons. Nfs are composed of at least three polypeptide subunits such as neurofilament light chain (NfL, 68 kDa), neurofilament medium chain (NfM, 150 kDa) and neurofilament heavy chain (NfH, 200 kDa) [23]. One of the known factors of the histopathological mechanisms of ALS includes alteration of axonal transport and protein aggregation [14]. It has been suggested that cerebrospinal fluid (CSF) NfL levels are increased in ALS patients compared to patients with other neurodegenerative diseases, and phosphorylation of Nfs is indicative of the pathogenesis of some neurodegenerative diseases [23]. Studies suggest that an up-regulation of phosphorylated NfH levels in plasma has a correlation with the later stages of disease progression in SOD1 mouse model of ALS. Their inverse correlation proved to be statistically significant; this includes 90% sensitivity and 87% specificity [24-26]. There-fore, NfH levels may be used as a marker to diagnose and facilitate treatment of ALS.
Coupled with NfH levels, other potential biomarkers have been identified, such as Cytoplasmic FMR Interacting Protein (CyFIP2) and/or Retinoblastoma Binding Protein 9 (RbBP9) in ALS patients [27]. It was found that mesenchymal stem cells (MSCs) of ALS patients exhibited a change in the expression levels and RNA editing of CyFIP2 and RbBP9 genes compared to non-ALS patients [27].
ALS pathophysiology has provided insights to investigate several potential biomarkers. For instance, transglutamine enzymes activity (TGases) in the CSF of ALS patients is elevated at early stages of the disease, while it is decreased at the terminal stages [26]. Furthermore, mutations in the transactive response DNA-binding protein 43 (TDP-43) is considered to be a cause of protein deposits in either familial or sporadic ALS [26, 28]. TDP-43 may be a biomarker of the early stages of the disease since the amount of abnormal TDP-43 expression found at the onset of the disease is elevated. Interestingly, a patent relates to novel proteins, the SOD1-dimer interface antibody (SODI antibody), which may recognize an epitope specific to misfolded or monomeric SOD1 but not the native protein [29]. As more knowledge is obtained about the genetic and environmental factors that determine the risk and pathophysiology of ALS, biomarkers or diagnostic tools would provide better diagnosis, prognosis and treatment of ALS.
3. THE ROLE OF NEUROTROPHIC FACTOR IN THE TREATMENT OF ALS
Studies have shown that the application of neurotrophic factors delays its onset and progression [reviewed by [1]]. Therefore, it is suggested that the loss of neurotrophic factors may be a cause of ALS. There are a number of neurotrophic factors that have been reported to promote neuroprotective and regenerative processes in mouse models of ALS (Tables 2 and 3). Specifically, insulin-like growth factor (IGF-1) and glial-cell-line-derived neurotrophic factor (GDNF) regulate survival and differentiation, maintains neuronal structural integrity, leading to extended lifespan of SOD1 transgenic mice. In addition, vascular endothelial growth factor (VEGF) exhibits neuroprotective action of delaying disease progression and prolonging survival in transgenic rodents. Alternatively, there is existence of derived peptides from activity-dependent neurotrophic factor (ADNF) that have been revealed to ameliorate neurodegeneration in ALS-SOD1 mice models [reviewed by [1]]. The effects of ADNF-9 in ALS models have been investigated and shown that this peptide suppressed SOD-1-mediated cell death. It is noteworthy that a deficiency of neurotrophic factors is critical in ALS pathogenesis; consequently, administration of neurotrophic factors as monotherapy or combination therapy may be considered a potential therapy for the treatment of ALS.
Table 2.
Neuroprotective Effects of Selective Neurotrophic Factors in ALS
| Neurotrophic Factors | Amyotrophic Lateral Sclerosis |
|---|---|
|
| |
| ADNF |
++ |
| ADNP |
− |
| BDNF |
− |
| FGF |
− |
| GDNF |
− |
| IGF |
− |
| NGF |
− |
| VEGF | − |
Abbreviations: ADNF: Activity-dependent neurotrophic factor; ADNP: Activity-dependent neuroprotective protein; BDNF: Brain derived neurotrophic factor; FGF: Fibroblast growth factor; GDNF: glial cell line-derived neurotrophic factor; IGF: Insulin-like growth factor; NGF: Nerve growth factor; VEGF: Vascular endothelial growth factor. (−) Unknown or not determined effect; (+) Neurotrophic factor is efficient; (++) Neurotrophic factor is considerably efficient.
Table 3.
Summary of Findings Investigated Potential Therapies for ALS
| Potential Therapies | Mechanism of Action | References |
|---|---|---|
| Insulin-like growth factor-I (IGF-I) | Role in cell growth, survival, apoptosis and differentiation | [30, 32, 34, 35, 37, 38, 41, 44] |
| Glial-cell-line-derived neurotrophic factor (GDNF) |
Involved in the survival of degenerative motor neurons in SOD1 mice | [45-49, 51-53] |
| Vascular endothelial growth factor (VEGF) |
Suggested to promote the survival of motoneuron in ALS models | [56, 57, 59, 60] |
| Colivelin and ADNF derived peptide | Improved motor performance in SOD-1 mice | [67, 70] |
| Arimoclomol | Protects against neuronal dysfunction and cell death | [72-75] |
| Talampanel | Beneficial effects on the rate of functional decline and the progression of symptoms. |
[76] |
| Beta-lactam antibiotics | May reduce glutamate excitotoxicity | [83, 84] |
| Bromocriptine | May delay deterioration of motor function and prolongs the survival of ALS- SOD1 transgenic mice |
[86] |
| Pramipexole | May prolong the life span of ALS-SOD1 transgenic mice | [89-91] |
| Nimesulide | Delays the onset of motor neuron symptoms in SOD1 mice | [92] |
| Diazoxide | Extend the survival rate in transgenic mouse models for ALS | [93] |
| Stem cell therapy | Potential to be grown to slow the progression of ALS | [96-101] |
3.1. Insulin-like Growth Factor-I (IGF-I)
Insulin-like growth factor is considered a potent neurotrophic factor in ALS due to its role in cell growth, survival, apoptosis and differentiation [30]. A study demonstrated that ALS patients have a decreased expression of IGF-I and three binding proteins in skeletal muscle [31]. The IGF-I system in neurons constitutes three ligands mediated by their specific receptors and IGF-I binding proteins (IGFBP) [32, 33]. Stimulation of IGF-I receptor follows a downstream signaling process, which includes the activation of insulin receptor substrate 1 and 2 (IRS1 and IRS2), followed by the PI3K/Akt and p44/42 MAPK pathways [32, 33]. These signaling mechanisms result in the neuroprotective effects of IGF-I [34]. In addition, there is an association between the signaling actions produced by the IGF-I, cell survival, and the increase in the rate of axonal outgrowth [34, 35]. In vivo studies also demonstrated that the direct application of IGF-I to the motoneurons in G93A-SOD1 mouse models activates the PI3K/Akt, p44/42 MAPK and downregulated IGF-IR and IRS-1 expression. Consequently, beneficial effects on the survival of motor neurons and other cell types such as astrocytes were observed. IGF-I prolonged cell survival by up to 11% [36, 37]. However, clinical trials with ALS patients failed to demonstrate significant effects when subcutaneous administration of IGF-I was initiated. It is suggested that these results may be due to the application of the neurotrophic factor at a late stage of the disease. Furthermore, the route of administration provided a limited delivery of the IGF-I to the target neurons, or an insufficient dose was provided [37].
In vitro and in vivo studies demonstrated that IGF promotes the survival of motor neurons by increasing the levels of phosphorylated Akt and decreasing caspase-9 cleavage [reviewed by [38]]. In models of transgenic mice that express the G93A SOD1 transgene, intramuscular injections of adeno-associated virus (AAV) expressing IGF-1 delay the disease’s progression and prolong the lives of ALS mice [39]. AAV is retrogradely transported through the axon to the cell body, a type of transport that may be responsible for the successful delivery of IGF-I to the target. This study showed that IGF-I treatment has positive outcomes against muscle atrophy. It also demonstrated a reduction of astrogliosis as well as a delayed loss of motor neurons and increased muscle mass [40, 41].
Interestingly, a combination of the delivery of adenoassociated virus expressing the IGF-1 gene and moderate exercise has been found to have profound effects on survival and function in the ALS-SOD1 mice models [40]. The mechanism of action of exercise is that it increases the IGF binding proteins (IGFBPs), which increases the IGF-I bioavailability. Therefore, muscle strength, muscle power and muscle capillary density are increased [40]. This procedure of delivery involves retrograde transport of IGF-1 along the axon of the motor neuron (Fig. 1). Thus, IGF-1 possesses potential clinical applications because it is found to affect spinal cord motoneurons through the muscle fiber [reviewed by [42]]. A patent involves the treatment of ALS by providing intracerebroventricular (ICV) delivery of IGF-I [43]. Another patent indicates the administration of compounds that elevate the concentration of IGF-I as a method to prevent or treat ALS, among other diseases. Two compounds were identified, an IGF-I derivative and anti-IGF-II antibodies, which inhibit the binding of IGF-I and IGFBP, thereby increasing the biological activity of IGF-I [44].
Fig. (1).
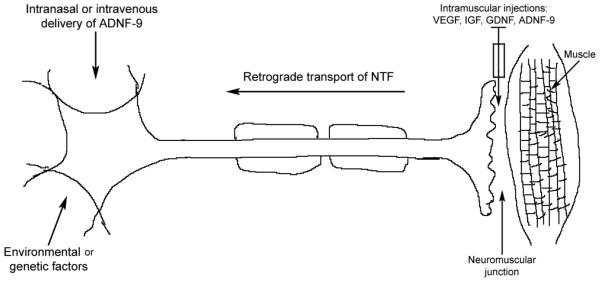
Model of delivery of neurotrophic factors in the prevention of motoneuron degeneration in Amyotrophic Lateral Sclerosis (ALS). ALS is caused by environmental or genetic factors. There are two possibilities for delivery of neurotrophic factors for neuroprotection of cortical, brainstem or spinal cord motoneurons: 1) VEGF, IGF, GDNF and ADNF-9 might be efficient if injected intramuscularly to undergo retrograde transport, 2) ADNF-9 is suggested to be efficient if it is delivered intranasally or intravenously, which primarily might affect the cell bodies of the motoneurons.
3.2. Glial Cell line-derived Neurotrophic Factor
Studies have demonstrated that intramuscular injections of adeno-associated virus that express GDNF (AAV-GDNF) prevent the degeneration of motor neurons and extend survival [45-47]. Studies have assessed the effects of AAV-GDNF on male transgenic mice with the G93A human SOD1 mutation. Interestingly, it was demonstrated that the delivery of GDNF inhibited motor neuron atrophy, maintained axonal projections, prolonged lifespan and slow the progression of disease in ALS transgenic mice [47, 48]. These studies also indicated that the mechanism of action of exogenous GDNF was through the prevention of apoptosis by the preservation of the Akt signaling pathway [48, 49]. GDNF is retrogradely transported from muscle-injected sites to the motoneurons’ projected axons (Fig. 1) [47, 50]. This type of delivery may have preventive role against degeneration of motoneurons and their targets, since it may mimic the transport of endogenous neurotrophic factors. However, the mechanism leading to neuroprotection of the motor neurons through retrograde transport is still unknown.
The efficiency of neuroprotective effects that involve retrograde transport depends on the types of neurotrophic factors. For instance, GDNF has been found less efficient than IGF-1 in the protection of motoneurons in SOD1 mice [41, 46, 47]. This suggests that IGF is more potent in neuroprotection than GDNF. Furthermore, the use of the C-terminal fragment of tetanus toxin (TTC) as a carrier has been shown to promote signaling pathways, which inhibit apoptosis in Neuro2A cells. Consequently, the delivery of TTC in SOD1ALS mouse models has been shown to produce an increased life span and improved quality of life [51]. Similar to TTC, the ex vivo gene therapy approach through the application of human mesenchymal stem cells engineered to secrete GDNF has been shown to improve survival and function of motor neurons in mouse models [52].
3.3. Vascular Endothelial Growth Factor
VEGF has several biological activities that differ from other neurotrophic factors. Similarly to IGF-1, VEGF stimulates neurogenesis, possesses a potential antero- and retrograde transport, and stimulates neuronal perfusion [53-55]. These effects have been suggested to promote the survival of motoneurons in ALS models [reviewed by [56]]. It is noteworthy that mice lacking hypoxia-response element at the VEGF promoter showed decreased level of VEGF and degeneration of motoneurons [57]. In humans, VEGF levels have been revealed to be low at early stages of ALS [58]. Thus, the cause of motoneuron degeneration may result from both the loss of VEGF in the brain and also in the vascular system of the spinal cord.
There are several neuropathological features in VEGF-transgenic mice, including the loss of cholineacetyltransferase in motoneurons, degeneration of motoneurons, and loss of myelinated axons of motoneurons, similar to those found in SOD1 mouse models of ALS [reviewed by [56]]. The intramuscular injections of lentivector that express VEGF significantly improved the pathological state in ALS mice [59]. Thus, the retrograde transport of VEGF plays an important role in preventing or slowing the progression of the disease. Furthermore, it has been shown that exercise has therapeutic benefits in the ALS mouse model [40]. This coincides perfectly with the fact that exercise increases the production of VEGF [60, 61]. We suggest here that the combination of both exercise and VEGF is considered a procedure for the improvement or treatment of ALS.
3.4. Colivelin and ADNF Derived Peptide in ALS
New directions have been taken toward the synthetic of hybrid peptide named colivelin, which constituted of ADNF-9 and AGA-(C8R)HNG17 (PAGASRLLLLTGEIDLP), a humanin derivative [62]. ADNF-9 is a nine-amino-acid peptide (ADNF-9 or SAL, SALLRSIPA) that possesses a protective effect against cell death associated with oxidative stress [63, 64]. Humanin was identified as an endogenous neuroprotective peptide, and it was suggested to protect against AD (Alzheimer’s disease)-related toxicity and cytotoxicity [65, 66]. Colivelin completely suppressed neuronal cell death against β-amyloid peptide in vitro. In vivo experiments demonstrated that colivelin has neuroprotective effects against the insult of β-amyloid [62].
The protective effect of derived peptide (ADNF-9) from ADNF has been investigated in the SOD-1 transgenic mice. ICV administration of ADNF-9 improved motor performance in SOD-1 mice [67]. However, ADNF-9 was not efficient in prolonging the survival of SOD-1 mice. It was suggested from this study that ADNF-9 preserved the functionality of neurons but was not sufficient to prolong neuronal survival. It is possible that ADNF-9 might be metabolized rapidly, as has been shown in vitro [63], which suggests its degradation in the in vivo paradigm. The synthetic hybrid peptide colivelin improved motor performance and prolonged survival in the SOD-1 mice [68]. In contrast to neurotrophic factor proteins, colivelin can be administered intravenously (Fig. 1). Thus, ADNF-9-humanin may have potential therapeutic effects for the treatment of ALS.
Alternatively, angiogenin (ANG) is a polypeptide that induces cell proliferation and enhances motor muscular function, neurogenesis, and survival of motor neurons, among other characteristics [69]. A patent indicates the potential role of angiogenin for treating ALS [70]. It indicates that the spinal cord of ALS patients who lack ANG mutations have a decreased ANG expression. Similarly, ALS-SOD1 mouse models have lower ANG levels compared to wild type mice. In addition, muscle strength, motor coordination, muscle function and life survival of ANG-treated mice were improved. Intraperitoneal injection of ANG was shown to be an efficient route of administration; however, other forms of delivery might be assessed in order to increase effectiveness and minimize side effects.
4. POTENTIAL DRUGS, IMMUNOTHERAPY AND STEM CELLS THERAPY FOR THE TREATMENT OF ALS
4.1. Drug Therapy
ALS is characterized by abnormal protein aggregation. Hence, there is a therapeutic approach that may target heat shock proteins (HSPs), which are cellular defense mechanisms with neuroprotective effects against these aggregates [71, 72]. Heat shock transcription factor 1 (HSF1) controls the induction of several HSPs. Therefore, the effect of heat shock protein inducers for ALS has been investigated. Arimoclomol, a hydroxylamine derivative, is a drug that acts as a heat shock response inducer in motor neurons (Table 3). It prolongs the activation of HSF1; as a result, it protects against neuronal dysfunction and cell death. In terms of arimoclomol’s effects on ALS, studies demonstrated that this drug therapy improved muscle function in SOD1 mouse models at both early and late stages of the disease [73, 74]. Therefore, arimoclomol might be a promising therapy at early stages. Another study showed that daily treatment with arimoclomol improved muscle function and increased the lifespan of ALS mice at the late stage of the disease [72]. Furthermore, clinical trials showed that arimoclomol is safe and tolerable for ALS patients [75]; however, further studies are warranted to determine its efficacy.
Talampanel is an orally active non-competitive antagonist of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors [76]. These receptors mediate glutamate-induced excitotoxicity to motor neurons. AMPA receptors exert a synaptic transmission followed by the neurotransmitter re-uptake [77]. Overstimulation leads to an excessive influx of calcium, causing toxic effects and damage within the cell. There is evidence that excess extracellular glutamate, especially via AMPA receptors, is a key factor in ALS neuropathogenesis [78]. In vivo studies of SOD1 mice models support the beneficial effects of AMPA antagonists such as talampanel [79]. However, it was demonstrated that the neuroprotective effects of talampanel were only present when it was applied during the early stage of the disease. Likewise, a phase II study was conducted in which 60 patients with ALS participated. The goal of the study was to determine its efficacy, safety and tolerability. Although the efficacy measures did not show significant differences, ALS Functional Rating Scale, muscle strength, and timed hand movements declined at a slower rate in talampanel-treated patients [76]. In addition, a patent disclosed evidence of the efficacy, tolerability and safety of 150 mg daily doses of talampanel in patients afflicted with ALS [80]. It showed that talampanel has beneficial effects on the rate of functional decline and the progression of symptoms.
Similar to talampanel, beta-lactam antibiotics are a potential drug therapy for the treatment of ALS. The reuptake of synaptic glutamate requires normal levels of the major glial glutamate transporter 1 (GLT1) [81]. There is evidence that patients with ALS have reduced GLT1 levels, which might lead to decreased elimination of glutamate from neuromuscular synapses [82]. Interestingly, it has been indicated that many beta-lactam antibiotics, such as penicillin and its derivatives and cephalosporin, upregulated the levels of GLT1 [83]. In a screening study of 1040 medications approved by the FDA, beta-lactam antibiotics had a relevant role in the pathogenesis of ALS. Rothstein and colleagues reported that ceftriaxone’s neuroprotective properties involve reducing glutamate excitotoxicity via the stimulation of the promoter sequence for GLT1 in SOD1 toxicity [83]. This effect maintained muscle stability and weight for a longer period of time; and moderately increased life span. Thus, phase I-II clinical trials were conducted in order to determine pharmacokinetics, safety, tolerability and efficacy of ceftriaxone in this neurodegenerative disease, ALS [84]. High and low doses of ceftriaxone achieved the target cerebrospinal fluid concentrations. Besides that, ceftriaxone met the tolerability criteria at dosages of up to 4 grams/day.
Likewise, another etiology of ALS includes oxidative stress-cell death of motor neurons [15, 85]. Thus, a patent provides the use of an FDA approved medication, bromocriptine mesylate, to treat ALS [86]. Bromocriptine is a free-radical scavenger that may inhibit oxidative stress-induced cell death. This patent indicates that this medication delays deterioration of motor function and prolongs the survival of ALS-SOD1 transgenic mice. Similarly, another patent relates to the use of a 1,3-diphenylurea derivative or multikinase inhibitor to treat ALS by reducing the amount of SOD1 gene expression. Although the mechanisms of action of ceftriaxone and bromocriptine are different for the treatment of ALS, both drugs show similar core structures. These drugs are heterocyclic and contain the exocyclic amide groups. In addition, bromocriptine has a gamma-lactam ring and a saturated six-membered ring while ceftriaxone has a beta-lactam ring with an unsaturated six-membered ring (Fig. 2). These similarities suggest that the lactam ring might be a functional site. It is warranted to determine whether the functional site is associated with lactam or exocyclic amide group.
Fig. (2).
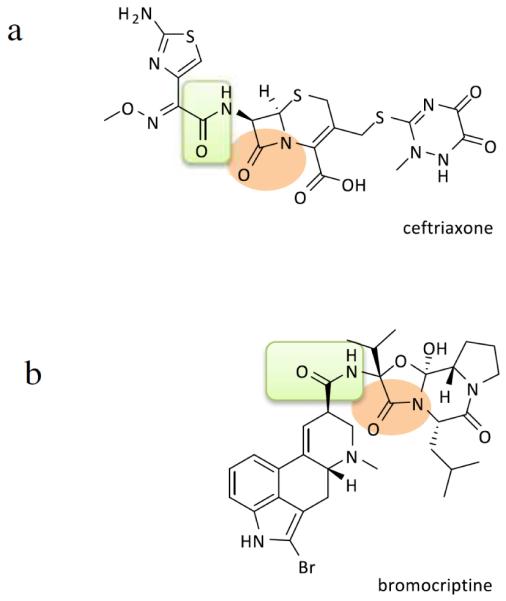
Diagrams show chemical structures of Ceftriaxone (a) and Bromocriptine (b). Both compounds are polycyclic and have lactam ring as shown in circle. The two compounds also show exocyclic amid groups as shown in rectangle.
The occurrence of cellular oxidative stress due to mitochondrial dysfunction is another proposed mechanism responsible for motor neuron degeneration in ALS [14, 81]. The mitochondrial alteration leads to abnormalities in the energy production pathways, resulting in generation of oxygen free radicals among others reactive oxygen species (ROS). In vitro and in vivo studies of both forms of ALS have been shown to generate ROS, as detected by increased levels of markers of oxidative stress, 2,3 DHBA [87, 88]. Pramipexole is a dopamine agonist, which has a free-radical scavenging action. Therefore, it may have a role ameliorating the oxidative response [88]. Dexpramipexole, the optical enantiomer of pramipexole, has less affinity to dopamine receptors; therefore, it can be given at much higher doses. It has been demonstrated that this drug therapy prolongs the life span of ALS-SOD1 transgenic mice [89, 90]. When evaluated in clinical trials, dexpramipexole met the safety and tolerability criteria [91]. Pramipexole and its enantiomer dexpramipexole have common structure, moiety 2-aminothiazole ring, with riluzole (Fig. 3). However, both pramipexole and dexpramipexole play a role in mitochondrial dysfunction; thus, affecting the oxidative stress, while riluzole is a glutamate antagonist involved in activation of voltage-dependent sodium channels. Thus, it is unclear whether this common structure is a key player in the functionality of the compounds. Further investigation should be established to determine the structural relationship of these potential compounds.
Fig. (3).
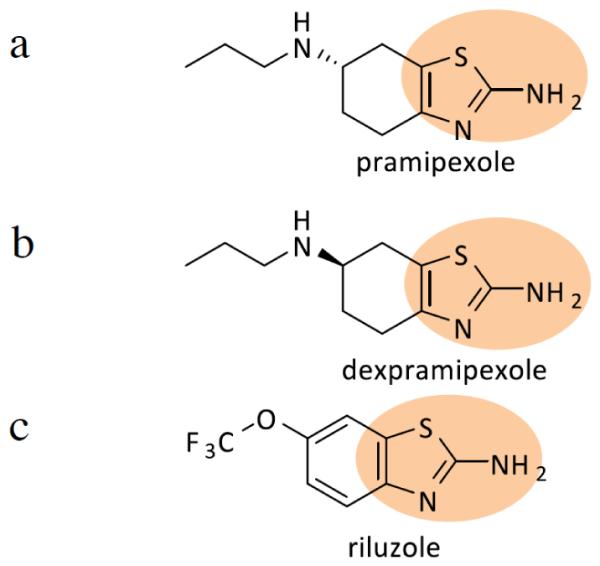
Diagrams show chemical structures of Pramipexole (a), Dexpramipexole (b), and Riluzole (c). (a) and b) compounds are structurally similar, except both of these compounds are enantiomers. Compound c) is structurally similar to compounds (a) and (b) with a common structural moiety a such as 2-aminothiazole ring as shown in circle. However, the side chain is different and the six-membrane ring is aromatic.
Another possible target includes cyclooxygenase enzymes. A patent suggests a method for delaying the onset of motor neuron symptoms in SOD1 mice models by administering nimesulide, which is a non-selective cyclooxygenase inhibiting non-steroidal anti-inflammatory drug (NSAIDs) [92]. This patent indicates that nimesulide may be provided as prophylaxis if the patient is at risk of developing ALS or if environmental exposure exists. It may also be administered at other stages during progression of the disease. The methods consist of daily oral administration of nimesulide as a single dose or as divided doses. In terms of KATP channels, proteins involved in one of the disease pathways of ALS, there is a patent that indicates that diazoxide acts as a KATP channel opener [93]. This patent indicates that low doses of diazoxide improve symptoms of ALS and extend the survival rate in transgenic mouse models for ALS. In regards to their chemical structures, both compounds feature bicyclic ring systems. Nimesulide contains a phenoxyphenyl group, while diazoxide features a benzothiadiazine ring. It is noteworthy that they have sulfonamides sides, which may suggest their efficacy in ALS (Fig. 4).
Fig. (4).
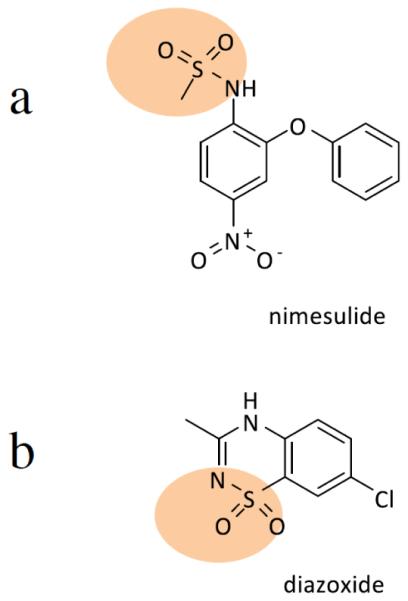
Diagrams show chemical structures of Nimesulide (a) and Diazoxide (b). Both compounds have sulfonamides sides. In addition, both compounds have bicyclic ring systems. Rings are fused in (b) but in (a).
Another patent indicates the use of the active ingredient, 3-methyl-1-phenyl-2-pirazoline-5-on, which may be used for the treatment of degeneration of motor neurons in neurodegenerative disease, including ALS [94]. It acts as a free-radical scavenger by suppressing lipid hyperoxidation; consequently, muscle spasticity and weakness were diminished, while the biceps muscle mass was increased in the motor neuron disease mouse model. Regarding the invention mentioned above, another patent has been disclosed for methods examining the optimum administration period of a pyrazolone derivative to treat ALS or symptoms caused by ALS [95]. The pyrazolone derivative used in this patent was 3-methyl-1-phenyl-2-pirazoline-5-on, which was shown to be effective in suppressing the decrease of % Forced Vital Capacity (FVC) and the increase of PaCO2 in ALS patients; hence, respiratory function was improved.
4.2. Stem Cell Therapy
Stem cell therapy is promising in the treatment of ALS since stem cells have the potential to be grown to slow the progression of motor neuron disease or even replace motor neurons [28, 96]. Interestingly, mouse embryonic stem cells are converted into specific neuronal subtypes such as motor neurons. These stem cells generate spinal motor neurons, extend axons and receive and make synapses with the muscle [97]. Stem cells are derived from multiple sources. Embryonic stem cells give rise to pluripotent cells, which generate all differentiated cell type tissues [98]. Embryonic neural tissue can be converted into neural stem cells (NSC), which would differentiate into astrocytes. A study showed that a tissue graft from a human fetal spinal neural stem cells (hNSCs) into the spinal cords of SOD1 mutant mice could be beneficial [99]. It was shown to provide a neuroprotective effect by forming synapses and increasing the number of motor neurons; however, survival rate was not affected. Alternatively, another study demonstrated that NSC extended the survival rate of ALS-SOD1 mice [100, 101]. Moreover, multipotent neural progenitor cells (NPC), which are derived from neural tissue, have been demonstrated to have the property to secrete GDNF and BDNF [102].
Mesenchymal stem cells (MSCs) are derived from adult stem cells. These cells are bone marrow (BM) cells that are differentiated into mesodermal cell derivatives [103]. It has been indicated that MSCs lead to the release of trophic factors, anti-inflammatory cytokines, and immunomodulatory chemokines to delay disease progression. A clinical trial was performed, in which the I.V. administration of MSCs in ALS patients was shown to be safe and induced immediate immunomodulatory activity [104]. Studies reported that the application of human skeletal muscle-derived stem cells (SkmSCs) with MSC-like characteristics in the Wobbler mouse (Wr), which shares the ALS disease process and symptoms, delayed ALS symptoms and improved motor activity [105]. In addition to BM or Skm-derived stem cells, adipose-derived MSC (ASC) has also been utilized. ASC was found to migrate into damaged tissues and exert immunomodulatory activity by inhibiting both in vitro and in vivo T cell proliferation [103, 106]. Studies have also shown that the systemic injection of MSC after disease onset on mSOD1 mice led to a high number of motor neurons and, consequently, a delay of symptoms and better motor performance [107]. In regard to stem cell differentiation, it was reported that CD133+ stem cells from ALS patients differentiate into neuron-like cells [108]. A patent provides methods for inducing cell differentiation from placental stem cells to treat ALS [109]. The patent indicates that this therapy promoted differentiation to adipocyte, chondrocyte and osteocyte lineages.
Induced pluripotent stem (iPS) cells are somatic cells derived; that is, they are produced by converting adult fibroblasts into pluripotent stem cells by being changed genetically to express critical ES genes and factors [28, 50]. The cells produced by iPS could be used to better explore the pathogenesis, disease modeling and to test potential compounds. Furthermore, a study reported that human iPSC-NPs induced survival and differentiation to mature neurons in the spinal cord in the ALS-SOD1 mouse model [110]. Another study also demonstrated that iPS cells could be derived from fibroblast cells of a senior patient diagnosed with ALS and be differentiated into motor neurons [111].
4.3. Immunotherapy
One of the factors that contribute to the progression of ALS includes oxidative stress or accumulation of free radicals as a consequence of the mutation of the SOD1 enzyme. The acquisition of a cytotoxic activity is associated with protein misfolding/aggregation [112]. The identification of this pathogenic process has led to a focus on immunotherapy in ALS. Specifically, mouse monoclonal antibodies against misfolded forms of mutant SOD1 (mSOD1) were produced to determine its effectiveness as a passive immunization for ALS [113]. It was shown that infusing D3H5 antibody through the ICV route for 42 days reduced levels of toxic SOD1 species in the spinal cord. It was also demonstrated that it maintained weight for a longer period of time, and extended the lifespan of ALS-SOD1 mice [113]. Another study used the recombinant WT-apo SOD1, which has similar characteristics to those of SOD1 mutant molecules. The application of WT-apo SOD1 vaccine induced a protective immunity, which extended the lifespan and delayed the onset of paralysis in transgenic mice models [114].
5. CONCLUSION AND CLOSING THOUGHTS
Several studies have demonstrated a small to moderate lifespan increase and motor integrity improvement in ALS-SOD1 mouse models with the uses of neurotrophic factors, certain FDA-approved drugs, stem cell therapy, and immunotherapy technologies. The route of administration plays a key role in obtaining a clinical effect. Specifically, neurotrophic factors IGF-1, GDNF and VEGF were retrogradely transported into motor neurons through viral vector injections, while colivelin might be injected intravenously since it has the ability to cross the blood-brain barrier. These trophic factors were found to provide one of the best results in regard to progression of the disease and survival of ALS mouse models. Additionally, several existing drugs such as talampanel, beta-lactam antibiotics, pramipexole, dexpramipexole and arimoclomol are currently considered as potential therapeutic compounds in the treatment of ALS. Alternatively, studies are testing the uses of stem cell therapy and immunotherapy for the potential treatment of ALS. However, several promising results obtained in ALS animal models have shown not to be effective in humans. Crossing the human blood-brain barrier is crucial for a drug to reach its target; thus, activating the appropriate site of action to produce the expected drug effects. Moreover, ALS biomarkers would be crucial in identifying an effective therapy that impacts disease progression of ALS patients. It is noteworthy that a combination of drugs or methods might provide effective therapy for patients with ALS.
ACKNOWLEDGEMENTS
This review article was supported by Award Number R01AA019458 (Y.S.) from the National Institutes on Alcohol Abuse and Alcoholism. We would like to thank Mrs. Charisse Montgomery for editing this review article.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Ciesler J, Sari Y. Neurotrophic Peptides: Potential Drugs for Treatment of Amyotrophic Lateral Sclerosis and Alzheimer's disease. Open journal of neuroscience. 2013:3. doi: 10.13055/ojns_3_1_2.130408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- [3].Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, Sapp P, Hung WY, Bebout J, McKenna-Yasek D, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 1991;324(20):1381–1384. doi: 10.1056/NEJM199105163242001. [DOI] [PubMed] [Google Scholar]
- [4].Siddique T, Pericak-Vance MA, Brooks BR, Roos RP, Hung WY, Antel JP, Munsat TL, Phillips K, Warner K, Speer M, et al. Linkage analysis in familial amyotrophic lateral sclerosis. Neurology. 1989;39(7):919–925. doi: 10.1212/wnl.39.7.919. [DOI] [PubMed] [Google Scholar]
- [5].Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, Griffin JW, Cornblath DR. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet. 1998;62(3):633–640. doi: 10.1086/301769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- [7].Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurro MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Consortium I, Galassi G, Scholz SW, Taylor JP, Restagno G, Chio A, Traynor BJ. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Berger MM, Kopp N, Vital C, Redl B, Aymard M, Lina B. Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS. Neurology. 2000;54(1):20–25. doi: 10.1212/wnl.54.1.20. [DOI] [PubMed] [Google Scholar]
- [11].Ceroni M, Malaspina A, Poloni TE, Alimonti D, Rognoni F, Habgood J, Imbesi F, Antonelli P, Alfonsi E, Curti D, deBelleroche J. Clustering of ALS patients in central Italy due to the occurrence of the L84F SOD1 gene mutation. Neurology. 1999;53(5):1064–1071. doi: 10.1212/wnl.53.5.1064. [DOI] [PubMed] [Google Scholar]
- [12].Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Daube JR, Nance M, Fan C, Kaplan J, Hung WY, McKenna-Yasek D, Haines JL, Pericak-Vance MA, Horvitz HR, Brown RH., Jr. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. Jama. 2000;284(13):1664–1669. doi: 10.1001/jama.284.13.1664. [DOI] [PubMed] [Google Scholar]
- [13].Majoor-Krakauer D, Ottman R, Johnson WG, Rowland LP. Familial aggregation of amyotrophic lateral sclerosis, dementia, and Parkinson's disease: evidence of shared genetic susceptibility. Neurology. 1994;44(10):1872–1877. doi: 10.1212/wnl.44.10.1872. [DOI] [PubMed] [Google Scholar]
- [14].Jackson M, Llado J, Rothstein JD. Therapeutic developments in the treatment of amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2002;11(10):1343–1364. doi: 10.1517/13543784.11.10.1343. [DOI] [PubMed] [Google Scholar]
- [15].McGeer EG, McGeer PL. Pharmacologic approaches to the treatment of amyotrophic lateral sclerosis. BioDrugs. 2005;19(1):31–37. doi: 10.2165/00063030-200519010-00004. [DOI] [PubMed] [Google Scholar]
- [16].Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38(1):73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- [17].Cleveland DW. From Charcot to SOD1: mechanisms of selective motor neuron death in ALS. Neuron. 1999;24(3):515–520. doi: 10.1016/s0896-6273(00)81108-3. [DOI] [PubMed] [Google Scholar]
- [18].Grieshammer U, Lewandoski M, Prevette D, Oppenheim RW, Martin GR. Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Dev Biol. 1998;197(2):234–247. doi: 10.1006/dbio.1997.8859. [DOI] [PubMed] [Google Scholar]
- [19].Riedel WJ, Jolles J. Cognition enhancers in age-related cognitive decline. Drugs Aging. 1996;8(4):245–274. doi: 10.2165/00002512-199608040-00003. [DOI] [PubMed] [Google Scholar]
- [20].Henderson CE, Yamamoto Y, Livet J, Arce V, Garces A, deLapeyriere O. Role of neurotrophic factors in motoneuron development. J Physiol Paris. 1998;92(3-4):279–281. doi: 10.1016/s0928-4257(98)80033-8. [DOI] [PubMed] [Google Scholar]
- [21].Sendtner M, Pei G, Beck M, Schweizer U, Wiese S. Developmental motoneuron cell death and neurotrophic factors. Cell Tissue Res. 2000;301(1):71–84. doi: 10.1007/s004410000217. [DOI] [PubMed] [Google Scholar]
- [22].Turner MR, Bowser R, Bruijn L, Dupuis L, Ludolph A, McGrath M, Manfredi G, Maragakis N, Miller RG, Pullman SL, Rutkove SB, Shaw PJ, Shefner PJ, Fischbeck KH. Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013:14. doi: 10.3109/21678421.2013.778554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Q, Xie F, Siedlak SL, Nunomura A, Honda K, Moreira PI, Zhua X, Smith MA, Perry G. Neurofilament proteins in neurodegenerative diseases. Cell Mol Life Sci. 2004;61(24):3057–3075. doi: 10.1007/s00018-004-4268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lu C, Petzold A, Kalmar B, Dick J, Malaspina A, Greensmith L. Plasma neurofilament heavy chain levels correlate to markers of late stage disease progression and treatment response in SOD1(G93A) mice that model ALS. PLoS One. 2012;7(7) doi: 10.1371/journal.pone.0040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ganesalingam J, An J, Bowser R, Andersen PM, Shaw CE. pNfH is a promising biomarker for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(2):146–149. doi: 10.3109/21678421.2012.729596. [DOI] [PubMed] [Google Scholar]
- [26].Tarasiuk J, Kułlakowska A, Drozdowski W, Kornhuber J, Lewczuk P. CSF markers in amyotrophic lateral sclerosis. J Neural Transm Suppl. 2012;119(7):747–757. doi: 10.1007/s00702-012-0806-y. [DOI] [PubMed] [Google Scholar]
- [27].Nachmany H, Wald S, Abekasis M, Bulvik S, Weil M. Two potential biomarkers identified in mesenchymal stem cells and leukocytes of patients with sporadic amyotrophic lateral sclerosis. Dis Markers. 2012;32(4):211–220. doi: 10.3233/DMA-2011-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Traub R, Mitsumoto H, Rowland LP. Research Advances in Amyotrophic Lateral Sclerosis, 2009 to 2010. Curr Neurol Neurosci Rep. 2011;11(1):67–77. doi: 10.1007/s11910-010-0160-0. [DOI] [PubMed] [Google Scholar]
- [29].Chakrabartty A, Cashman N, Rakhit R. Methods and Compositions for Detecting Amyotrophic Lateral Sclerosis. 2010 US7794692. [Google Scholar]
- [30].Dupont J, Dunn SE, Barrett JC, LeRoith D. Microarray analysis and identification of novel molescules involved in insulin-like growth factor-1 receptor signaling and gene expression. Recent Prog Horm Res. 2003;58:325–342. doi: 10.1210/rp.58.1.325. [DOI] [PubMed] [Google Scholar]
- [31].Lunetta C, Serafini M, Prelle A, Magni P, Dozio E, Ruscica M, Sassone J, Colciago C, Moggio M, Corbo C, Silani V. Impaired Expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve. 2012;45(2):200–208. doi: 10.1002/mus.22288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Duan C. Specifying the cellular responses to IGF signals: roles of IGF-binding proteins. J Endocrinol. 2002;175(1):41–54. doi: 10.1677/joe.0.1750041. [DOI] [PubMed] [Google Scholar]
- [33].De Meyts P, Wallach B, Christoffersen CT, Urso B, Gronskov K, Latus LJ, Yakushiji F, Ilondo MM, Shymko RM. The insulin-like growth factor-I receptor. Structure, ligand-binding mechanism and signal transduction. Horm Res. 1994;42(4-5):152–169. doi: 10.1159/000184188. [DOI] [PubMed] [Google Scholar]
- [34].Sakowski SA, Schuyler AD, Feldman EL. Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(2):63–73. doi: 10.1080/17482960802160370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9(11):1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- [36].Narai H, Nagano I, Ilieva H, Shiote M, Nagata T, Hayashi T, Shoji M, Abe K. Prevention of spinal motor neuron death by insulin-like growth factor-1 associating with the signal transduction systems in SODG93A transgenic mice. J Neurosci Res. 2005;82(4):452–457. doi: 10.1002/jnr.20668. [DOI] [PubMed] [Google Scholar]
- [37].Nagano I, Ilieva H, Shiote M, Murukami T, Yokoyama M, Shoji M, Abe K. Therapeutic benefit of intrathecal injection of insulin-like growth factor-1 in a mouse model of Amyotrophic Lateral Sclerosis. J Neuro Sci. 2005;235(1-2):61–68. doi: 10.1016/j.jns.2005.04.011. [DOI] [PubMed] [Google Scholar]
- [38].Wilczak N, de Keyser J. Insulin-like growth factor system in amyotrophic lateral sclerosis. Endocr Dev. 2005;9:160–169. doi: 10.1159/000085764. [DOI] [PubMed] [Google Scholar]
- [39].Vincent AM, Feldman EL, Song DK, Jung V, Schild A, Zhang W, Imperiale MJ, Boulis NM. Adeno-associated viral-mediated insulin-like growth factor delivery protects motor neurons in vitro. Neuromolecular medicine. 2004;6(2-3):79–85. doi: 10.1385/NMM:6:2-3:079. [DOI] [PubMed] [Google Scholar]
- [40].Kaspar BK, Frost LM, Christian L, Umapathi P, Gage FH. Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann Neurol. 2005;57(5):649–655. doi: 10.1002/ana.20451. [DOI] [PubMed] [Google Scholar]
- [41].Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- [42].Festoff BW. The preclinical rationale for the use of insulin-like growth factor-I in amyotrophic lateral sclerosis. Drugs Today (Barc) 1998;34(1):65–77. doi: 10.1358/dot.1998.34.1.485201. [DOI] [PubMed] [Google Scholar]
- [43].Dodge J, Scheule R. Intraventricular protein delivery for amyotrophic lateral sclerosis. 2007 WO084743 a3. [Google Scholar]
- [44].Sakano K, Higashihashi N, Hashimoto R. Composition of an endogenous insulin-like growth II factor derivative. 2002 US6428781. [Google Scholar]
- [45].Acsadi G, Anguelov RA, Yang H, Toth G, Thomas R, Jani A, Wang Y, Ianakova E, Mohammad S, Lewis RA, Shy ME. Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum Gene Ther. 2002;13(9):1047–1059. doi: 10.1089/104303402753812458. [DOI] [PubMed] [Google Scholar]
- [46].Lu YY, Wang LJ, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Matsushita T, Hanazono Y, Kume A, Nagatsu T, Ozawa K, Nakano I. Intramuscular injection of AAV-GDNF results in sustained expression of transgenic GDNF, and its delivery to spinal motoneurons by retrograde transport. Neurosci Res. 2003;45(1):33–40. doi: 10.1016/s0168-0102(02)00195-5. [DOI] [PubMed] [Google Scholar]
- [47].Wang LJ, Lu YY, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Matsushita T, Hanazono Y, Kume A, Nagatsu T, Ozawa K, Nakano I. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci. 2002;22(16):6920–6928. doi: 10.1523/JNEUROSCI.22-16-06920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Manabe Y, Nagano I, Gazi MSA, Murakami T, Shiote M, Shoji M, Kitagawa H, Setoguchi Y, Abe K. Adenovirus-mediated gene transfer of glial cell line-derived neurotrophic factor prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Apoptosis. 2002;7(4) doi: 10.1023/a:1016123413038. [DOI] [PubMed] [Google Scholar]
- [49].Manabe Y, Nagano I, Gazi MSA, Murakami T, Shiote M, Shoji M, Kitagawa H, Abe K. Glial cell line-derived neurotrophic factor protein prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Neurol Res. 2003;25(2):195–200. doi: 10.1179/016164103101201193. [DOI] [PubMed] [Google Scholar]
- [50].O'Connor DM, Boulis NM. Cellular and molecular approaches to motor neuron therapy in amyotrophic lateral sclerosis and spinal muscular atrophy. Neurosci Lett. 2012;527(2):78–84. doi: 10.1016/j.neulet.2012.04.079. [DOI] [PubMed] [Google Scholar]
- [51].Ciriza J, Moreno-Igoa M, Calvo AC, Yaque G, Palacio J, Miana-Mena FJ, Munoz MJ, Zaraqoza P, Brulet P, Osta R. A genetic fusion GDNF-C fragment of tetanus toxin prolongs survival in a symptomatic mouse ALS model. Restor Neurol Neurosci. 2008;26(6):459–465. [PubMed] [Google Scholar]
- [52].Suzuki M, McHugh J, Tork C, Shelley B, Hayes A, Bellantuono I, Aebischer P, Svendsen CN. Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol Ther. 2008;16(12):2002–2010. doi: 10.1038/mt.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rosenstein JM, Mani N, Khaibullina A, Krum JM. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci. 2003;23(35):11036–11044. doi: 10.1523/JNEUROSCI.23-35-11036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26(9):943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- [55].Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8(1):85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- [56].Lambrechts D, Carmeliet P. VEGF at the neurovascular interface: Therapeutic implications for motor neuron disease. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbadis.2006.04.005. [DOI] [PubMed] [Google Scholar]
- [57].Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D, Carmeliet P. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28(2):131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- [58].Devos D, Moreau C, Lassalle P, Perez T, De Seze J, Brunaud-Danel V, Destee A, Tonnel AB, Just N. Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 2004;62(11):2127–2129. doi: 10.1212/01.wnl.0000129913.44351.a3. [DOI] [PubMed] [Google Scholar]
- [59].Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429(6990):413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- [60].Fabel K, Tam B, Kaufer D, Baiker A, Simmons N, Kuo CJ, Palmer TD. VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur J Neurosci. 2003;18(10):2803–2812. doi: 10.1111/j.1460-9568.2003.03041.x. [DOI] [PubMed] [Google Scholar]
- [61].Powrozek TA, Sari Y, Singh RP, Zhou FC. Neurotransmitters and substances of abuse: effects on adult neurogenesis. Curr Neurovasc Res. 2004;1(3):251–260. doi: 10.2174/1567202043362225. [DOI] [PubMed] [Google Scholar]
- [62].Chiba T, Yamada M, Hashimoto Y, Sato M, Sasabe J, Kita Y, Terashita K, Aiso S, Nishimoto I, Matsuoka M. Development of a femtomolar-acting humanin derivative named colivelin by attaching activity-dependent neurotrophic factor to its N terminus: characterization of colivelin-mediated neuroprotection against Alzheimer's disease-relevant insults in vitro and in vivo. J Neurosci. 2005;25(44):10252–10261. doi: 10.1523/JNEUROSCI.3348-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brenneman DE, Hauser J, Neale E, Rubinraut S, Fridkin M, Davidson A, Gozes I. Activity-dependent neurotrophic factor: structure-activity relationships of femtomolar-acting peptides. J Pharmacol Exp Ther. 1998;285(2):619–627. [PubMed] [Google Scholar]
- [64].Glazner GW, Gressens P, Lee SJ, Gibney G, Gozes I, Gozes Y, Brenneman DE, Hill JM. Activity-dependent neurotrophic factor: a potent regulator of embryonic growth and development. Anat Embryol (Berl) 1999;200(1):65–71. doi: 10.1007/s004290050260. [DOI] [PubMed] [Google Scholar]
- [65].Chiba T, Nishimoto I, Aiso S, Matsuoka M. Neuroprotection neuroprotective peptide Colivelin. Molecular neurobiology. 2007;35(1):55–84. [PubMed] [Google Scholar]
- [66].Luciano F, Zhai D, Zhu X, Bailly-Maitre B, Ricci JE, Satterthwait AC, Reed JC. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. The Journal of biological chemistry. 2005;280(16):15825–15835. doi: 10.1074/jbc.M413062200. [DOI] [PubMed] [Google Scholar]
- [67].Chiba T, Hashimoto Y, Tajima H, Yamada M, Kato R, Niikura T, Terashita K, Schulman H, Aiso S, Kita Y, Matsuoka M, Nishimoto I. Neuroprotective effect of activity-dependent neurotrophic factor against toxicity from familial amyotrophic lateral sclerosis-linked mutant SOD1 in vitro and in vivo. J Neurosci Res. 2004;78(4):542–552. doi: 10.1002/jnr.20305. [DOI] [PubMed] [Google Scholar]
- [68].Chiba T, Yamada M, Sasabe J, Terashita K, Aiso S, Matsuoka M, Nishimoto I. Colivelin prolongs survival of an ALS model mouse. Biochem Biophys Res Commun. 2006;343(3):793–798. doi: 10.1016/j.bbrc.2006.02.184. [DOI] [PubMed] [Google Scholar]
- [69].Padhi AK, Vasaikar SV, Jayaram B, Gomes J. Fast prediction of deleterious angiogenin mutations causing amyotrophic lateral sclerosis. FEBS Lett. 2013;587(12):1762–1766. doi: 10.1016/j.febslet.2013.04.022. [DOI] [PubMed] [Google Scholar]
- [70].Hu G, Kishikawa H. Angiogenin and Amyotrophic Lateral Sclerosis. 2009 WO146178. [Google Scholar]
- [71].Brown IR. Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- [72].Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10(4):402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- [73].Kalmar B, Novoselov S, Gray A, Cheetham ME, Margulis B, Greensmith L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem. 2008;107(2):339–350. doi: 10.1111/j.1471-4159.2008.05595.x. [DOI] [PubMed] [Google Scholar]
- [74].Gordon PH. Amyotrophic lateral sclerosis: pathophysiology, diagnosis and management. CNS Drugs. 2011;25(11):1–15. doi: 10.2165/11586000-000000000-00000. [DOI] [PubMed] [Google Scholar]
- [75].Cudkowicz ME, Shefner JM, Simpson E, Grasso D, Yu H, Zhang H, Shui A, Schoenfeld D, Brown RH, Wieland S, Barber JR, Consortium NA. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in Amyotrophic Lateral Sclerosis. Muscle Nerve. 2008;38(1):837–844. doi: 10.1002/mus.21059. [DOI] [PubMed] [Google Scholar]
- [76].Pascuzzi RM, Shefner J, Chappell AS, Bjerke JS, Tamura R, Chaudhry V, Clawson L, Haas L, Rothstein JD. A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(3):266–271. doi: 10.3109/17482960903307805. [DOI] [PubMed] [Google Scholar]
- [77].L. VDB, W. V, Klaassen H, Van Houtte E, Robberecht W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J Neurol Sci. 2000;180(1-2):29–34. doi: 10.1016/s0022-510x(00)00414-7. [DOI] [PubMed] [Google Scholar]
- [78].Tortarolo M, Grignaschi G, Calvaresi N, Zennaro E, Spaltro G, Colovic M, Fracasso C, Guiso G, Elger B, Schneider H, Seilheimer B, Caccia S, Bendotti C. Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J Neurosci Res. 2006;83(1):134–146. doi: 10.1002/jnr.20715. [DOI] [PubMed] [Google Scholar]
- [79].Paizs M, Tortarolo M, Bendotti C, Engelhardt JI, Siklós L. Talampanel reduces the level of motoneuronal calcium in transgenic mutant SOD1 mice only if applied presymptomatically. Amyotroph Lateral Scler. 2011;12(5):340–344. doi: 10.3109/17482968.2011.584627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ben-Ami M, Bassan M, Sorani K. Method for Treatment of Amyotrophic Lateral Sclerosis using Talampanel. 2009 US0258863. [Google Scholar]
- [81].Carrì MT, Grignaschi G, Bendotti C. Targets in ALS: designing multidrug therapies. Trends Pharmacol Sci. 2006;27(5):267–273. doi: 10.1016/j.tips.2006.03.009. [DOI] [PubMed] [Google Scholar]
- [82].Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12(19):2519–2532. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- [83].Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- [84].Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, Felsenstein D, Krivickas L, David WS, Vriesendorp F, Pestronk A, Caress JB, J. K, Simpson E, Rosenfeld J, Pascuzzi R, Glass J, Rezania K, Rothstein JD, Greenblatt DJ, Cudkowicz ME, Consortium. NA. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Contestabile A. Amyotrophic lateral sclerosis: from research to therapeutic attempts and therapeutic perspectives. Curr Med Chem. 2011;18(36):5655–5665. doi: 10.2174/092986711798347289. [DOI] [PubMed] [Google Scholar]
- [86].Ikeda JE, Osuga I, Kasunori T. Therapeutic Agent for Amyotrophic lateral Sclerosis. 2011 US0105517. [Google Scholar]
- [87].Carrí MT, Ferri A, Cozzolino M, Calabrese L, Rotilio G. Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull. 2003;61(4):365–374. doi: 10.1016/s0361-9230(03)00179-5. [DOI] [PubMed] [Google Scholar]
- [88].Pattee GL, Post GR, Gerber RE, Bennett JPJ. Reduction of oxidative stress in amyotrophic lateral sclerosis following pramipexole treatment. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4(2):90–95. doi: 10.1080/14660820310012736. [DOI] [PubMed] [Google Scholar]
- [89].Danzeisen R, Schwalenstoecker B, Gillardon F, Buerger E, Krzykalla V, Klinder K, Schild L, Hengerer B, Ludolph AC, Dorner-Ciossek C, Kussmaul L. Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2-amino-4,5,6,7-tetrahydro-6-Lpropylamino-benzathiazole dihydrochloride] J Pharmacol Exp Ther. 2006;316(1):189–199. doi: 10.1124/jpet.105.092312. [DOI] [PubMed] [Google Scholar]
- [90].Gribkoff VK, Bozik ME. KNS-760704 [(6R)-4,5,6,7-tetrahydro-N6-propyl-2, 6-benzothiazole-diamine dihydrochloride monohydrate] for the treatment of amyotrophic lateral sclerosis. CNS Neurosci Ther. 2008;14(3):215–226. doi: 10.1111/j.1755-5949.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wang H, Larriviere KS, Keller KE, Ware KA, Burns TM, Conaway MA, Lacomis D, Pattee GL, Phillips L. H. n., Solenski NJ, Zivkovic SA, Bennett JPJ. R+ pramipexole as a mitochondrially focused neuroprotectant: initial early phase studies in ALS. Amyotroph Lateral Scler. 2008;9(1):50–58. doi: 10.1080/17482960701791234. [DOI] [PubMed] [Google Scholar]
- [92].Pasinetti GM. Treatment of Amyotrophic Lateral Sclerosis with nimesulide. 2006 US0041022 a1. [Google Scholar]
- [93].Pugliese M, Espinosa Parrilla J, Virgili Treserres N, Mancera Aroca P, Pasten Zamorano A, Mahy Gehenne J, Rodriguez Allue M. Diazoxide for use in the Treatment of Amyotrophic Laterla Sclerosis (ALS) 2012 US022730 a1. [Google Scholar]
- [94].Ikeda K. Therapeutic Agent for Amyotrophic Lateral Sclerosis (ALS) 2005 US6,933,310 B1. [Google Scholar]
- [95].Yoshino H, Yoneoka T. Novel therapeutic agent for amyotrophic lateral sclerosis (ALS) or disease attributable to ALS. 2006 WO075434. [Google Scholar]
- [96].Harper JM, Krishnan C, Darman JS, Deshpande DM, Peck S, Shats I, Backovic S, Rothstein JD, Kerr DA. Axonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron-injured adult rats. Proc Natl Acad Sci U S A. 2004;101(18):7123–7128. doi: 10.1073/pnas.0401103101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110(3):385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- [98].Pandya RS, Mao LL, Zhou EW, Bowser R, Zhu Z, Zhu Y, Wang X. Neuroprotection for amyotrophic lateral sclerosis: role of stem cells, growth factors, and gene therapy. Cent Nerv Syst Agents Med Chem. 2012;12(1):15–27. doi: 10.2174/187152412800229152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hefferan MP, Galik J, Kakinohana O, Sekerkova G, Santucci C, Marsala S, Navarro R, Hruska-Plochan M, Johe K, Feldman E, Cleveland DW, Marsala M. Human neural stem cell replacement therapy for amyotrophic lateral sclerosis by spinal transplantation. PLoS One. 2012;7(8) doi: 10.1371/journal.pone.0042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Xu L, Yan J, Chen D, Welsh AM, Hazel T, Johe K, Hatfield G, Koliatsos VE. Human neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation. 2006;82(7):865–875. doi: 10.1097/01.tp.0000235532.00920.7a. [DOI] [PubMed] [Google Scholar]
- [101].Lunn JS, Hefferan MP, Marsala M, Feldman EL. Stem cells: comprehensive treatments for amyotrophic lateral sclerosis in conjunction with growth factor delivery. Growth Factors. 2009;27(3):133–140. doi: 10.1080/08977190902814855. [DOI] [PubMed] [Google Scholar]
- [102].Klein SM, Behrstock S, McHugh J, Hoffmann K, Wallace K, Suzuki M, Aebischer P, Svendsen CN. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther. 2005;16(4):509–521. doi: 10.1089/hum.2005.16.509. [DOI] [PubMed] [Google Scholar]
- [103].Mazzini L, Vercelli A, Ferrero I, Boido M, Cantello R, Fagioli F. Transplantation of mesenchymal stem cells in ALS. Prog Brain Res. 2012;201:333–359. doi: 10.1016/B978-0-444-59544-7.00016-0. [DOI] [PubMed] [Google Scholar]
- [104].Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, Bulte JW, Petrou P, Ben-Hur T, Abramsky O, Slavin S. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67(10):1187–1194. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Canzi L, Castellaneta V, Navone S, Nava S, Dossena M, Zucca I, Mennini T, Bigini P, Parati EA. Human skeletal muscle stem cell antiinflammatory activity ameliorates clinical outcome in amyotrophic lateral sclerosis models. Mol Med. 2012;18:401–411. doi: 10.2119/molmed.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Marconi S, Bonaconsa M, Scambi I, Squintani GM, Rui W, Turano E, Ungaro D, D'Agostino S, Barbieri F, Angiari S, Farinazzo A, Constantin G, Del Carro U, Bonetti B, Mariotti R. Systemic treatment with adipose-derived mesenchymal stem cells ameliorates clinical and pathological features in the amyotrophic lateral sclerosis murine model. Neuroscience. 2013;248C:333–343. doi: 10.1016/j.neuroscience.2013.05.034. [DOI] [PubMed] [Google Scholar]
- [107].Uccelli A, Milanese M, Principato MC, Morando S, Bonifacino T, Vergani L, Giunti D, Voci A, Carminati E, Giribaldi F, Caponnetto C, Bonanno G. Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis. Mol Med. 2012;18:794–804. doi: 10.2119/molmed.2011.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].González-Garza MT, Martínez HR, Caro-Osorio E, Cruz-Vega DE, Hernández-Torre M, Moreno-Cuevas JE. Differentiation of CD133+ stem cells from amyotrophic lateral sclerosis patients into preneuron cells. Stem Cells Transl Med. 2013;2(2):129–135. doi: 10.5966/sctm.2012-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Hariri RJ, Gurney JP. Treatment of Amyotrophi Lateral Sclerosis Using Placental Stem Cells. 2013 WO05547. [Google Scholar]
- [110].Popescu IR, Nicaise C, Liu S, Bisch G, Knippenberg S, Daubie V, Bohl D, Pochet R. Neural progenitors derived from human induced pluripotent stem cells survive and differentiate upon transplantation into a rat model of amyotrophic lateral sclerosis. Stem Cells Transl Med. 2013;2(3):167–174. doi: 10.5966/sctm.2012-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- [112].Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8(6):933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- [113].Gros-Louis F, Soucy G, Larivière R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113(5):1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x. [DOI] [PubMed] [Google Scholar]
- [114].Takeuchi S, Fujiwara N, Ido A, Oono M, Takeuchi Y, Tateno M, Suzuki K, Takahashi R, Tooyama I, Taniguchi N, Julien JP, Urushitani M. Induction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. 2010;69(10):1044–1056. doi: 10.1097/NEN.0b013e3181f4a90a. [DOI] [PubMed] [Google Scholar]