

Abstract
The de novo design of catalysts that mimic the affinity and specificity of natural enzymes remains one of the Holy Grails of chemistry. Despite decades of concerted effort we are still unable to design catalysts as efficient as enzymes. Here we critically evaluate approaches to (re)design of novel catalytic function in proteins using two test cases: Kemp elimination and ester hydrolysis. We show that the degree of success thus far has been modest when the rate enhancements seen for the designed proteins are compared with the rate enhancements by small molecule catalysts in solvents with properties similar to the active site. Nevertheless, there are reasons for optimism: the design methods are ever improving and the resulting catalyst can be efficiently improved using directed evolution.
Introduction
Enzymes catalyze kinetically challenging reactions at remarkable rates with exquisite control over the mechanism, regiochemistry and stereochemistry of the molecules upon which they act. Over the last half century, chemists have tried to design molecules from first principles that emulate enzymes for both fundamental and practical purposes. Clearly, if we truly understand enzymes we should be able to design them from scratch — and if we can design enzymes from scratch we should be able to design catalysts to act on substrates and surfaces that are not even remotely similar to the ones found in nature. However, between the thought and the action falls the shadow [1]. Successive generations have turned to molecules of ever-increasing size and functional diversity to achieve this goal. Early studies focused on micelles, membranes [2], cyclodextrins [3], and macrocyclic ‘molecular hosts’ [4] prepared by synthetic chemistry to engulf substrates and present reactive groups. Next, researchers began to explore ‘catalytic antibodies’ whose binding sites could be programmed to bind tightly to molecules that resemble high-energy intermediates in organic reactions [5]. As the mechanisms by which proteins fold became clearer, it next became possible to design proteins entirely from scratch, and modest progress has been made in the design of metalloproteins that catalyze various redox reactions [6]. Finally, in the last decade, computational methods to redesign the sequences of natural enzymes have been devised to catalyze reactions not related to the starting catalyst [7]. After a half century of intense efforts it is safe to say that we have not yet achieved our objectives. By comparison to natural enzymes the various mimics that have so far been devised generally have low catalytic efficiencies (with the exception of catalysts of the Kemp elimination, but more about that later), particularly for reactions that have even modest energy barriers or complex reaction mechanisms.
Looking at the previous enzyme-mimetic literature, one observes cyclic trends of: (1) initial exciting discoveries that promise to enable design enzymes from scratch; (2) a flurry of publications of ever-increasing sophistication representing improvements on the initial findings; (3) a plateau that inevitably falls far short of natural enzymes. However, our understanding has advanced through careful and well-designed half-successes or even failures when the experiments are well constructed and analyzed. We have learned that binding, general acid/base catalysis, and proximity effects all contribute to catalysis, but they are rarely if ever sufficient — otherwise modern day enzymes would not need to be as complex and the designers of enzyme mimics would have succeeded decades ago.
The latest wave in enzyme mimetic design — de novo computational enzyme design — has now been the topic of very intense efforts over the last half decade, so it is timely to ask how well it is working. In what ways are we recapitulating the past versus, or are we at the inflection point of a field poised for unprecedented progress? To address these questions we will focus on two simple reactions that have been studied over the years; the ‘Kemp elimination’ of benzisoxazoles and ester hydrolysis. We will show that proteins designed to catalyze these reactions are roughly on par with those obtained through earlier approaches such as catalytic antibodies. However, there is reason for optimism. Computationally designed proteins can be evolved to greater efficiency using in vitro evolution, resulting in substantial improvements relative to previous studies.
Although there remains gap between designed/evolved enzyme mimics and true enzymes, we feel the field is positioned to make impressive progress in the next decade. To progress, however, we argue that it is important to define benchmarks that allow workers in the field to understand how impressive a given enzyme mimic is relative to a set of appropriate small molecule controls. Furthermore, we argue that progress will depend on concurrent technical improvements that are discussed in more detail in other articles in this issue. One example is the improvement of potential functions to define the energetics of small molecule binding; the design of proteins that bind highly functionalized small molecules remains in its infancy, and without mastery of the ability to bind small molecules it will be difficult to design catalysts that act on small molecules! Moreover, the dynamics of catalysis needs to be considered. As a reaction progresses through a series of intermediates on an enzyme, the shape of the substrate changes, the positions of protons on general acids/bases need to move. The dynamics of the enzyme is therefore tuned to appropriately position functional groups and transiently stabilize high-energy intermediates and transition states while not stabilizing alternative states that could lead to side product formation. Recapitulation of this fine balance in a designed enzyme will require strategies for stabilization of multiple closely related conformational states, including negative design strategies to destabilize alternative pathways [8]. As these and other methods discussed in this volume bear fruit we should be able to ask evermore sophisticated questions concerning the mechanism of enzymatic catalysis, which in turn will enable the design of useful catalysts of nonbiological reactions. Let us now look at the accomplishments of the field to date from this perspective.
The Kemp elimination
The Kemp elimination (Figure 1) is a much studied reaction in which a catalytic base abstracts a proton from the heterocyclic ring of a benzisoxazole. This abstraction cleaves the isoxazole ring and simultaneously forms an acidic 2-cyano-phenol. This popular reaction is a good benchmark for computational enzyme design, because it has been so thoroughly studied and evaluated in antibody, cyclodextrin-based, micelle and bilayer catalysts. However, it is also a very facile reaction, quite different from most biological C–H abstractions (such as that in triose phosphate isomerase) in that charge is highly delocalized throughout the ring systems in the transition state and product. Thus, the reaction is enhanced by removing the substrate from water, and there is minimal if any requirement for protonation of the leaving group to achieve an impressive rate constant [9]. Carboxylic acids dissolved in organic solvents are excellent catalysts for this reaction, in part because this environment increases the basicity of the carboxylate group making it more able to remove a proton from the isoxazole. The reaction is first order in carboxylate and first order in substrate (Eqn 1), and the second order rate constant (k2) for elimination of the 5-nitrobenisoxazole derivative, 1, is 2800 m−1 s−1 in acetonitrile [10].
Figure 1.

Reactions and common substrates used to test various protein design methodologies.
| (1) |
| (2) |
| (3) |
One of the most difficult problems in enzyme mimetic design is expressing a ‘rate enhancement’ and relating an observed rate to what might be expected from a small molecule control. On the one hand, we might compare to the reaction rate in water — after all that is the medium in which the enzyme mimic and substrate are dissolved. And, for a reaction that is slow in water such as the Kemp elimination this comparator certainly gives large enhancements that helps get papers published! However, if we wish to understand the molecular nature of the catalysis, the more appropriate benchmark might be the rate in an environment that more resembles the intended active site, which are generally designed to sequester the substrate from bulk water. From this perspective, the rate for the acetate-catalyzed reaction in acetonitrile represents what might be achieved in an enzyme-like environment, but in which the acetate and substrate were randomly colliding with no fixed orientation [10]. Michaelis-Menten equation (2) for an enzyme working at substrate concentrations significantly below KM is simplified and best described by Eqn 3, and the value k2 is equivalent to kcat/KM, allowing a direct comparison between the small molecule and protein catalyst. Thus, an enzyme mimic that did nothing more than to create an environment similar to acetate in acetonitrile would be expected to have an equivalent second order rate constant if the free energy for transfer from water to the active site were zero and there were no large barriers to binding or dissociation. Of course, a well-designed protein should show much larger rate enhancements by binding the substrate and bringing it into proximity with a base that is not randomly oriented (as for acetate's collisions with substrate in acetonitrile), but rather oriented for efficient proton abstraction. Indeed, one might expect that it would be relatively easy to achieve rate enhancements of 104–106 over acetate in acetonitrile to approach 108–109 m−1 s−1, the limit for diffusion of a small molecule to a protein. It is therefore profoundly humbling that designed proteins have not reached the value seen for acetate in acetonitrile, although it is perhaps reassuring that it can be reached and exceeded after repeated rounds of optimization by directed evolution [11••, 12•].
A second metric by which designed proteins can be judged is their turnover number (kcat), which is the rate observed when the protein is fully bound by substrate. Because small molecule catalysts do not show saturation kinetics it is harder to compare directly to kcat, but we might consider the rate that would result if, for example, acetate were at a sufficiently high concentration such that it at any given time one acetate would be in contact with one substrate in acetonitrile, as when the acetate made up about 10 mol% of the solvent. The pseudo-first order rate computed from Eqn 1 would then be 5600 s−1 (assuming 2 m concentration of acetate, approximately equivalent to 10 molar percent in acetonitrile). This value provides an order of magnitude estimate of the rate we would expect for kcat for a ‘Kemp eliminase’ with an acetonitrile-like active site and a catalytic base that was randomly oriented in its collisions with substrate. This value represents a benchmark for protein design to beat with functional group pre-organization and introduction of additional interactions. Again, orientation effects could greatly enhance the reaction. Let us now explore how these expectations match up with off-the-shelf non-catalytic proteins, catalytic antibodies, and protein design (Figure 2).
Figure 2.

Comparison of the catalytic efficiencies (left) and turnover numbers (right) of Kemp eliminases obtained by various approaches. The efficiencies of the computationally (re)designed catalyst are shown in green and blue, the subsequent improvement by directed evolution is shown in red.
At near neutral pH values the Kemp elimination is catalyzed by lysine side chains of proteins such as serum albumins (kcat = 0.012 s−1 and kcat/KM = 8.6 s−1m−1) [10], setting an expectation for a protein with a hydrophobic binding cavity. After much optimization catalytic antibodies reached kcat/KM = 5500 m−1 s−1 [13–15], slightly exceeding the value expected for acetate in acetonitrile. The corresponding kcat = 0.66 s−1 is modest illustrating the contribution of binding (KM = 0.12 mM) to the overall value of kcat/KM.
Of the various strategies employed to redesign natural proteins to catalyze the Kemp elimination, the minimalist approach (outlined in Figure 3) taken by Korendovych et al. is by far the simplest [16]. Knowing that the minimal requirement for activity is a substrate-sized hydrophobic cavity with an appropriately placed glutamate with an elevated pKa we chose calmodulin (CaM), which has a very hydrophobic cavity, lined only by apolar residues. This protein was also chosen because it lacks enzymatic activity and its natural substrates are not small molecules, providing a neutral starting point that was not genetically optimized for a catalytic function. Moreover its conformation is allosterically regulated by binding Ca2+ at a site distant from the hydrophobic binding site. Docking calculations indicated that the substrate could be well accommodated in the binding pocket. We next tested whether a catalytic group might be introduced without redesigning the other sidechains in the active site, expecting that inclusion of a Glu or Asp in an apolar environment would be sufficient to increase its basicity. A computational repacking algorithm identified a single position that could be appropriately positioned in a low-energy rotamer to interact with the docked substrate. The initially designed protein, called AlleyCat is an allosterically regulated Kemp eliminase with modest activity (kcat/KM = 6 m−1 s−1). This value falls short of the acetate in acetonitrile benchmarks, but is nevertheless within the range of the first catalytic antibodies and the most successful Kemp eliminases designed using far more complex algorithms, discussed below. As a control, Glu/Asp was scanned in other positions within apolar cavity of CaM, leading to entirely inactive proteins. Although the control proteins were well expressed and folded they were far less efficient catalysts of the Kemp elimination reaction. Thus, the appropriate positioning of a carboxylate in the appropriate geometry was important for activity.
Figure 3.
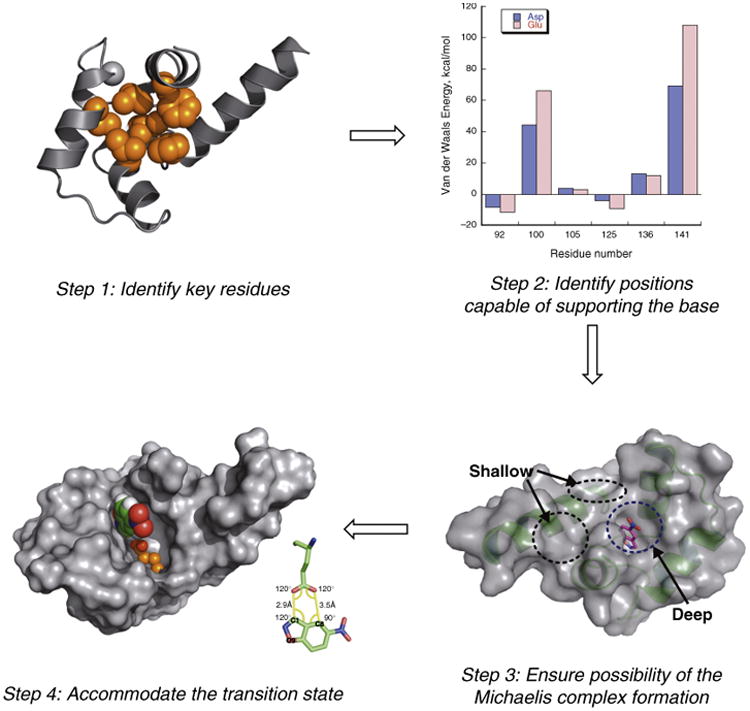
Minimalistic approach to designing a Kemp eliminase [16].
The NMR structure of AlleyCat was consistent with the design, but also showed regions of dynamics that might impede catalytic efficiency, which might be improved by directed evolution. Despite its small size, AlleyCat was further improved by directed evolution to reach the turnover number of 3.2 s−1 and enzymatic efficiency of 1280 m−1 s−1 in just seven rounds [17•]. The allosteric nature of AlleyCat provides additional opportunities in sensing [18]. Conferring the ability to convert colorless substrate into yellow product onto a calcium-modulated protein creates a catalytically amplified sensor for calcium. Moreover, the EF-hands of AlleyCat were reprogrammed to preferentially respond to lanthanides [19]. Ultimately, directed evolution may be applied to the metal-binding domain to develop sensors for a variety of different metals.
The ability of proteins to adopt catalytic activity by careful positioning of active residues in hydrophobic cavities is by no means unique to calmodulin. Using simple modeling Merski and Shoichet introduced a histidine residue into an engineered mutant of a T4 lysozyme to create a Kemp eliminase [20]. Subsequent rational improvement of the catalyst guided by structural information yielded an enzyme with a catalytic efficiency of 1.8 m−1s−1 at pH 5.0, and the structure of the protein was confirmed by X-ray crystallography.
Baker and coworkers used a computational approach based on constructing a theoretical transition state, theozyme [21], optimized by high level computation that relies on multiple residues to reproduce a transition state for a particular reaction. While high level computational approach may not be necessary for this simple reaction, such degree of complexity will be needed as we begin to progress to ever-more energetically demanding reactions where multiple groups must contribute to catalysis. The theozyme is then incorporated into a rigid protein scaffold obtained from the Protein Data Bank. For Kemp eliminase design, a theozyme constructed with an active base (Asp/Glu or His), a H-bond donor and an aromatic residue for π-stacking was constructed based on quantum mechanical calculations. A Rosetta Match search protocol was run to identify whether 87 various protein scaffolds could accommodate the theozyme. After the search and optimization a total of 59 designs were generated and 8 of them showed activity ranging from 6 m−1 s−1 to ∼160 m−1 s−1, the activity of the most active design, KE59, could be only estimated due to protein misfolding [22]. Interestingly, all of the active designs came from just four TIM barrel enzymes, even though only 25% of the total 87 different scaffolds that were considered in the search belonged to that class. Three designs (KE07, KE70 and KE59) were selected for directed evolution [12•, 23, 24] and the most active catalyst KE59 R13-3/11H reached the catalytic efficiency of 60 000 m−1 s−1 with the turnover number of 9.5 s−1.
Mayo and coworkers approached the problem of creating an efficient Kemp eliminase by constructing a transition state that is similar to that of Baker et al. by using a different computational algorithm. The initial design, named HG-1, despite its structural similarity to the target model, turned out to be inactive. However, structurally guided, iterative, rational redesign of HG-1 produced active catalysts HG-2 and HG-3 [25]. The catalytic efficiency of 430 s−1 m−1 for HG-3 was further improved by 17 rounds of directed evolution to reach 230 000 s−1 m−1, which is the highest reported catalytic efficiency of any designed Kemp eliminase to date [11••]. Importantly, the increase in catalytic efficiency was achieved predominantly by increase in the turnover number (reaching 700 s−1) and not by lowering the KM. Comparing kcat to the pseudo first order rate expected for 2 m acetate in acetonitrile (5600 s−1), we see that the field has not yet progressed to reach this benchmark, but this is nevertheless a very important step forward. The structure of the protein in complex with a substrate analog showed the importance of positioning the general base appropriately for proton abstraction. Also, systematic studies with substrates of differing substitutions showed that the observed rate varied in the expected manner with electronic effects, indicating that the chemical step, rather than substrate binding or release, determined the rate of the reaction. The authors suggested that the protein might accelerate the rate by stabilizing the anionic leaving group, although it is difficult to assess the contribution of this effect for a reaction where protonation of the leaving group does not appear to be required for catalysis [9].
The vast array of structural and functional data for the original and optimized designs obtained by Baker, Tawfik et al. provide good insight to strengths and deficiencies of the theozyme methodology. Three major conclusions can be derived from this work.
Introduction of the catalytic base was quite successful in every case, however, the properties of the active residue(s) almost universally needed further improvement by subsequent directed evolution. Simple geometric matching seems to have a hard time creating a proper pKa of the active residue base, solvent accessibility and the electrostatics at the reactive site. It seems the simplest strategy for altering the pKa of the Glu is to place it in a hydrophobic pocket with few interactions that would stabilize its ionized state (that would decrease the pKa).
Generally, introduction of π-stacking for proper substrate positioning and lowering the KM is quite successful. Structural analysis of the evolved designs shows that Trp and Tyr residues introduced to fulfill this goal are preserved in the directed evolution studies (although it is not always clear if they were allowed to be mutated in this process) and in van der Waals contact in the structures of the designs with the substrate mimics.
Introduction of polar functional groups for additional interactions (e.g. proton transfer or transition state stabilization) into the theozymes designed to aid the catalysis is almost invariably unsuccessful. Removal of these rationally placed interactions in subsequent directed evolution improves catalysis and could even lead to complete reshaping of the active-site cavity with completely new interactions.
Ester and phosphoester hydrolysis
These practically important reactions are very diverse in terms of potential substrates and most of the design work focused on molecules that produce colored or fluorescent product that can be easily identified in a high-throughput fashion (Figure 1). Using the rationale described above for setting a benchmark for Kemp eliminases, we can establish similar points for esterases. Imidazole is an effective nucleophilic catalyst of pNPA hydrolysis, and the second order rate constant for hydrolysis by the free base form of acetyl-histidine in water is on the order of 0.2 m−1s−1, [26] providing a benchmark for this relatively facile reaction. For designed metalloesterases, the corresponding benchmark is the pseudo first order rate constant of pNPA hydrolysis at 10 molar percent of OH−, ∼56 s−1 [27]. Notably, this value is very similar to the activities shown by carbonic anhydrase in pNPA hydrolysis (kcat = 53 s−1) [28] and alkaline phosphatase in the hydrolysis of 6 (kcat = 35 s−1) [29]. Both pNPA and 6 are not the normal substrates for the enzymes but the metal ion is perfectly tuned to create an active nucleophile for ester hydrolysis.
Early on it was shown that catalytic antibody 43C9 hydrolyzes p-nitrophenol ester 4 with an impressive turnover number of 25 s−1 and catalytic efficiency of 430 000 m−1 s−1 but suffers from substantial product inhibition (KI ∼ 1 μm) [30, 31]. Subsequent rational design of esterases showed that simple peptides are also capable of catalyzing this reaction, albeit no enzyme-like saturation is observed and the corresponding second rate order constants are modest (k2 < 0.3 m−1 s−1) [32, 33]. The advent of computational tools for protein design made possible introduction of new functionalities into existing scaffolds. A single histidine residue insertion conferred esterase activity onto bacterial thioredoxin with a kcat of 5 × 10 −4 s−1 and a kcat/KM of 3 m−1 s −1 [34]. More recently, by introducing a single histidine into calmodulin Moroz et al. (Moroz YS, Dunston TT, Moroz OV, Wu Y, Yoon JH, Olsen AB, McLaughlin JM, Mack KL, Gosavi PM, van Nuland NAJ, et al.: Single mutations in a non-enzymatic protein give rise to various catalytic activities, submitted for publication) were able to convert this non-enzymatic protein into an allosterically regulated esterase AlleyCatE with a catalytic efficiency significantly greater (kcat = 9 ×10−3 s−1, kcat/KM = 150 m−1 s−1) than those of the rational and computational designs [32,34] as well as the catalysts identified from various libraries [35].
The design efforts were not limited to the use of histidine as the nucleophile. Richter et al. have successfully designed several catalysts with a catalytic dyad that consists of a cysteine, whose nucleophilicity is modulated by a neighboring histidine. The best of the designs (ECH13) showed kcat of 1.8 × 10−2 s−1 and kcat/KM of 309 m−1 s−1 for hydrolysis of 2 [36].
Creating a strong nucleophile using just side chains of the genetically encoded 20 amino acids is often difficult, but metal ions substantially broaden the repertoire of possibilities. Metal ions have also been used as a design element in ester hydrolysis. Zinc is often a metal of choice due to its hydrolytic robustness, tight binding to common amino acid side chains, simple coordination sphere and diamagnetism that simplifies NMR characterization. Moreover, the activity of zinc esterases is not hindered by slow hydrolysis of the acyl intermediate common in designs with histidine and cysteine as nucleophiles. Several examples of metal-loesterases have been recently reported. Der et al showed that a simple cleft together with a metal coordination site formed at the interface of two designed proteins is capable of efficient hydrolysis of pNPA (kcat = 0.30 s−1, kcat/KM = 630 m−1 s−1) and less reactive phosphoester 6 (kcat = 2 × 10−4 s−1 and kcat/KM = 14 m−1 s−1) [38]. Zastrow et al. designed a zinc coordination site in a trimeric coiled coil for CO2 hydration [39•]. The resulting protein can also catalyze pNPA hydrolysis with kcat of 4 × 10−2 s−1 and kcat/KM of 23 m−1 s−1. Rufo et al. took a minimalistic approach to the next level (Figure 4) and used self-assembly of short 7-residue peptides to create amyloid fibrils capable of efficient hydrolysis of pNPA (kcat ∼ 0.1 s−1and kcat/KM = 360 m−1 s−1) [37••]. The ability of the fibrils to hydrolyze pNPA is higher than that of carbonic anhydrase on a weight basis. Finally, Khare et al. designed PT3 a catalyst for hydrolysis of nerve agent analog 5 (kcat = 0.2 × 10−3 s −1 and kcat/KM = 4 m−1 s−1). Over just three rounds of directed evolution activity of PT3 was improved to reach kcat of 0.35 s−1 and kcat/KM of 10 000 m−1 s−1 [40].
Figure 4.
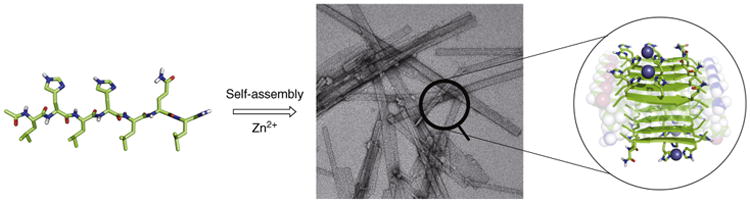
De novo designed peptides self-assemble to form amyloid-like fibrils that catalyze ester hydrolysis [37••].
The designs utilizing histidines as nucleophiles appear very successful compared to imidazole suggesting that we are able to constrain the active residue in a productive conformation to facilitate catalysis. However, the designed esterases are still largely inferior to natural enzymes both in terms of enzymatic efficiency and turnover number. Moreover, reported kcat and kcat/KM often refer to ‘fast’ or ‘burst’ phase of the reaction and not to the acyl intermediate hydrolysis step, where the turnover occurs. De novo designed metalloesterases, whose kinetic parameters can be directly compared to that of the hydroxide ion, show characteristics similar to those of Kemp eliminases. While kcat/KM values for the designed enzymes show substantial improvement over the background rate, the corresponding kcat values still cannot match the pseudo first order rate constant of the proximal hydroxide ion.
Conclusions
We are still far away from being able to create efficient enzyme-like catalysts from scratch. So far, most successes can be ascribed to bringing the substrate into close proximity with one single catalytic group. The most successful strategies to date have focused on functional tuning of the active site and stability of the protein scaffold to maximize chances of success in subsequent directed evolution studies.
Consideration of a transition state alone appears to be insufficient for effective catalysis. All parts of the catalytic cycle (Michaelis complex, transition state(s), product dissociation) will ultimately need to be considered, and dynamics optimized so the reaction pathway does not have large energy barriers and/or deep energy wells. Several designs addressing parts of the puzzle separately may need to be done and then combined together iteratively to create an efficient catalyst with high turnover.
Acknowledgments
This work was supported in part by a grant number 1332349 from NSF-EFRI, ORAU Ralph E Powe Junior Faculty Enhancement award and a Humboldt Research Fellowship to IVK and NIH grant GM54616 to WFD.
Footnotes
Conflict of interest: None declared
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Eliot TS. The Waste Land. New York: Horace Liveright; 1922. [Google Scholar]
- 2.Kunitake T, Shinkai S. Catalysis by micelles, membranes and other aqueous aggregates as models of enzyme action. Adv Phys Org Chem. 1981;17:435–487. [Google Scholar]
- 3.Breslow R, Dong SD. Biomimetic reactions catalyzed by cyclodextrins and their derivatives. Chem Rev. 1998;98:1997–2012. doi: 10.1021/cr970011j. [DOI] [PubMed] [Google Scholar]
- 4.Cram DJ. The design of molecular hosts, guest and their complexes. Angew Chem Int Ed Engl. 2003;27:1009–1020. [Google Scholar]
- 5.Hilvert D. Critical analysis of antibody catalysis. Annu Rev Biochem. 2000;69:751–793. doi: 10.1146/annurev.biochem.69.1.751. [DOI] [PubMed] [Google Scholar]
- 6.Nanda V, Koder RL. Designing artificial enzymes by intuition and computation. Nat Chem. 2010;2:15–24. doi: 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiss G, Cçelebi-O¨lcçu¨m N, Moretti R, Baker D, Houk KN. Computational enzyme design. Angew Chem Int Ed. 2013;52:5700–5725. doi: 10.1002/anie.201204077. [DOI] [PubMed] [Google Scholar]
- 8.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective βSZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemp DS, Casey ML. Physical organic chemistry of benzisoxazoles. II Linearity of the Bronsted free energy relationship for the base-catalyzed decomposition of benzisoxazoles. J Am Chem Soc. 1973;95:6670–6680. [Google Scholar]
- 10.Hollfelder F, Kirby AJ, Tawfik DS. Off-the-shelf proteins that rival tailor-made antibodies as catalysts. Nature. 1996;383:60–63. doi: 10.1038/383060a0. [DOI] [PubMed] [Google Scholar]
- 11••.Blomberg R, Kries H, Pinkas DM, Mittl PRE, Gru¨tter MG, Privett HK, Mayo SL, Hilvert D. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature. 2013;503:418–421. doi: 10.1038/nature12623. Iterative semi-rational design approach enchanced by subsequent directed evolution yielded highly efficient Kemp eliminase with turnover number of 700 s−1 approaching enzymatic levels of activity. [DOI] [PubMed] [Google Scholar]
- 12•.Khersonsky O, Kiss G, Ro¨thlisberger D, Dym O, Albeck S, Houk KN, Baker D, Tawfik DS. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc Natl Acad Sci USA. 2012;109:10358–10363. doi: 10.1073/pnas.1121063109. Extensive biochemical, structural and molecular dynamics simulation data on Kemp eliminase KE59 and its evolved variants provides insight into the factors important for computational design and directed evolution of enzymes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorn SN, Daniels RG. Large rate accelerations in antibody catalysis by strategic use of haptenic charge. Nature. 1995;373:228–230. doi: 10.1038/373228a0. [DOI] [PubMed] [Google Scholar]
- 14.Debler EW, Muller R, Hilvert D, Wilson IA. An aspartate and a water molecule mediate efficient acid–base catalysis in a talored antibody pocket. Proc Natl Acad Sci USA. 2009;106:18539–18544. doi: 10.1073/pnas.0902700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genre-Grandpierre A, Tellier C. Catalysis of the Kemp elimination by antibodies elicitied against a cationic hapten. Bioorg Med Chem Lett. 1997;7:2497–2502. [Google Scholar]
- 16.Korendovych IV, Kulp DW, Wu Y, Cheng H, Roder H, DeGrado WF. Design of a switchable eliminase. Proc Natl Acad Sci USA. 2011;108:6823–6827. doi: 10.1073/pnas.1018191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Moroz OV, Moroz YS, Wu Y, Olsen AB, Cheng H, Mack KL, McLaughlin JM, Raymond EA, Zhezherya K, Roder H, et al. A single mutation in a regulatory protein produces evolvable allosterically regulated catalyst of unnatural reaction. Angew Chem Int Ed. 2013;52:6246–6249. doi: 10.1002/anie.201302339. Given enough stability even a small non-enzymatic 74-residue protein domain of vcalmodulin can be evolved to become an efficient, allosterically regulated catalyst of Kemp elimination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makhlynets OV, Korendovych IV. Design of catalytically amplified sensors for small molecules. Biomolecules. 2014;4:402–418. doi: 10.3390/biom4020402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mack KL, Moroz OV, Moroz YS, Olsen AB, McLaughlin JM, Korendovych IV. Reprogramming EF-hands for design of catalytically amplified lanthanide sensors. J Biol Inorg Chem. 2013;18:411–418. doi: 10.1007/s00775-013-0985-5. [DOI] [PubMed] [Google Scholar]
- 20.Merski M, Shoichet BK. Engineering a model protein cavity to catalyze the Kemp elimination. Proc Natl Acad Sci USA. 2012;109:16179–16183. doi: 10.1073/pnas.1208076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tantillo DJ, Chen J, Houk KN. Theozymes and compuzymes: theoretical models for biological catalysis. Curr Opin Chem Biol. 1998;2:743–750. doi: 10.1016/s1367-5931(98)80112-9. [DOI] [PubMed] [Google Scholar]
- 22.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 23.Khersonsky O, Rothlisberger D, Dym O, Albeck S, Jackson CJ, Baker D, Tawfik DS. Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series. J Mol Biol. 2010;396:1025–1042. doi: 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 24.Khersonsky O, Rothlisberger D, Wollacott AM, Murphy P, Dym O, Albeck S, Kiss G, Houk KN, Baker D, Tawfik DS. Optimization of the in-silico-designed Kemp eliminase KE70 by computational design and directed evolution. J Mol Biol. 2011;407:391–412. doi: 10.1016/j.jmb.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Privett HK, Kiss G, Lee TM, Blomberg R, Chica RA, Thomas LM, Hilvert D, Houk KN, Mayo SL. Iterative approach to computational enzyme design. Proc Natl Acad Sci U S A. 2012;109:3790–3795. doi: 10.1073/pnas.1118082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruice TC, Benkovic S. Bioorganic Mechanisms. New York: W.A. Benjamine, Inc; 1966. [Google Scholar]
- 27.Jencks WP, Gilchrist M. Nonlinear structure–reactivity correlations. Reactivity of nucleophilic reagents toward esters. J Am Chem Soc. 1968;90:2622–2637. [Google Scholar]
- 28.Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem. 1967;242:4221–4229. [PubMed] [Google Scholar]
- 29.Sun L, Martin DC, Kantrowitz ER. Rate-determining step of Escherichia coli alkaline phosphatase altered by the removal of a positive charge at the active center. Biochemistry. 1999;38:2842–2848. doi: 10.1021/bi981996h. [DOI] [PubMed] [Google Scholar]
- 30.Gibbs RA, Benkovic PA, Janda KD, Lerner RA, Benkovic SJ. Substituent effects on an antibody-catalyzed hydrolysis of phenyl-esters: further evidence for an acyl-antibody intermediate. J Am Chem Soc. 1992;114:3528–3534. [Google Scholar]
- 31.Janda KD, Schloeder D, Benkovic SJ, Lerner RA. Induction of an antibody that catalyzes the hydrolysis of an amide bond. Science. 1988;241:1188–1191. doi: 10.1126/science.3413482. [DOI] [PubMed] [Google Scholar]
- 32.Broo KS, Brive L, Ahlberg P, Baltzer L. Catalysis of hydrolysis and transesterification reactions of p-nitrophenyl esters by a designed helix–loop–helix dimer. J Am Chem Soc. 1997;119:11362–11372. [Google Scholar]
- 33.Nilsson J, Baltzer L. Reactive-site design in folded-polypeptide catalysts — the leaving group pKa of reactive esters sets the stage for cooperativity in nucleophilic and general-acid catalysis. Chemistry. 2000;6:2214–2220. doi: 10.1002/1521-3765(20000616)6:12<2214::aid-chem2214>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 34.Bolon DN, Mayo S. Enzyme-like proteins by computational design. Proc Natl Acad Sci USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei Y, Hecht MH. Enzyme-like proteins from an unselected library of designed amino acid sequences. Protein Eng Des Sel. 2004;17:67–75. doi: 10.1093/protein/gzh007. [DOI] [PubMed] [Google Scholar]
- 36.Richter F, Blomberg R, Khare SD, Kiss G, Kuzin AP, Smith AJT, Gallaher J, Pianowski Z, Helgeson RC, Grjasnow A, et al. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J Am Chem Soc. 2012;134:16197–16206. doi: 10.1021/ja3037367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Rufo CM, Moroz YS, Moroz OV, Stöhr J, DeGrado WF, Korendovych IV. Short peptides self-assemble to produce catalytic amyloids. Nat Chem. 2014;6:303–309. doi: 10.1038/nchem.1894. Amyloidogenic peptides can catalyze not only their own assembly but chemical reactions as well. Small 7-residue peptides were designed to catalyze ester hydrolysis upon self-assembly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Der BS, Edwards DR, Kuhlman B. Catalysis by a de novo zinc-mediated protein interface: implications for natural enzyme evolution and rational enzyme engineering. Biochemistry. 2012;51:3933–3940. doi: 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat Chem. 2012;4:118–123. doi: 10.1038/nchem.1201. A zinc binding site was introduced into a de novo designed trimeric coiled coil creating a higly efficient catalyst for carbon dioxide hydration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khare SD, Kipnis Y, Greisen P, Jr, Takeuchi R, Ashani Y, Goldsmith M, Song Y, Gallaher JL, Silman I, Leader H, et al. Computational redesign of a mononuclear zinc metalloenzyme for organophosphate hydrolysis. Nat Chem Biol. 2012;8:294–300. doi: 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]