

Abstract
The murine cell surface protein Crry is a key complement regulator with similar activities to human membrane cofactor protein (MCP) and decay-accelerating factor. MCP plays a critical role in preventing complement-mediated tissue injury and its mutation has been implicated in several human kidney diseases. Study of Crry in mice has relevance to understanding MCP activity in human diseases but such efforts have been hampered by the embryonic lethality phenotype of Crry gene knockout. Here we used a conditional gene targeting approach and deleted Crry from the mouse proximal tubular epithelial cells where Crry is prominently expressed. Absence of Crry from proximal tubular epithelial cells resulted in spontaneous C3 deposition on the basolateral surface but no apparent renal disease in unchallenged mice. However, mice deficient in Crry on proximal tubular epithelial cells developed exacerbated renal injury when subjected to renal ischemia reperfusion showing increased blood urea nitrogen levels, higher tubular injury scores, more tubular epithelial cell apoptosis, and inflammatory infiltrates. Renal ischemia reperfusion injury in the Crry conditional knockout mice was prevented by blocking C3 and C5 activation using an anti-properdin or anti-C5 mAb, respectively. Thus, Crry has a critical role in protecting proximal tubular epithelial cells during ischemia reperfusion challenge. Our results highlight the latent risk for inflammatory kidney injury associated with defects in membrane complement regulators.
Introduction
The complement system is a finely tuned innate immune system that, while playing a critical role in host defense, has the potential to cause significant tissue injury if not properly controlled.1, 2 The kidney appears to be especially vulnerable to complement attack and clinical studies have long revealed the importance of complement as an inflammatory pathway in the pathogenesis of various human kidney diseases.3–5 More recently, several rare and genetically predisposed kidney pathologies including C3 glomerulopathy and atypical hemolytic uremic syndrome (aHUS) have been linked to dysregulation of complement, often arising from defects in complement regulators such as factor H (fH) and membrane cofactor protein (MCP, CD46).6 Complement regulator insufficiency can be caused by mutations in the genes encoding these proteins or by autoantibodies against them. In addition to C3 glomerulopathy and aHUS, complement has also been implicated in the outcomes of kidney transplantation and contributes to tubular injury during renal ischemia reperfusion.3, 7–10 Thus, understanding how complement is regulated in the various compartments of the kidney has relevance to the prevention and treatment of rare as well as common kidney pathologies.
Apart from MCP and fH, host cells are also protected by other membrane complement regulators including decay-accelerating factor (DAF) and CD59. While all these regulators are expressed in the human kidney, MCP appears to be the only membrane regulator of C3 activation present in abundance on human renal tubular epithelial cells.3, 11, 12 MCP inhibits complement activation by acting as a cofactor for factor I-mediated cleavage of C4b and C3b to prevent the formation of both classical and alternative pathway (AP) C3 convertases.13 Although a MCP gene exists in mice and rats, its expression in these rodent species is rather limited, being detected primarily in the testis.14–17 Crry (complement receptor 1-related protein/gene y) is a rodent transmembrane protein with both DAF and MCP activities. Because Crry has MCP activity and is expressed in many mouse and rat tissues where MCP is absent,14, 18 the study of Crry bears relevance to understanding human MCP in health and disease. However, investigation of the physiological role of Crry in vivo has been hampered by the fact that global gene knockout of the Crry gene in mice is embryonically lethal.19 Nevertheless, previous studies have provided evidence that Crry plays a critical role in protecting the mouse kidney from AP complement attack.20–24
To circumvent the embryonic lethality phenotype of global Crry gene deficiency and directly assess the physiological role of Crry on PTECs, we used conditional gene targeting and selectively deleted Crry expression in mouse PTECs. We found that Crry deficiency from PTECs resulted in local C3 deposition on the cells and the presence of activated C3 fragments in plasma. Although no constitutive renal injury was observed, the mutant mice developed exacerbated tubular injury when subjected to renal IR stress. Importantly, blocking AP complement activation or terminal complement pathway by using an anti-properdin or anti-C5 mAb, respectively, prevented renal IR injury, suggesting that IR injury in the mutant mice was caused by increased sensitivity to AP complement attack. Our results provide direct evidence for an important role of Crry in protecting PTECs from AP complement injury during IR stress and suggest that defects in membrane complement regulators can be a latent risk factor for complement-mediated kidney pathologies.
Results
PTEC-specific Crry gene deletion in mice
We screened homozygous Crry floxed mice (Crryflox/flox) with or without Cre transgene expression by PCR analysis of tail DNA. Bands for WT or floxed Crry alleles were observed at approximately 970 bp and 1100 bp, respectively, while expression of the Cre transgene was indicated by a 500 bp PCR product (Fig 1A). Previous characterization of the PEPCK-Cre transgenic mouse established that Cre expression is restricted to the proximal tubules in the renal cortex and medulla and a subset of periportal hepatocytes.25 Cre-mediated deletion of exon 5 from the floxed Crry allele could be indicated by the presence of a 350 bp band. As shown in Fig 1B, mutant Crry allele was detected in the kidney, but not the spleen, of PEPCK-Cre+-Crryflox/flox mice, consistent with expected Cre expression driven by the PEPCK gene promoter.25 To confirm Crry gene deletion at the protein level, we first produced, purified and validated a rabbit anti-Crry SCR3/4 antibody. Fig 1C and D show that on Western blot this antibody reacted strongly with the recombinant Crry SCR3/4, and detected endogenous Crry protein in the kidney of WT but not Crry−/−C3−/− mice. By immunofluorescence, the same antibody detected abundant Crry expression in the glomeruli and basolateral surfaces of PTECs of WT but not Crry−/−C3−/− mice (Fig 1E), consistent with previously reported Crry expression pattern in the mouse kidney.22, 26, 27 We next used immunofluorescence to compare Crry expression in the kidneys of Cre−- and Cre+-Crryflox/flox mice by co-staining with megalin, a specific marker for PTECs.28, 29 While megalin and glomerular Crry expression were unchanged, the basolateral expression of Crry on PTECs normally observed in Cre (−) mice was markedly reduced, and in some areas was almost absent, in Cre (+) mice (Fig 1F and Supplemental Fig 1). These results confirmed Crry deletion from PTECs of the mutant mice.
Figure 1. Generation and confirmation of a proximal TEC-specific Crry knockout mouse.

(A) PCR genotyping of wild type (WT) and floxed Crry gene alleles using primers flanking exon 5 of the Crry gene. WT Crry alleles produced a 970bp product while the floxed allele produced an 1100bp product (upper panel). Mice positive for the Cre transgene (+) showed a PCR product of approximately 500bp (lower panel). (B) PCR confirmed the specific deletion of Crry allele in Cre+ kidney tissue (kid) by the presence of a 350 bp band that was not observed in spleen tissue (spl) from the same mouse. (C) SDS-PAGE shows the expression and purification of a His-tagged recombinant Crry SCR3-4 protein (in “elution 1” fraction) on Ni+ column. (D) Western blot showing that a rabbit anti-Crry (SCR3-4) antibody recognized recombinant Crry (SCR3-4) and endogenous Crry protein in the kidney homogenate from a WT mouse. As expected, no signal was detectable in the kidney homogenate of a Crry−/−C3−/− mouse (Crry/C3 DKO), validating the specificity of the antibody. (E) Immunofluorescence staining was also used to confirm specificity of rabbit anti-Crry (SCR3-4) antibody in tissues. Positive staining was observed in WT but not Crry−/−C3−/− mouse kidneys. Original magnification 200x, scale bar = 50 μm. (F) Co-staining of megalin (red color) and Crry (green) in Cre− (upper row) and Cre+ (lower row) mouse kidneys shows specificity of Crry deletion to PTECs. Megalin is expressed on the apical surface of PTECs and no difference in its expression was observed between Cre− and Cre+ mice. Crry is expressed in both the glomeruli and TECs of Cre− mice. No difference in glomerular Crry expression (arrowhead) was observed between Cre− and Cre+ mice. In contrast, Crry expression on PTECs of Cre+ mice was dramatically reduced, and nearly absent in some areas. Representative images from two different mice are shown for each genotype (Cre+ and Cre−). Original magnification 200x for each panel on left (scale bars = 50 μm); box indicates section of tissue shown in corresponding right panel at 5x greater magnification.
Spontaneous complement activation on PTECs of Crry mutant mice
Global Crry gene knockout is embryonically lethal19 and Crry−/− mice rescued by maternal complement inhibition displayed spontaneous plasma complement activation and consumption after birth.30, 31 To assess the impact of Crry deletion from PTECs on complement activation, we measured circulating levels of intact and activated C3 by ELISA and examined C3 deposition on PTECs. We found that deletion of Crry from PTECs had no discernible effect on plasma intact C3 levels but slightly increased activated C3 fragments in circulation (data not shown). Furthermore, immunofluorescence staining of C3 showed spontaneous C3 deposition on the basolateral surface of PTECs in Cre+-Crryflox/flox mice where Crry had been deleted (Fig 2), suggesting that there was local complement activation on these cells and some activated C3 fragments may have been released into the systemic circulation. Despite the clear evidence of increased complement activation on PTECs, however, we found no functional or histological signs of kidney injury in Cre+-Crryflox/flox mice up to 24 months of age (Fig 3 and data not shown). The Crry mutant mice had normal blood urea nitrogen (BUN) levels and showed no proteinuria (Fig 3A, B). PAS staining revealed normal kidney architecture with prominent brush borders in the proximal tubules in both the cortex and outer medulla (Fig 3C).
Figure 2. Effect of Crry deletion from PTECs on local complement activation.

Representative immunofluorescence pictures showing that Crry deletion from PTECs caused increased C3 deposition on the basolateral surface. In the Cre− mouse kidney, segmental linear staining of C3 was detectible along the basement membrane, possibly reflecting local C3 synthesis.47 In the Cre+ mouse kidney, thicker and ribbon-like C3 staining was seen on basolateral portions of the tubular cells. Images from three Cre− and four Cre+ mice are shown. Original magnification for top panels, 200x. Boxed areas are shown underneath at 2x greater magnification. All scale bars indicate 100 μm length.
Figure 3. Crry deletion from PTECs does not cause spontaneous renal injury.

Compared with Cre− mice, no abnormal albuminuria (A) or elevated BUN (B) was detected in Cre+ mice. (NS: no significance, n=8 mice per group, approximately 3 months of age). In both Cre− and Cre+ mice, PAS staining showed normal kidney architecture with prominent brush border in the proximal tubules of the cortex and outer medulla (C). Original magnification 200x. Inset panels show 2x higher magnification of the area indicated by arrow. Pictures in panel C are representative of 8 kidneys in each genotype examined. All scale bars = 50 μm.
Deficiency of Crry in PTECs increases sensitivity to renal IRI
We next subjected Cre+-Crryflox/flox mice and their Cre− littermates to renal IR challenge to determine if Crry deficiency from PTECs may render the mutant mice more susceptible to renal injury. Compared with 0h time point, BUN levels at 24h after reperfusion increased both in Cre−-Crryflox/flox (0h: 24.50 ±0.80 mg/dl vs. 24h: 45.19 ±5.31 mg/dl, p=0.0014) and Cre+-Crryflox/flox mice (0h: 24.29 ±0.60 mg/dl vs. 24h: 75.93 ±12.28 mg/dl, p=0.0007). Notably, at 24h after reperfusion, BUN levels in Cre+-Crryflox/flox mice were significantly higher than that in Cre−-Crryflox/flox mice (p<0.05) (Fig 4A). Increased kidney injury in Cre+-Crryflox/flox mice was corroborated by histological findings. At 1h post-reperfusion, kidney tubular injury was not seen in Cre−-Crryflox/flox mice, but cast formation was observed in some tubular lumen in the cortex and medulla of Cre+-Crryflox/flox mice (Fig 4B). The difference between Cre− and Cre+ mice became more pronounced at 24h after reperfusion. Compared with Cre−-Crryflox/flox mice, Cre+-Crryflox/flox mice had markedly more severe tubular injury with pronounced tubular necrosis, brush border disruption, cast formation, tubular dilatation and inflammatory cell infiltration (Fig 4B and Supplemental Fig 2). The significant increase in tubular injury in the Cre+-Crryflox/flox group was also confirmed by a semi-quantitative pathology grading (4.36±0.28 vs. 2.27±0.26, p<0.001) (Fig 4C). It is interesting that IR-induced renal injury in Cre+-Crryflox/flox mice was not correlated with more tubular C3 deposition at either 1h or 24 hr (Fig 5). In fact, tubular C3 staining in Cre+-Crryflox/flox mice at 24 h after IR was significantly decreased compared with that detected at 1 h or 0 h (before IR challenge) (Fig 5). Nevertheless, it is apparent by visual scoring or quantitative image analysis of fluorescence intensity (data not shown) that at all time points there was more tubular C3 deposition in PEPCK-Cre+-Crryflox/flox mice than in Cre− mice (Fig 5).
Figure 4. Crry deletion from PTECs renders mice more susceptible to renal IRI.

(A) While BUN was higher in both Cre− and Cre+ mice 24h post IR challenge compared with 0h, BUN in Cre+ mice at 24 hr was significantly higher than in Cre− mice (p<0.05, n=9 mice per group). (B) Representative histology of kidneys harvested at 1h and 24 h after renal IR challenge. PAS staining of Cre+ mice showed more severe tubular injury than Cre− mice both in the cortex and outer medulla. Arrows and arrowheads indicate tubules with necrotic debris and cast formation, respectively, and inflammatory infiltrate is denoted with asterisks. Images from one mouse at 1h and three mice at 24h per genotype are shown to illustrate range of injury severity. Original magnification, 200x. Scale bars = 100 μm. (C) Semi-quantitative analysis confirmed that tubular injury in Cre+ mice was significantly more severe than in Cre− mice (p<0.001, n=9 mice per group). Mice were approximately 3 months of age with body weight of 25–30g.
Figure 5. Tubular C3 deposition in Cre− and Cre+ mice after renal IRI challenge.
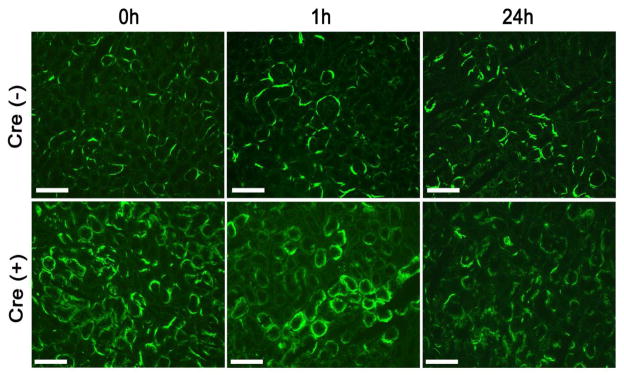
Tubular C3 deposition was more prominent in Cre+ mice than in Cre− mice at all time points examined. However, there was no significant difference between 0h and 1h or 24h after IR in Cre− mice. There was also no difference between 0h and 1h in Cre+ mice, but tubular C3 deposition at 24h after IR was significantly reduced in Cre+ mice compared with that at 0h or 1hr. Original magnification, 200x. Scale bars = 100 μm.
Exacerbated renal IRI in PTEC-specific Crry knockout mice correlates with increased mononuclear phagocyte infiltration and PTEC apoptosis
To assess and compare inflammatory infiltrate into the kidney after IR challenge, we stained mononuclear phagocytes by immunofluorescence with an F4/80 antibody. IR challenge caused a noticeable but only modest increase in F4/80+ mononuclear phagocyte infiltration in the kidneys of Cre−-Crryflox/flox mice at 24 hrs (0h: 12.20±0.8602, 1h: 12.60±1.435, 24h: 18.40±1.208) (Fig 6A). In comparison, infiltration of F4/80+ mononuclear phagocytes at 24 hr after IR treatment was much more striking in Cre+-Crryflox/flox mice (0h: 12.60±1.502, 1h: 16±0.707, 24h: 50.8±2.289) (Fig 6A). Increased infiltration of mononuclear phagocytes in the kidneys of Cre+-Crryflox/flox mice was unlikely caused by any intrinsic phenotype differences in these cells, as Crry expression on bone marrow cells of Cre− and Cre+ mice was the same (data not shown).
Figure 6. Deficiency of Crry from PTECs exacerbates IR-induced inflammation and tubular apoptosis.

(A) Representative tissue sections from kidneys harvested before (0h) and 1h or 24h after IR were stained for mononuclear phagocyte infiltration with an F4/80 antibody (red). Few F4/80+ cells were observed per high power field (hpf) in the Cre− mouse kidneys and showed only modest increase after IR. Although there was no significant difference between 0h and 1h in Cre+ mice, the number of mononuclear phagocytes in the medulla of Cre+ mice had dramatically increased by 24h post IR (p<0.001, n=5 mice per group). Scale bars = 20 μm. (B) Apoptotic cells in outer medulla of the kidney were identified by TUNEL assay and counted. Representative tissue sections from Cre- and Cre+ mice before IR (0h) and 24h after IR challenge are shown. Original magnification 400x. Insets show 2x higher magnification of the areas indicated by arrows. All scale bars = 25 μm. The number of TUNEL-positive cells had significantly increased by 24h in both in Cre− and Cre+ mice compared to 0h (p<0.05 and p<0.001, respectively), but there were still significantly more apoptotic cells in the Cre+ mouse kidney than in the Cre− mouse kidney (p<0.001, n=5 mice per group).
Apoptosis is also a common feature of inflammatory organ damage. Therefore we next determined the degree of apoptosis in the kidneys of IR challenged Cre− and Cre+ mice using the TUNEL assay. In both groups of mice, we observed increased numbers of apoptotic tubular cells at 24 h after IR compared with naïve mice (TUNEL-positive cells in Cre− mice: 6.4 ±0.8124 at 0h vs. 17.40 ±2.293 at 24 h, p<0.05; Cre+ mice: 8.200±1.281 at 0h vs. 66.00 ±4.301 at 24 h, p<0.001). The degree of apoptosis at 24h post-reperfusion was significantly greater in Cre+-Crryflox/flox mice compared with Cre−-Crryflox/flox mice (p<0.001) (Fig 6B), indicating that Cre+ mice had greater sensitivity to IR-induced tissue damage.
IR-induced renal injury in PEPCK-Cre+-Crryflox/flox mice is ameliorated by blocking alternative pathway or terminal pathway of complement activation
To confirm that exacerbated renal IR injury in PTEC-specific Crry knockout mice was caused by increased complement activation, we blocked the alternative pathway or terminal pathway of complement activation with an anti-properdin or anti-C5 mAb, respectively. Fig 7A shows that pretreatment with either anti-properdin or anti-C5 mAb significantly reduced BUN levels in Cre+-Crryflox/flox mice (p<0.001 for both groups, compared to control mAb). The reduction in BUN in the anti-complement mAb-treated mice correlated with significant improvement in renal pathology. In control mAb-treated Cre+-Crryflox/flox mice, both the cortex and medulla architecture was distorted and tubules were filled with hyaline casts and sloughed cells/necrotic debris with inflammatory cells detected prominently in the tubular interstitium (Fig 7B and Supplemental Fig 3). On the other hand, mice pretreated with either anti-C5 mAb or anti-properdin mAb had little necrosis or cast formation in the cortex and only focal necrotic and cast areas in the medulla (Fig 7B and Supplemental Fig 3). Semi-quantitative scoring of tubular damage confirmed the protective effect of anti-C5 and anti-properdin mAbs (Fig 7C). As expected, anti-properdin but not anti-C5 mAb significantly reduced the intensity of tubular C3 staining (Fig 8A, B). Furthermore, compared with control mAb-treated mice, macrophage infiltration in the kidney at 24 hr was significantly reduced in both anti-C5 and anti-properdin mAb-pretreated mice (control mAb: 53.60±1.965; anti-C5: 22.00±2.168; anti-properdin: 19.60±2.249; p<0.001 vs. control mAb) (Fig 8C, D).
Figure 7. Complement inhibition ameliorates renal IRI in PTEC-specific Crry knockout mice.

(A) Compared with control mAb (Control Ab), pre-treatment of Cre+-Crryflox/flox mice (2 h before IR) with anti-properdin mAb (Properdin Ab) or anti-C5 mAb (C5 Ab) significantly reduced BUN levels 24h post-IR injury (p<0.001, n=5–6 mice per group). (B) Representative PAS-stained kidney sections from mAb pre-treated mice. In mice pre-treated with the control mAb, the cortex and medulla architecture was greatly distorted and tubules were filled with hyaline casts (arrowhead) and sloughed cells/necrotic debris (arrow), and inflammatory infiltrate was prominent within the tubular interstitium (asterisk). In mice pre-treated with either anti-C5 mAb or anti-properdin mAb, there was little necrosis or cast formation in the cortex and only focal necrotic areas and casts were visible in the medulla. Original magnification 200x. Scale bars = 100 μm. (C) Semi-quantitative scoring of tubular injury showed significant (p<0.05, n=5–6 mice per group) protection from renal IRI by C5 mAb or Properdin mAb pre-treatment compared with pre-treatment with the control mAb.
Figure 8. Effects of anti-properdin and anti-C5 mAbs on tubular C3 deposition and mononuclear phagocyte infiltration in response to IR challenge in Crry mutant mice.

(A) Compared with control mAb treatment, pre-treatment of Cre+-Crryflox/flox mice with anti-properdin mAb significantly reduced tubular C3 staining at 24h after IR challenge while anti-C5 mAb treated kidneys were unaffected. Original magnification 200x. Scale bars = 100 μm. (B) Semi-quantitative image analysis of C3 deposition confirmed tubular C3 staining intensity to be significantly reduced in anti-properdin mAb-treated mice compared to control or anti-C5 mAb treatment groups (p<0.001, n=3 mice per group). (C) Immunofluorescence staining showed mononuclear phagocyte infiltration in the kidney of control mAb-treated Cre+-Crryflox/flox mice at 24h after IR was largely prevented by anti-properdin mAb or anti-C5 mAb (red=F4/80, blue=DAPI). (D) Quantification of mononuclear phagocyte infiltration. Compared with control mAb-treated mice, the number of mononuclear phagocytes per high power field (hpf) in the kidneys of anti-C5 or anti-properdin mAb-treated mice was significantly (p<0.001, n=5 mice per group) reduced. Original magnification 400x. Scale bars = 20 μm.
Discussion
In this study, we have used a conditional gene targeting approach to evaluate the physiological role of Crry on PTECs of the kidney. Crry is a key membrane complement regulator in rodent species and its study bears relevance to understanding the role of human MCP in health and disease. It is the only membrane-bound C3 regulator expressed on PTECs within the mouse kidney.3, 11, 27 Previous studies have shown that Crry is localized to the basolateral aspect of PTECs and loss of polarity in Crry expression on these cells preceded AP complement activation in a renal IR injury model.27 Inhibition of Crry in cultured primary murine PTECs also rendered these cells susceptible to AP complement attack upon exposure to mouse serum.22 Similar sensitivity to AP complement activation was observed when PTECs from Crry and factor B double knockout mice were treated with normal mouse serum.22 In other studies, antibody-mediated neutralization of Crry in rat kidneys allowed C3 activation in the tubules and peri-vascular capillaries and transient glomerular C3 staining.32 Crry−/−/C3−/− mouse kidneys developed remarkable inflammatory cell infiltration, tubular damage, and interstitial fibrosis when transplanted into syngeneic wild-type mice, a phenomenon that was not observed in transplanted wild-type kidneys.20
Despite these earlier evidences of Crry function in the kidney, a direct evaluation of the in vivo role of Crry on tubular epithelial cells has not been possible until now due to the embryonic lethality phenotype of global Crry deletion19. In two recent studies, live Crry−/− mice were obtained by inhibiting the complement system during gestation30, 31. However, such mice were observed to have systemic complement activation and depletion, thus also preventing analysis of Crry function in vivo in the context of normal complement activity.30, 31 In the present study, we circumvented these limitations by using the Cre/loxP system to selectively inactivate the Crry gene in renal PTECs. We confirmed Crry gene deletion in PTECs and established that Cre+ mutant mice had normal plasma C3, indicating that no systemic complement depletion and secondary complement insufficiency has occurred. The latter finding was consistent with data from two other tissue-specific Crry gene knockout studies. Using the same Crryflox/flox mouse strain crossed to Cre-transgenic mice, we found in earlier experiments that Crry deletion from mouse T cells or platelets did not affect systemic complement levels and activity.33, 34
By immunofluorescence staining, we detected significant C3 deposition on PTECs of naïve Cre+ mice. This suggested that lack of Crry on these cells rendered them susceptible to spontaneous complement attack. While the degree of complement activation on PTECs did not significantly affect plasma levels of intact C3, we did observe increased C3b fragments in the plasma, thus providing an independent marker of spontaneous complement activation in the mutant mice. Previous studies of Crry−/− mice likewise showed the presence of activated C3b fragments in the circulation,30, 31 presumably reflecting an excess of activated C3 fragments from uncontrolled surface convertase activity that did not immediately link covalently to cell surface hydroxyl or amine groups. Surprisingly, despite clear signs of complement activation on PTECs, we did not detect any appreciable renal injury in either young or aged Cre+-Crryflox/flox mice by functional (BUN) or histopathological evaluation. However, when Cre− and Cre+ mice were subjected to IR challenge, we found that Cre+ mice incurred significantly more renal injury than Cre− mice. We noted considerable spread in the BUN levels in challenged Cre+-Crryflox/flox mice, which may reflect variations in Crry deletion efficiency in different animals. Of interest, increased sensitivity to renal IRI in Cre+ mice was not correlated with more intense tubular C3 staining at 1h or 24h after reperfusion. In fact, tubular C3 staining at 24h after reperfusion in the kidneys of Cre+ mice was significantly reduced when compared with that in naïve mice without IR challenge, potentially a consequence of cellular apoptosis and necrosis observed by this time point which might have limited local complement availability. A similar finding of reduced tubular C3 staining at 24 h after reperfusion was observed previously in the DAF−/−CD59−/− mouse model of renal IRI.35 Thus, depending on the disease model and time course, C3 deposition on tissues may not always be a meaningful marker of complement injury.
Although not directly correlated with tubular C3 deposition, exacerbated renal IRI in Cre+-Crryflox/flox mice was clearly mediated by complement activation, as anti-properdin or anti-C5 mAb ameliorated renal injury. IR challenge may augment complement activation, particularly the terminal pathway, on tubular cells by impairing the activity of factor H (fH). In addition to Crry, previous studies have suggested that the fluid phase complement regulator fH also plays a role in protecting tubular cells of the kidney.22, 36 By interacting with surface deposited C3b and host cell-specific glycosaminoglycans (GAG), fH can act as a potent inhibitor of AP complement activation on host cells. Tubular epithelial cells of the kidney are known to express GAGs,37 which together with spontaneously deposited C3b may attract fH binding to PTECs in Cre+-Crryflox/flox mice. Prevention by fH of excessive AP and terminal pathway complement activation on PTECs would explain the lack of F4/80+ mononuclear phagocyte infiltration and associated renal injury in naïve Cre+ mice despite an appreciable degree of AP complement activation on Crry-deficient PTECs. During IR challenge, tubular cell membrane remodeling may lead to the loss or reduction of GAGs and other fH ligands and this may decrease fH binding and protection, resulting in unchecked complement-dependent renal injury.
The finding that anti-properdin and anti-C5 mAbs are effective in ameliorating renal IRI in Crry mutant mice is consistent with our previous data on IR-challenged DAF/CD59 knockout mice.38 It however contrasted with the finding from recent C3 glomerulopathy models where properdin deficiency was shown to exacerbate kidney injury.39, 40 This difference may relate to differential requirement of properdin in AP complement activation on certain host cells (e.g. endothelial and renal tubular cells) versus other cellular structures (e.g. the GBM) or in the fluid phase.39, 41 Thus, although anti-properdin therapy may be ineffective or counterproductive in disorders involving fluid phase complement activation, it is expected to benefit other, cell surface-driven AP complement-mediated pathologies. Our data here showed that one such setting where prophylactic anti-properdin or anti-C5 therapy can be considered could be transplantation-related IRI.
In summary, this study has provided direct evidence for a role of Crry in controlling spontaneous AP complement activation on renal tubular epithelial cells. It also demonstrates that a defect in AP complement control on target cells may not always produce a spontaneous pathological phenotype but may instead pose an increased risk for injury in settings where complement regulation may be further compromised. This relates to the partial penetrance of complement gene defects observed in human patients. Not all individuals carrying fH or MCP mutations develop complement-mediated disorders and those who do often have their disease onset or flare-ups triggered by potentially complement-activating events such as infection, surgery or trauma.42, 43 In addition, our results showing that renal IRI in Cre+-Crryflox/flox mice was correlated with F4/80+ mononuclear phagocyte infiltration and PTEC apoptosis provide mechanistic insight on the pathogenesis of this condition. Finally, the finding that renal IRI was ameliorated by anti-properdin and anti-C5 mAbs suggests that these treatments may benefit human patients in settings of complement-mediated IRI.
Materials and methods
Generation of PTEC-specific Crry knockout mice
To generate PTEC-specific Crry knockout (KO) mice, Crryflox/+ mice on a mixed 129/C57BL6 background33 were crossed with a PEPCK-Cre+ transgenic mouse (kindly provided by Dr. Volker Haase, Vanderbilt University). PEPCK-Cre+ transgenic mice were generated by using a modified portion of the phosphoenolpyruvate carboxykinase (PEPCK) gene promoter to selectively drive Cre expression in PTECs, although Cre expression was also noted in the medulla and a subset of hepatocytes.25 The resulting PEPCK-Cre+-Crryflox/+ mice were then intercrossed to obtain PEPCK-Cre+-Crryflox/flox and Crryflox/flox mice as breeders to produce experimental mice. In all experiments, Cre-negative littermates were used as controls for PEPCK-Cre+-Crryflox/flox mice. Mouse genotyping was performed by PCR of tail and/or kidney DNAs. For the WT, floxed and deleted Crry allele, a fragment of 970bp, 1100bp and 350 bp, respectively, was expected with the use of the following pair of primers, TX-1 (5′-CAGAGTAATCTACAGTTTCACC-3′) and E5S2 (5′-GTTCACTGTATTCCCTCATCCAGA-3′). PEPCK-Cre transgenic mice were identified using a pair of Cre-specific primers, 5′-ATTCTCCCACCGTCAGTACG-3′ and 5′-CGTTTTCTGAGCATACCTGGA-3′. Experiments were conducted by following established guidelines for animal care and all protocols were approved by the appropriate institutional committees.
Generation and purification of rabbit anti-mouse Crry SCR3/4 antibody
The targeting strategy for Crry was to delete exon 5 which encodes short consensus repeat (SCRs) 3 and 4 critical for Crry function.19, 44, 45 Because a small amount of mutant, non-functional Crry protein was still made in the Crry gene targeted mice,33 we prepared a SCR3/4-specific rabbit polyclonal antibody to distinguish WT and mutant Crry proteins. We expressed and purified a 6xHis-tagged mouse Crry SCR3/4 fragment using the pCAGGS vector and HEK293 cells as described previously.34 This recombinant Crry protein was then used as an antigen to produce a polyclonal antibody in rabbits by Cocalico Biologicals, Inc (Reamstown, PA). Total IgGs were purified from rabbit serum by Protein A affinity column and Crry SCR3/4-specific IgGs were purified on a second affinity column constructed by conjugating recombinant Crry SCR3/4 protein onto CNBr-Sepharose beads (Amersham). The specificity of this antibody was confirmed by Western blot and immunostaining experiments.
C3 ELISA assays
ELISA assays to measure intact and activated C3 levels in the mouse plasma were performed as described previously.39
Functional evaluation of renal injury
Urinary albumin was quantified using a mouse albumin ELISA kit according to manufacturer’s instructions (Bethyl Laboratories). Urine samples were collected in metabolic cages for 16h and volumes were recorded. Total urinary albumin was determined by multiplying albumin concentration (determined by ELISA) by total urine volume of 16h. Blood urea nitrogen (BUN) levels were measured using urea nitrogen reagents (Sigma-Aldrich) as described previously.35
Induction of renal ischemia reperfusion injury
Renal ischemia reperfusion was performed as described previously.35 In brief, both renal pedicles were clamped for 25 min using microaneurysm clamps, followed by reperfusion for 1 or 24 hours. Mice were sacrificed at the end of the reperfusion experiment and kidneys were harvested for histological analysis. Blood samples were collected before and after IR surgery.
Renal pathology grading
Kidneys were fixed in 10% formalin-PBS before processing and paraffin embedding. Samples were cut in 4μm sections and stained with periodic acid Schiff (PAS) reagent. As described previously35, tubular injury was scored by estimating the percentage of tubules in the cortex and in the outer medulla that showed epithelial necrosis or had necrotic debris or cast as follows: 0, none; 1+, <10%; 2+, 10–25%; 3+, 26–45%; 4+, 46–75%; 5+, >75%. Ten viewing fields, randomly selected from the cortex and outer medulla on each slide section, were examined at 200x magnification.
Immunofluorescence for C3 and F4/80
Cryostat sections (4μm) of frozen kidneys were fixed with ice-cold acetone for 10min, and blocked with 10% goat serum to decrease background staining. FITC-conjugated goat anti-mouse C3 Ab (4.0 mg/ml, MP Biomedicals) was used directly at 1:500 dilution. Under ×200 magnification, 10 viewing fields from sections of each animal were photographed. Areas of positive staining were highlighted and the fluorescence intensity of C3 was determined using the plugin “Measure particles” of ImageJ software. F4/80-positive mononuclear phagocytes were visualized by staining with a rat anti-mouse F4/80 Ab (1.0 mg/ml, AbD seroTEC) used at 1:50 dilution followed by Alexa Fluor 555-goat anti-rat IgG (2.0 mg/ml, Invitrogen) used at 1:2000 dilution. Under 400x magnification, F4/80+ cells were counted by examining ten viewing fields randomly selected from the outer medulla on each slide.
TUNEL Assay for Apoptosis
Deparaffinized and rehydrated tissue sections were prepared according to standard protocols. Slides were incubated with proteinase K for 20 minutes at RT. TUNEL labeling was carried out using an in situ cell death detection kit (Roche, Indianapolis, IN) according to the manufacturer’s instructions, and color was developed using the diaminobenzidine substrate kit (Vector, Burlingame, CA). The number of apoptotic cells in the outer medulla was counted from ten different fields at 400x magnification for each sample and were averaged.
Administration of anti-properdin and anti-C5 mAbs
Mice were pretreated with neutralizing monoclonal anti-C5 antibody BB5.1, anti-properdin antibody 14E138 or isotype control mouse IgG1, MOPC-31C (all at a dose of 1mg/mouse) by intraperitoneal injection 2 hours before the induction of ischemia. Selection of mAbs dosages was based on previously published studies.38, 46
Statistical analysis
Statistical comparisons were performed using GraphPad Prism 4.0 software. All data are reported as the mean ± SEM. The difference between two groups was calculated using unpaired t-test for normally distributed data. For data with nonparametric distributions, Mann-Whitney test was applied for two groups. For multiple group comparisons, one-way ANOVA with a Tukey test was used. A p value of less than 0.05 was considered significant.
Supplementary Material
Acknowledgments
This study was supported by NIH grants AI044970, AI049344, AI085596 and GM092108. We thank Drs David Gasser and Volker Haase for providing the PEPCK-Cre transgenic mice.
Footnotes
Disclosure statement
The authors have nothing to disclose.
References
- 1.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 3.Lesher AM, Song WC. Review: Complement and its regulatory proteins in kidney diseases. Nephrology (Carlton) 2010;15:663–675. doi: 10.1111/j.1440-1797.2010.01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sacks S, Zhou W. New boundaries for complement in renal disease. J Am Soc Nephrol. 2008;19:1865–1869. doi: 10.1681/ASN.2007101121. [DOI] [PubMed] [Google Scholar]
- 5.Brown KM, Sacks SH, Sheerin NS. Mechanisms of disease: the complement system in renal injury--new ways of looking at an old foe. Nat Clin Pract Nephrol. 2007;3:277–286. doi: 10.1038/ncpneph0465. [DOI] [PubMed] [Google Scholar]
- 6.Pickering M, Cook HT. Complement and glomerular disease: new insights. Curr Opin Nephrol Hypertens. 2011;20:271–277. doi: 10.1097/MNH.0b013e328345848b. [DOI] [PubMed] [Google Scholar]
- 7.Thurman JM. Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol. 2007;123:7–13. doi: 10.1016/j.clim.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lien YH, Lai LW, Silva AL. Pathogenesis of renal ischemia/reperfusion injury: lessons from knockout mice. Life Sci. 2003;74:543–552. doi: 10.1016/j.lfs.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Arumugam TV, Shiels IA, Woodruff TM, et al. The role of the complement system in ischemia-reperfusion injury. Shock. 2004;21:401–409. doi: 10.1097/00024382-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Chowdhury P, Zhou W, Sacks SH. Complement in renal transplantation. Nephron Clin Pract. 2003;95:c3–8. doi: 10.1159/000073012. [DOI] [PubMed] [Google Scholar]
- 11.Puri TS, Quigg RJ. The many effects of complement C3- and C5-binding proteins in renal injury. Semin Nephrol. 2007;27:321–337. doi: 10.1016/j.semnephrol.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Ichida S, Yuzawa Y, Okada H, et al. Localization of the complement regulatory proteins in the normal human kidney. Kidney Int. 1994;46:89–96. doi: 10.1038/ki.1994.247. [DOI] [PubMed] [Google Scholar]
- 13.Seya T, Turner JR, Atkinson JP. Purification and characterization of a membrane protein (gp45–70) that is a cofactor for cleavage of C3b and C4b. J Exp Med. 1986;163:837–855. doi: 10.1084/jem.163.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Miwa T, Song WC. Membrane complement regulatory proteins: insight from animal studies and relevance to human diseases. Int Immunopharmacol. 2001;1:445–459. doi: 10.1016/s1567-5769(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 16.Mizuno M, Harris CL, Suzuki N, et al. Expression of CD46 in developing rat spermatozoa: ultrastructural localization and utility as a marker of the various stages of the seminiferous tubuli. Biol Reprod. 2005;72:908–915. doi: 10.1095/biolreprod.104.035485. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno M, Harris CL, Johnson PM, et al. Rat membrane cofactor protein (MCP; CD46) is expressed only in the acrosome of developing and mature spermatozoa and mediates binding to immobilized activated C3. Biol Reprod. 2004;71:1374–1383. doi: 10.1095/biolreprod.104.030114. [DOI] [PubMed] [Google Scholar]
- 18.Holers VM, Kinoshita T, Molina H. The evolution of mouse and human complement C3-binding proteins: divergence of form but conservation of function. Immunol Today. 1992;13:231–236. doi: 10.1016/0167-5699(92)90160-9. [DOI] [PubMed] [Google Scholar]
- 19.Xu C, Mao D, Holers VM, et al. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- 20.Bao L, Wang Y, Chang A, et al. Unrestricted C3 activation occurs in Crry-deficient kidneys and rapidly leads to chronic renal failure. J Am Soc Nephrol. 2007;18:811–822. doi: 10.1681/ASN.2006101176. [DOI] [PubMed] [Google Scholar]
- 21.Miwa T, Zhou L, Tudoran R, et al. DAF/Crry double deficiency in mice exacerbates nephrotoxic serum-induced proteinuria despite markedly reduced systemic complement activity. Mol Immunol. 2007;44:139–146. doi: 10.1016/j.molimm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 22.Renner B, Coleman K, Goldberg R, et al. The complement inhibitors Crry and factor H are critical for preventing autologous complement activation on renal tubular epithelial cells. J Immunol. 2010;185:3086–3094. doi: 10.4049/jimmunol.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe M, Morita Y, Mizuno M, et al. CD59 protects rat kidney from complement mediated injury in collaboration with crry. Kidney Int. 2000;58:1569–1579. doi: 10.1046/j.1523-1755.2000.00318.x. [DOI] [PubMed] [Google Scholar]
- 24.Hori Y, Yamada K, Hanafusa N, et al. Crry, a complement regulatory protein, modulates renal interstitial disease induced by proteinuria. Kidney Int. 1999;56:2096–2106. doi: 10.1046/j.1523-1755.1999.00765.x. [DOI] [PubMed] [Google Scholar]
- 25.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B, Sallee C, Dehoff M, et al. Mouse Crry/p65. Characterization of monoclonal antibodies and the tissue distribution of a functional homologue of human MCP and DAF. J Immunol. 1993;151:4295–4305. [PubMed] [Google Scholar]
- 27.Thurman JM, Ljubanovia D, Royer PA, et al. Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J Clin Invest. 2006;116:357. doi: 10.1172/JCI24521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christensen EI, Gliemann J, Moestrup SK. Renal tubule gp330 is a calcium binding receptor for endocytic uptake of protein. J Histochem Cytochem. 1992;40:1481–1490. doi: 10.1177/40.10.1382088. [DOI] [PubMed] [Google Scholar]
- 29.Lundgren S, Carling T, Hjalm G, et al. Tissue distribution of human gp330/megalin, a putative Ca(2+)-sensing protein. J Histochem Cytochem. 1997;45:383–392. doi: 10.1177/002215549704500306. [DOI] [PubMed] [Google Scholar]
- 30.Ruseva MM, Hughes TR, Donev RM, et al. Crry deficiency in complement sufficient mice: C3 consumption occurs without associated renal injury. Mol Immunol. 2009;46:803–811. doi: 10.1016/j.molimm.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Spitzer D, Mao D, et al. Membrane protein Crry maintains homeostasis of the complement system. J Immunol. 2008;181:2732–2740. doi: 10.4049/jimmunol.181.4.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nomura A, Nishikawa K, Yuzawa Y, et al. Tubulointerstitial injury induced in rats by a monoclonal antibody that inhibits function of a membrane inhibitor of complement. J Clin Invest. 1995;96:2348–2356. doi: 10.1172/JCI118291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miwa T, Zhou L, Kimura Y, et al. Complement-dependent T cell lymphopenia caused by thymocyte deletion of the membrane complement regulator Crry. Blood. 2009;113:2684–2694. doi: 10.1182/blood-2008-05-157966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barata L, Miwa T, Sato S, et al. Deletion of Crry and DAF on murine platelets stimulates thrombopoiesis and increases factor H-dependent resistance of peripheral platelets to complement attack. J Immunol. 2013;190:2886–2895. doi: 10.4049/jimmunol.1202536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada K, Miwa T, Liu J, et al. Critical protection from renal ischemia reperfusion injury by CD55 and CD59. J Immunol. 2004;172:3869–3875. doi: 10.4049/jimmunol.172.6.3869. [DOI] [PubMed] [Google Scholar]
- 36.Renner B, Ferreira VP, Cortes C, et al. Binding of factor H to tubular epithelial cells limits interstitial complement activation in ischemic injury. Kidney Int. 2011;80:165–173. doi: 10.1038/ki.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaferani A, Vives RR, van der Pol P, et al. Factor h and properdin recognize different epitopes on renal tubular epithelial heparan sulfate. J Biol Chem. 2012;287:31471–31481. doi: 10.1074/jbc.M112.380386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miwa T, Sato S, Gullipalli D, et al. Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol. 2013;190:3552–3559. doi: 10.4049/jimmunol.1202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesher AM, Zhou L, Kimura Y, et al. Combination of factor H mutation and properdin deficiency causes severe C3 glomerulopathy. J Am Soc Nephrol. 2013;24:53–65. doi: 10.1681/ASN.2012060570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruseva MM, Vernon KA, Lesher AM, et al. Loss of properdin exacerbates C3 glomerulopathy resulting from factor H deficiency. J Am Soc Nephrol. 2013;24:43–52. doi: 10.1681/ASN.2012060571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol. 2013;56:191–198. doi: 10.1016/j.molimm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703–712. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- 43.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–1859. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hepburn NJ, Chamberlain-Banoub JL, Williams AS, et al. Prevention of experimental autoimmune myasthenia gravis by rat Crry-Ig: A model agent for long-term complement inhibition in vivo. Mol Immunol. 2008;45:395–405. doi: 10.1016/j.molimm.2007.06.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paul MS, Aegerter M, Cepek K, et al. The murine complement receptor gene family. III. The genomic and transcriptional complexity of the Crry and Crry-ps genes. J Immunol. 1990;144:1988–1996. [PubMed] [Google Scholar]
- 46.De Vries B, Matthijsen RA, Wolfs TG, et al. Inhibition of complement factor C5 protects against renal ischemia-reperfusion injury: inhibition of late apoptosis and inflammation. Transplantation. 2003;75:375–382. doi: 10.1097/01.TP.0000044455.05584.2A. [DOI] [PubMed] [Google Scholar]
- 47.Farrar CA, Zhou W, Lin T, et al. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J. 2006;20:217–226. doi: 10.1096/fj.05-4747com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.