

Abstract
Understanding the mechanisms that sustain pluripotency in human embryonic stem cells (hESCs) is an active area of research that may prove useful in regenerative medicine and will provide fundamental information relevant to development and cancer. hESCs and cancer cells share the unique ability to proliferate indefinitely and rapidly. Because the protein survivin is uniquely overexpressed in virtually all human cancers and in hESCs, we sought to investigate its role in supporting the distinctive capabilities of these cell types. Results presented here suggest that survivin contributes to the maintenance of pluripotency and that post-transcriptional control of survivin isoform expression is selectively regulated by microRNAs. miR-203 has been extensively studied in human tumors, but has not been characterized in hESCs. We show that miR-203 expression and activity is consistent with the expression and subcellular localization of survivin isoforms that in turn modulate expression of the Oct4 and Nanog transcription factors to sustain pluripotency. This study contributes to understanding of the complex regulatory mechanisms that govern whether hESCs proliferate or commit to lineages.
Keywords: Survivin, miRNA-203, ΔEx3 survivin isoform, Pluripotency, Embyonic stem cells
INTRODUCTION
Pluripotent human embryonic stem cells (hESCs) and cancer cells both have the ability to rapidly and continuously proliferate, including in harsh environments. In addition, they share common transcriptional regulatory networks (Kim et al., 2010) which may contribute to their rapid, unrestricted, proliferative phenotype. In hESCs, the cell cycle is characterized by a short G1 phase, which promotes rapid proliferation and is tightly regulated (reviewed in (Kapinas et al., 2013). Given the similarities between hESCs and cancer cells, understanding the mechanisms that control proliferation and differentiation in hESCs is instructive for identifying potential molecular targets for cancer treatment.
Survivin is abundantly expressed in cancer, embryos, and hESCs, but not in differentiated cells
In this context, survivin protein (BIRC5) can be particularly informative because it exhibits high expression levels almost exclusively in hESCs and cancer cells; it is a member of the baculovirus inhibitor of apoptosis containing repeat (BIRC) family of proteins (Ambrosini et al., 1997; Vaux and Silke, 2005). To date, survivin has been largely studied in the context of cancer, where it is involved in control of mitosis, apoptosis regulation, and stress responses. Overexpression of survivin is a hallmark of virtually every human tumor, and is prognostic for poor outcomes and disease recurrence (Altieri, 2013). Survivin expression is nearly absent from normal differentiated cells, but is high in proliferative areas of normal tissues (Ambrosini et al., 1997; Conway et al., 2003; Fukuda and Pelus, 2002; Pennartz et al., 2004).
Survivin is also abundantly expressed in embryonic tissues and hESCs (Adida et al., 1998; Filion et al., 2009). In hESCs, survivin and its two major splice variants exhibit differential cellular localization, and survivin inhibition using shRNAs results in decreased levels of OCT4 and NANOG pluripotency markers (Mull et al., 2014). These results, in the context of earlier results showing that survivin is downregulated during embryoid body (EB) formation (Blum et al., 2009), suggest that survivin and/or its variants have a role in maintaining pluripotency in hESCs. Homozygous knockout of the survivin gene in mice results in embryonic lethality (Conway et al., 2002; Uren et al., 2000) with embryos exhibiting morphological abnormalities as early as embryonic day 2.5. Note that, since the blastocyst develops on embryonic day 5 in mice and ESCs are derived from the inner cell mass of the blastocyst, this survivin-null embryo lethality suggests that survivin expression is necessary for, and precedes, the development of ESCs.
Survivin function is correlated to its cellular location
Five survivin isoforms have been identified. Most of what is known about the expression, localization, and functions of survivin isoforms comes from studies conducted in cancer cell lines. For example, in HeLa cells, 80% of the survivin pool is cytoplasmic and 20% is nuclear. Cytoplasmic and nuclear survivin are immunochemically distinct, independently modulated during cell cycle progression, and have different binding partners (Fortugno et al., 2002). Numerous studies propose that the functions of survivin isoforms in cancer are dependent on the subcellular localization (Colnaghi et al., 2006; Fortugno et al., 2002; Knauer et al., 2007; Mahotka et al., 1999). Isoforms with a nuclear export signal (NES) are actively transported to the cytoplasm by Crm1. Canonical survivin and survivin-2B contain NESs and are primarily expressed in the cytoplasm (Colnaghi et al., 2006; Mahotka et al., 2002). In contrast, the ΔEx3 isoform lacks a specific NES and is localized in the nucleus (Mahotka et al., 2002).
Overall the data suggest that, in the nucleus, survivin promotes mitosis and in the cytoplasm, it prevents apoptosis. It has been observed that nuclear survivin associates with a protein complex at centromeres and is necessary for mitosis, whereas cytoplasmic survivin prevents apoptosis in most cases. Canonical survivin (cytoplasmic) and the ΔEx3 isoform (nuclear) have typically been shown to prevent apoptosis. In contrast, survivin-2B typically increases apoptosis (Mahotka et al., 1999; Zhu et al., 2004). Notably, selective overexpression of survivin isoforms in different types of cancers has been observed (Altieri, 2013; Coumar et al., 2013).
Although survivin isoforms have been intensely studied in cancer, little is known about the expression, localization, and functions of survivin isoforms in hESCs. However a recent report confirms that all survivin isoforms are more highly expressed in hESCs than in differentiated cell types, with canonical survivin showing the highest expression levels (Mull et al., 2014).
Tight control of survivin expression could be facilitated by miRNAs in hESCs
In cancer cells, regulation of survivin expression is complex and occurs at the transcriptional, post-transcriptional, and post-translational levels (Altieri, 2013; Coumar et al., 2013). This multimodal control of expression is likely important in embryonic development, which requires rapid, tight regulation of survivin expression. Survivin RNA is very stable (Donahue et al., 2011; Ezponda et al., 2010; Vaira et al., 2007), therefore transcriptional regulation, though it plays a key role, is insufficient for the rapid survivin down-regulation required during critical stages of development.
Previous studies have shown that microRNAs are critical for the maintenance of pluripotency, differentiation, and function of hESCs (Altieri, 2003; Lian et al., 2012; Melton and Blelloch, 2010). Recent work documents that microRNA-203 (miR-203) post-transcriptionally regulates survivin in cancer cell lines, suppressing cell proliferation and suggesting that, in some cancer cells, miR-203 acts as an anti-oncomir (Wang et al., 2012; Xu et al., 2013). In this study, we examined whether survivin isoform expression in hESCs is likewise regulated by miR-203. In cancer, miR-203 expression is typically lost or downregulated (Ju et al., 2014; Taube et al., 2013; Wang et al., 2012; Zhao et al., 2013). One study found that a miR-203 feedback loop regulates the epithelial to mesenchymal transition, which contributes to epithelial cell plasticity during differentiation and tumor progression (Moes et al., 2012). Further, miR-203 expression is upregulated during mesenchymal stem cell differentiation (Nissan et al., 2011), suggesting a role for miR-203 in the normal differentiation process of adult stem cells. Given the shared properties of hESCs and tumor cells, and the reported role of miR-203 in differentiation, we hypothesized that miR-203 could functionally contribute to hESC differentiation. Consistent with this hypothesis, miR-203 targets survivin in cancers that include breast, pancreatic, prostate, and laryngeal carcinoma (Bian et al., 2012; Saini et al., 2011; Wang et al., 2012; Xu et al., 2013).
Here, we demonstrate that miR-203 regulates the expression of the ΔEx3 survivin isoform and that survivin knockdown decreases levels of pluripotency markers. Taken together, these findings suggest that miR-203 selectively regulates survivin isoform expression to sustain pluripotency of hESCs. Our results support a novel mechanism for the regulation of hESC pluripotency.
METHODS
Cell culture
H9 human embryonic stem cells (hESCs) were obtained from WiCell (Madison, WI). They were grown on Matrigel (BD Biosciences, San Jose, CA) and maintained in pluripotent conditions in mTeSR1 (Stem Cell Technologies, Vancouver, BC, Canada) at 37°C and 5% CO2, according to WiCell protocols.
For in vitro differentiation, H9 cells were subject to differentiation medium approximately 2 days post-plating (30% confluence). Differentiation medium consisted of DMEM/F12 (I :I), 1% non-essential amino acids (NEAA), 2 mM L-glutamine, 0.1 mM beta-mercaptoethanol, and 1 μM all-trans retinoic acid (RA, all from Sigma-Aldrich, St. Louis, MO).
Constructs
BIRC5 isoform 1 (canonical survivin, NM_001168.2) and BIRC5 isoform 2 (survivin-ΔEx3, NM_001012270.l) 3′ untranslated regions (UTRs) were amplified by PCR, using H9 genomic DNA as a template. UTR fragments were subcloned into the cytomegalovirus promoter-luciferase reporter, pMIR-REPORT (Ambion, Austin, TX) using Mlu1 and Sac1 restriction enzyme (New England Biolabs, Ipswich, MA) sites.
Transfections
H9 cells were transiently transfected 2 days post-plating (30% confluence) in 24-well cell culture plates. X-tremeGene siRNA transfection reagent (Roche, Nutley, NJ) 5 μL per well was used to co-transfect luciferase-3′UTR constructs and a constitutively active Renilla luciferase construct (Promega, Madison, WI) as a control for transfection efficiency. H9 cells were co-transfected with luciferase-3′UTR constructs, Renilla luciferase construct, and 100 nM Anti-miR miRNA inhibitors (Ambion) or 50 nM LNA inhibitors (Exiqon, Woburn, MA) using 6 μL X-tremeGene siRNA transfection reagent per well. 24 h post-transfection, cultures were either maintained in pluripotent conditions (mTeSR1 medium) or in differentiation conditions (RA medium) for 24 h. Cultures were harvested in 1X Reporter Lysis Buffer (Promega), according to the manufacturer’s instructions. All transfection experiments were performed at least 6 times, using n=3–4 for each experiment. Equal aliquots of cell lysate were used to determine luciferase activity (Dual Luciferase Assay System, Promega). To control for transfection efficiency, firefly luciferase activity was normalized to that of Renilla luciferase.
siRNAs
H9 cells were transiently transfected 2 days post-plating (30% confluence) in 12-well cell culture plates with 5 μL X-tremeGene siRNA transfection reagent (Roche) per well and survivin siRNA (Beltrami et al., 2004), SI: ggaccaccgcaucucuaca, S4: gagccaagaacaaaauugc.
Western blotting
After H9 cells reached ~30% confluence, they were cultured for up to 5 days under pluripotency or differentiation conditions. In some experiments, H9 cells in 12-well plates were transiently transfected with 100 nM Anti-miR miRNA inhibitors or 50 nM LNA inhibitors using 5 μL of X-tremeGENE siRNA Transfection Reagent per well, and then cultured for 48–72 h. Cells were harvested in IX Cell Culture Lysis Buffer (Promega), according to the manufacturer’s instructions. Protein content was measured using the Bio-Rad (Hercules, CA) DC protein assay kit, according to the manufacturer’s instructions. Equal amounts of lysate were electrophoretically separated through a 15% SDS-polyacrylamide gel under reducing conditions and transferred to a polyvinylidene difluoride membrane (Thermo Fisher Scientific, Waltham, MA) using semi-dry transfer conditions.
Membranes were blocked with 5% nonfat milk powder in PBST (phosphate-buffered saline, 0.1% Tween-20) and incubated with anti-survivin (NB500-201, NB500-205, Novus Biologicals, Littleton, CO), anti-Oct3/4 (sc-8628, Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-Nanog (#3580, Cell Signaling, Danvers, MA), anti-CDK2 (sc-163, Santa Cruz), or anti-actin (sc-1615, Santa Cruz) primary antibody. Appropriate secondary-HRP antibodies were used (Santa Cruz). Bands were visualized by chemiluminesence (Perkin Elmer, Waltham, MA) and fluorography. Blots were stripped and reprobed as necessary.
RT-PCR
RNA was isolated from H9 cells using TRizol reagent (Life Technologies, Carlsbad, CA) and quantified spectroscopically (NanoDrop, Thermo Fisher Scientific). Equal amounts of RNA were treated with RQl DNase I (Promega) to remove DNA. To detect mRNA levels in total RNA, DNase-treated RNA was reverse-transcribed with Moloney murine leukemia virus-reverse transcriptase (MMLV-RT, Invitrogen), followed by PCR amplification. The primer sets were based on those described previously (Mahotka et al., 1999; Zhu et al., 2004).
Real-time PCR (qPCR)
miRNA levels were quantified in total RNA by real-time PCR using the TaqMan miRNA qPCR Kit and primer/probe sets (Life Technologies), following the manufacturer’s instructions. Results were normalized to U6, using the relative quantitation (RQ) method.
Immunofluorescence
Cells were grown on Matrigel-coated coverslips and immunofluorescence microscopy analysis was carried out as described previously (Ghule et al., 2007); cells were fixed with 3.7% formaldehyde for 30 min, permeabilized with 0.25% Triton X-100 for 20 min, and then incubated with primary antibody for 1 h at 37°C, followed by detection using the appropriate fluorescence dye–tagged secondary antibody. The nuclei were counterstained with DAPI (4′,6-diamidino-2- phenylindole). Cells were viewed under an epifluorescence Zeiss Axioplan 2 microscope, and images were captured using a Hamamatsu (C4742-95) charge-coupled-device (CCD) camera and analyzed using Metamorph imaging software (Universal Imaging, West Chester, PA). The following antibodies were used: anti-survivin (NB500-201, NB500-205, Novus Biologicals) at 1:250 and appropriate secondary antibodies conjugated with Alexa Fluor dyes at 1:800.
Data analysis
The data were represented as mean ± S.E. and analyzed by one-way analysis of variance (ANOVA) with Bonferroni post-hoc test, or by Student’s t test (KaleidaGraph, Synergy Software, Reading, PA).
RESULTS AND DISCUSSION
Human embryonic stem cell differentiation has been extensively studied in the context of generating specific lineage-committed cells. However, the regulatory processes that maintain pluripotency are not fully understood. Our results support the hypothesis that survivin is functionally linked to sustaining hESC pluripotency and survivin isoform expression is selectively regulated by miR-302.
Characterization of culture conditions on hESC differentiation and pluripotency
We characterized pluripotency and differentiation markers in H9 hESCs to confirm that our culture conditions successfully maintain hESC pluripotency or trigger the onset of differentiation. Using brightfield microscopy, we found the expected cellular morphology and colony structure differences between cells cultured in mTeSR1 to maintain pluripotency, compared those treated with RA to induce differentiation (data not shown). Cells grown in differentiation culture-conditions became elongated, appeared to have an increased cytoplasmic to nuclear ratio, and established a monolayer rather than growing in colonies.
Biochemically, the pluripotency markers, Nanog and Oct3/4, were found to be abundantly expressed in cells grown in pluripotency medium for the entire time course of up to 5 days (Fig. 1A). Conversely, cells cultured in the presence of RA did not express detectable levels of Nanog and Oct3/4 protein after 3 days of culture. As expected, the differentiation markers Pax6 and Gata4 were more highly expressed in cells grown in RA-containing medium compared to those cultured to retain pluripotency (Fig. 1B). These data demonstrate that, as expected, H9 cells lose characteristics associated with pluripotency upon treatment with RA.
Fig. 1. hESC pluripotency is maintained in mTeSR1 culture medium and treating with retinoic acid triggers differentiation.
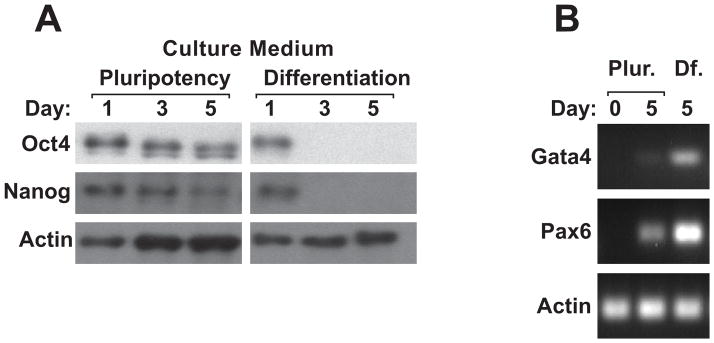
A: Western blot showing the presence of pluripotency markers, Oct4 and Nanog, in H9 hESC cultures grown for 1, 3, and 5 days in medium designed to maintain pluripotency, mTeSR1. The right side of the figure shows that Oct4 and Nanog protein expression is lost at 3 and 5 days in culture medium that contains retinoic acid, indicating that cells have begun to differentiate. B: RT-PCR for differentiation markers Gata4 and Pax6 shows that these genes are much more highly expressed in cells grown in RA-containing medium compared to those cultured to retain pluripotency.
Selective expression and subcellular localization of survivin isoforms in pluripotent cells
To address the role of survivin in maintenance of pluripotency, it is important to characterize expression of survivin isoforms in hESCs. In an initial experiment, we used RT-PCR to determine which survivin isoforms, if any, were expressed in H9 cells. The PCR primers were designed to amplify and resolve canonical survivin, and ΔEx3 and survivin-2B isoforms. Bands representing canonical survivin (380 bp) and ΔEx3 (240 bp) were detected in cultures grown in either pluripotency or differentiation culture conditions (Fig. 2A). Canonical survivin mRNA was not detected until 3 days of culture, whereas isoform ΔEx3 expression was seen on each day assayed. Survivin-2B was not detected. We also detected expression of BIRC4 (XIAP), another member of the BIRC family of proteins involved in inhibition of apoptosis (data not shown).
Fig. 2. Selective expression and subcellular localization of survivin isoforms shows discrepant mRNA and protein levels of nuclear/ΔEx3 survivin.

A: RT-PCR of RNA from H9 hESCs cultured in pluripotency or differentiation media for the indicated number of days, with PCR primers designed to differentiate canonical and ΔEx3 isoforms based on amplicon size. B: Fluorescence images of H9 hESCs cultured in pluripotency media for 0 days; top, DAPI nuclear staining; middle/bottom, immunofluorescence using with primary antibodies for cytoplasmic and nuclear survivin, respectively, detected with Alexa-fluor conjugated secondary antibodies. C: Western blot showing cytoplasmic and nuclear isoforms of survivin in H9 hESCs cultured as in panel A. D: Densitometric analysis of blot shown in panel C. Cytoplasmic/canonical survivin mRNA levels increased in pluripotency medium, but mRNA was not detected in cells grown in differentiation medium until day 3. Protein was likewise very low/not detectable at day 1, then levels increased over time in both media, with a more dramatic increase seen in pluripotency medium. Survivin ΔEx3 isoform protein levels did not correspond directly with mRNA levels. mRNA was seen in all cultures; levels decreased over time in cells grown in pluripotency medium and remained constant in differentiation medium. Nuclear/ΔEx3 survivin protein increased in pluripotency medium and decreased at day 5 in differentiation medium.
Next, we examined survivin protein levels in H9 cells in pluripotency or differentiation culture conditions. To discriminate nuclear and cytoplasmic survivin isoforms, we characterized two commercially available survivin antibodies in H9 cells, using immunofluorescence. We first established that the two antibodies recognize distinct nucleus- or cytoplasm-localized survivin protein (Fig. 2B). We then used these antibodies to quantify nuclear and cytoplasmic survivin protein levels using Western blotting (Fig. 2C). We found that cytoplasmic survivin protein expression increased over time in pluripotent and differentiating culture conditions. The nuclear survivin-protein level also increased in pluripotency medium, however, it decreased over time in differentiation medium (Fig. 2D). A recent study found that all survivin isoforms decrease rapidly in hESCs cultured in conditions that promote differentiation (Mull et al., 2014) and a separate previous study showed canonical survivin expression is downregulated in embryoid bodies compared to ESCs (Blum et al., 2009). These published results contrast with those presented here, which show selective expression of survivin isoforms at the onset of hESC differentiation and suggest that nuclear survivin is associated with pluripotency.
Based on the known subcellular localization of canonical and ΔEx3 survivin isoforms to cytoplasm and nucleus, respectively, and our own results showing that these isoforms were easily detectable, we infer that the cytoplasmic survivin antibody primarily detects canonical survivin and the nuclear survivin antibody primarily detects ΔEx3 survivin in these experiments. Interestingly, there was a discrepancy between the relative expression levels of the survivin isoforms at the mRNA and protein levels. Cytoplasmic/canonical survivin mRNA and protein levels were comparable. mRNA and protein expression were very low/not detectable at day 1, then increased over time in both media. Nuclear/ΔEx3 survivin protein levels did not correspond directly with mRNA levels. mRNA was detected in all cultures; protein levels increased over time in pluripotency medium and decreased at day 5 in differentiation medium.
3′ UTR control of survivin isoform expression
To gain insight into how selective expression of survivin isoforms in hESCs is mediated, we focused on post-transcriptional regulation by miRNAs. This mechanism is consistent with the data in Fig. 2 that show a discrepancy between mRNA and protein expression of the nuclear/ΔEx3 isoform. It also is indicated as a possibility by the unique 3′ end of the ΔEx3 survivin isoform. The mRNA does not include exon 3; exon 4 is present, but is frameshifted relative to the other isoforms. Because of this frameshift, ΔEx3 survivin lacks one of BIR domains present in canonical survivin. It also includes additional 3′ coding sequence that corresponds to 3′ UTR in the other isoforms. Canonical survivin has a longer 3′ UTR due to an earlier stop codon, compared to the ΔEx3 survivin transcript—this adds 104 bp at the proximal 3′ UTR (Fig. 3). The 3′ UTRs of both canonical and ΔEx3 survivin contain the same poly(A) signals, which confer the potential to express 3 different 3′ UTRs (short, medium, and long) for each isoform (Fig. 3A).
Fig. 3. 3′UTR control of survivin isoform expression.

A: Canonical and ΔEx3 survivin mRNA isoforms showing exons, possible polyadenylation sites, 3′ UTR lengths. B: RT-PCR showing RNA corresponding to all three survivin polyadenylation sites are detected in H9 cells cultured for 1 and 3 days in both media. Expression of medium- and long-poly(A) tail mRNAs is lower at day 1 in differentiating vs. pluripotency media. At day 3, only expression of the long-poly(A) tail mRNA is reduced in differentiation medium compared to in pluripotency medium. C: Luciferase reporter (pMIR-Report) constructs containing 3′ UTR sequences from canonical and ΔEx3 survivin isoforms (left). Normalized luciferase signal in cells transiently co-transfected with the constructs shown at left and a Renilla luciferase construct, cultured for 24 h post-transfection in either pluripotency or differentiation medium (right). Short 3′ UTR constructs from both survivin isoforms showed increased expression regardless of culture media, whereas constructs containing only medium and long poly(A) tail 3′ UTR constructs from canonical survivin showed upregulation in the presence of RA (differentiation medium). ΔEx3-derived medium and long poly(A) tail 3′ UTRs were not upregulated in the presence of RA.
We used RT-PCR to amplify the three 3′ UTRs (Fig. 3B). We were able to detect all 3′ UTRs in hESC cultures at day 1 and day 3. It appears that medium and long 3′ UTRs were expressed at lower levels in cells grown in differentiation medium (containing RA).
To determine whether isoform-specific 3′ UTRs could differentially regulate gene expression at the onset of cell fate commitment, we cloned the six potential 3′ UTRs into a luciferase reporter vector and transiently transfected the resulting reporter constructs into hESCs grown in either pluripotency or differentiation culture conditions (Fig. 3C). The short survivin (1.1 kb) and short ΔEx3 (1 kb) 3′ UTR constructs showed significantly increased luciferase expression when transfected cells were cultured in differentiation conditions (RA present), rather than in pluripotency media. With the medium-length constructs, this sharp difference in expression was seen only with the survivin construct (1.8 kb), not with the 3′ UTR from the ΔEx3 isoform (1.7 kb) (which showed no significant difference in luciferase expression when cells were cultured in differentiation medium). The long survivin 3′ UTR (2.1 kb) also increased luciferase expression in differentiation medium, while the long (2 kb) ΔEx3 3′ UTR did not. This finding suggests that there is a regulatory element present within both medium-length 3′ UTRs. The regulatory element was found to be repressive exclusively in the context of the ΔEx3 3′ UTR when hESCs are treated with RA to induce cell fate commitment. A mechanism for involvement of 3′ UTRs in regulation of survivin isoform-specific expression during hESC differentiation is proposed.
miRNAs selectively regulate survivin isoform expression
Recently, miRNAs have been implicated in the maintenance of pluripotency (Anokye-Danso et al., 2011) and have been shown to target survivin (Bian et al., 2012; Saini et al., 2011; Wang et al., 2012; Xu et al., 2013). We proposed miRNA-mediated control as a mechanism of regulation because the survivin 3′ UTR constructs in this study showed differential regulation of gene expression. We used TargetScan software to predict possible miRNA binding sites in the 3′ UTRs of survivin and its ΔEx3 isoform, then selected six potential miRNA candidates based on their validated expression during H1 hESC differentiation (Bar et al., 2008), (Fig. 4A). Some of these miRNAs have been shown to mediate gene expression during hESC differentiation. For example, miR-218 negatively regulates survivin and other genes to suppress nasopharyngeal cancer progression (Alajez et al., 2011). miR-542 negatively regulates survivin to induce growth arrest in A549 cells (Yoon et al., 2010). As mentioned previously, miR-203 targets survivin in diverse cancer cells.
Fig. 4. miRNAs selectively regulate survivin isoform expression.

A: Selected, predicted miRNA-binding sites in survivin isoform 3′ UTRs. B: qRT-PCR for selected microRNAs in H9 cells shows that miR-203, -218, -542-5p, and -335 are more highly expressed in h9 cells grown in differentiation medium compared to in pluripotency medium. miR-338 expression was equivalent in the two media and miR-135a expression was lower in differentiation vs. pluripotency medium. C: Western blot showing the effects of miRNA inhibitors on survivin isoforms and the pluripotency marker Nanog in cells grown in differentiation medium. Cells transfected with the miR-203 inhibitor displayed increased levels of nuclear, but not cytoplasmic survivin, relative to control-transfected cells; Nanog levels were also higher. Inhibition of miR-135a resulted in decreased levels of both nuclear and cytoplasmic survivin, as well as Nanog. D: Western blot showing that cells grown in pluripotency medium and transfected with miR-203 precursor, to mimic miR-203 overexpression, showed decreased levels of nuclear survivin and Nanog, but no change in cytoplasmic survivin levels. E: Normalized luciferase signal in cells transiently co-transfected with short- and medium-length 3′ UTR luciferase constructs and miR-203 inhibitor. Show that only the medium-length 3′ UTR from the ΔEx3/nuclear survivin isoform is affected by mir-302 inhibitor.
We used qRT-PCR to quantitate expression levels of selected miRNAs (Fig. 4B). miR-203, -218, -542, and -335 levels were significantly higher in H9 ESCs cultured in differentiation medium, compared to in cells cultured in pluripotency medium: 20- 10-, 8-, and 3-fold, respectively. This suggests that these miRNAs may be generally involved in the differentiation process. miR-338 expression was equivalent in the two media and miR-135a expression was 3-fold lower in the presence of RA. These miRNA expression patterns were largely comparable to those seen in H1 cells at the onset of differentiation. A slight discrepancy is the behavior of miR-338, which did not significantly change in our experiments with H9 cells, but was downregulated in a published characterization of H1 differentiation. This suggests that miR-338 may not be directly involved in controlling general differentiation processes—we did not include miR-338 in further experiments. To determine if these miRNAs could regulate endogenous survivin protein levels, H9 cells were transiently transfected with miRNA inhibitors (some were Anti-miRs and others were LNA miRNA inhibitors). Compared to the negative control inhibitor, cells transfected with the miR-203 inhibitor had increased levels of nuclear survivin, but not cytoplasmic survivin (Fig. 4C). Moreover, these cells also showed increased levels of Nanog protein. Importantly, miR-203 is not predicted to directly target Nanog. No effects were seen on survivin or Nanog protein levels in cells transfected with miR-218 and -335 inhibitors. Compared to the negative control LNA, cells transfected with the miR-135a LNA showed decreased levels of nuclear survivin, cytoplasmic survivin, and Nanog. Like miR-203, miR-135a is not predicted to target Nanog. We also tested overexpression of miR-203 by transfecting cells with miR-203 precursor; this resulted in a decrease in nuclear survivin levels, but no change in cytoplasmic survivin quantity (Fig. 4D). In these cells, Nanog protein levels also decreased, but Oct4 expression levels were not affected.
To determine if miR-203 can regulate survivin isoform expression mediated by the 3′UTR, we transiently co-transfected H9 cells with the luciferase-3′ UTR constructs and specific miRNA inhibitors. In agreement with the Western blots, the miR-203 inhibitor increased gene expression mediated by the ΔEx3 survivin isoform, but did not affect expression of the canonical survivin isoform, when compared to the negative control inhibitor (Fig. 4E). These data demonstrate that miR-203 negatively regulates the nuclear/ΔEx3 survivin isoform, which may contribute to the maintenance of pluripotency. This suggests a post-transcriptional mechanism for regulation of survivin isoform–specific expression during hESC differentiation.
Survivin knockdown results in decreased levels of key pluripotency markers
With data indicating that miR-203 selectively regulates survivin isoform expression and that survivin isoforms are differentially expressed during hESC differentiation, we conducted survivin knockdown experiments using siRNA to examine the contribution of survivin in the expression of core pluripotency transcription factors, Oct4 and Nanog. We transiently transfected H9 cells with survivin isoform-specific siRNAs: S1 targets ΔEx3 and S4 targets canonical survivin. Under brightfield microscopy, cultures transfected with survivin siRNAs exhibited morphological changes indicative of differentiation (e.g., larger cytoplasm and less structured colonies, data not shown). In pluripotency culture media, compared to the nontargeting control siRNA, cultures transfected with the S4 survivin siRNA had reduced levels of both nuclear and cytoplasmic survivin (Fig. 5). Importantly, S4-transfected cultures also showed dramatically reduced Oct4 and Nanog levels. The S1 siRNA had a similar effect, but to a much lesser extent.
Fig. 5. Survivin knockdown results in decreased levels of key pluripotency markers.

Western blot showing the effects of survivin knockdown using siRNA. Cells cultured in the indicated medium were either mock transfected (M), transfected with nontargeting siRNA (C), or transfected with siRNA targeting survivin exon 1 (S1) or exon 3 (S4). Knockdown of survivin resulted in reduced levels of pluripotency markers Oct4 and Nanog.
These results suggest that survivin is necessary to maintain pluripotency. Knocking down survivin caused morphological and growth habit changes indicative of a loss of pluripotency or the onset of differentiation. In addition, the levels of pluripotency markers Oct4 and Nanog were reduced. Nanog is not predicted to be a direct target of miR-203. Therefore, miR-203 may inhibit pluripotency by negatively regulating survivin expression.
Possible mechanisms
The most apparent mechanism by which miR-203 and survivin could affect pluripotency is through control of the cell cycle and proliferation. Survivin expression is necessary for cell cycle progression, thus positioning the cancer cell for proliferation (Altieri, 2008). In cancer cells, miR-203 expression generally results in cell cycle arrest, decreasing proliferation (Ju et al., 2014; Miao et al., 2014; Taube et al., 2013; Tian et al., 2014; Zhu et al., 2013). However, this has not been reported in hESCs and preliminary data from our laboratory indicates that miR-203 overexpression or survivin knockdown in hESCs has no observable effect on the cell cycle or parameters of proliferation. Therefore, in hESCs, miR-203 and survivin may not influence the cell cycle or proliferation.
Another possibility is that survivin functions as a transcription factor or co-regulatory protein to regulate pluripotency markers Oct4 and Nanog—this role for survivin has been recently described (Tang et al., 2012). Our findings provide a novel dimension to survivin-mediated control. The selective regulation of survivin isoform expression by miR-203 may contribute to mechanisms that maintain pluripotency of human embryonic stem cells and support fidelity of the abbreviated pluripotent cell cycle.
Acknowledgments
Contract Grant Sponsor: National Institutes of Health; Contract Grant Numbers: R01 CA78810, P01 CA82834 and R01 CA139322
The authors thank Jennifer Díaz and Patricia Bosley for editorial assistance with preparation of the manuscript.
References
- Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- Alajez NM, Lenarduzzi M, Ito E, Hui AB, Shi W, Bruce J, Yue S, Huang SH, Xu W, Waldron J, O’Sullivan B, Liu FF. MiR-218 suppresses nasopharyngeal cancer progression through downregulation of survivin and the SLIT2-ROBO1 pathway. Cancer Res. 2011;71:2381–2391. doi: 10.1158/0008-5472.CAN-10-2754. [DOI] [PubMed] [Google Scholar]
- Altieri DC. Survivin and apoptosis control. Adv Cancer Res. 2003;88:31–52. doi: 10.1016/s0065-230x(03)88303-3. [DOI] [PubMed] [Google Scholar]
- Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- Altieri DC. Targeting survivin in cancer. Cancer Lett. 2013;332:225–228. doi: 10.1016/j.canlet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y, Zhang Y, Yang W, Gruber PJ, Epstein JA, Morrisey EE. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, Ruzzo WL, Ware C, Radich JP, Gentleman R, Ruohola-Baker H, Tewari M. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrami E, Plescia J, Wilkinson JC, Duckett CS, Altieri DC. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J Biol Chem. 2004;279:2077–2084. doi: 10.1074/jbc.M309479200. [DOI] [PubMed] [Google Scholar]
- Bian K, Fan J, Zhang X, Yang XW, Zhu HY, Wang L, Sun JY, Meng YL, Cui PC, Cheng SY, Zhang J, Zhao J, Yang AG, Zhang R. MicroRNA-203 leads to G1 phase cell cycle arrest in laryngeal carcinoma cells by directly targeting survivin. FEBS Lett. 2012;586:804–809. doi: 10.1016/j.febslet.2012.01.050. [DOI] [PubMed] [Google Scholar]
- Blum B, Bar-Nur O, Golan-Lev T, Benvenisty N. The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat Biotechnol. 2009;27:281–287. doi: 10.1038/nbt.1527. [DOI] [PubMed] [Google Scholar]
- Colnaghi R, Connell CM, Barrett RM, Wheatley SP. Separating the anti-apoptotic and mitotic roles of survivin. J Biol Chem. 2006;281:33450–33456. doi: 10.1074/jbc.C600164200. [DOI] [PubMed] [Google Scholar]
- Conway EM, Pollefeyt S, Steiner-Mosonyi M, Luo W, Devriese A, Lupu F, Bono F, Leducq N, Dol F, Schaeffer P, Collen D, Herbert JM. Deficiency of survivin in transgenic mice exacerbates Fas-induced apoptosis via mitochondrial pathways. Gastroenterology. 2002;123:619–631. doi: 10.1053/gast.2002.34753. [DOI] [PubMed] [Google Scholar]
- Conway EM, Zwerts F, Van Eygen V, DeVriese A, Nagai N, Luo W, Collen D. Survivin-dependent angiogenesis in ischemic brain: molecular mechanisms of hypoxia-induced up-regulation. Am J Pathol. 2003;163:935–946. doi: 10.1016/S0002-9440(10)63453-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. Treat cancers by targeting survivin: just a dream or future reality? Cancer Treat Rev. 2013;39:802–811. doi: 10.1016/j.ctrv.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Donahue JM, Chang ET, Xiao L, Wang PY, Rao JN, Turner DJ, Wang JY, Battafarano RJ. The RNA-binding protein HuR stabilizes survivin mRNA in human oesophageal epithelial cells. Biochem J. 2011;437:89–96. doi: 10.1042/BJ20110028. [DOI] [PubMed] [Google Scholar]
- Ezponda T, Pajares MJ, Agorreta J, Echeveste JI, Lopez-Picazo JM, Torre W, Pio R, Montuenga LM. The oncoprotein SF2/ASF promotes non-small cell lung cancer survival by enhancing survivin expression. Clin Cancer Res. 2010;16:4113–4125. doi: 10.1158/1078-0432.CCR-10-0076. [DOI] [PubMed] [Google Scholar]
- Filion TM, Qiao M, Ghule PN, Mandeville M, van Wijnen AJ, Stein JL, Lian JB, Altieri DC, Stein GS. Survival responses of human embryonic stem cells to DNA damage. J Cell Physiol. 2009;220:586–592. doi: 10.1002/jcp.21735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortugno P, Wall NR, Giodini A, O’Connor DS, Plescia J, Padgett KM, Tognin S, Marchisio PC, Altieri DC. Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J Cell Sci. 2002;115:575–585. doi: 10.1242/jcs.115.3.575. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Pelus LM. Elevation of Survivin levels by hematopoietic growth factors occurs in quiescent CD34+ hematopoietic stem and progenitor cells before cell cycle entry. Cell Cycle. 2002;1:322–326. [PubMed] [Google Scholar]
- Ghule PN, Becker KA, Harper JW, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Cell cycle dependent phosphorylation and subnuclear organization of the histone gene regulator p220(NPAT) in human embryonic stem cells. J Cell Physiol. 2007;213:9–17. doi: 10.1002/jcp.21119. [DOI] [PubMed] [Google Scholar]
- Ju SY, Chiou SH, Su Y. Maintenance of the stemness in CD44(+) HCT-15 and HCT-116 human colon cancer cells requires miR-203 suppression. Stem Cell Res. 2014;12:86–100. doi: 10.1016/j.scr.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Kapinas K, Grandy R, Ghule P, Medina R, Becker K, Pardee A, Zaidi SK, Lian J, Stein J, van Wijnen A, Stein G. The abbreviated pluripotent cell cycle. J Cell Physiol. 2013;228:9–20. doi: 10.1002/jcp.24104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer SK, Kramer OH, Knosel T, Engels K, Rodel F, Kovacs AF, Dietmaier W, Klein-Hitpass L, Habtemichael N, Schweitzer A, Brieger J, Rodel C, Mann W, Petersen I, Heinzel T, Stauber RH. Nuclear export is essential for the tumor-promoting activity of survivin. FASEB J. 2007;21:207–216. doi: 10.1096/fj.06-5741com. [DOI] [PubMed] [Google Scholar]
- Lian JB, Stein GS, van Wijnen AJ, Stein JL, Hassan MQ, Gaur T, Zhang Y. MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol. 2012;8:212–227. doi: 10.1038/nrendo.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahotka C, Liebmann J, Wenzel M, Suschek CV, Schmitt M, Gabbert HE, Gerharz CD. Differential subcellular localization of functionally divergent survivin splice variants. Cell Death Differ. 2002;9:1334–1342. doi: 10.1038/sj.cdd.4401091. [DOI] [PubMed] [Google Scholar]
- Mahotka C, Wenzel M, Springer E, Gabbert HE, Gerharz CD. Survivin-deltaEx3 and survivin-2B: two novel splice variants of the apoptosis inhibitor survivin with different antiapoptotic properties. Cancer Res. 1999;59:6097–6102. [PubMed] [Google Scholar]
- Melton C, Blelloch R. MicroRNA Regulation of Embryonic Stem Cell Self-Renewal and Differentiation. Adv Exp Med Biol. 2010;695:105–117. doi: 10.1007/978-1-4419-7037-4_8. [DOI] [PubMed] [Google Scholar]
- Miao L, Xiong X, Lin Y, Cheng Y, Lu J, Zhang J, Cheng N. miR-203 inhibits tumor cell migration and invasion via caveolin-1 in pancreatic cancer cells. Oncol Lett. 2014;7:658–662. doi: 10.3892/ol.2014.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moes M, Le Bechec A, Crespo I, Laurini C, Halavatyi A, Vetter G, Del Sol A, Friederich E. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PLoS One. 2012;7:e35440. doi: 10.1371/journal.pone.0035440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mull AN, Klar A, Navara CS. Differential localization and high expression of SURVIVIN splice variants in human embryonic stem cells but not in differentiated cells implicate a role for SURVIVIN in pluripotency. Stem Cell Res. 2014;12:539–549. doi: 10.1016/j.scr.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Nissan X, Denis JA, Saidani M, Lemaitre G, Peschanski M, Baldeschi C. miR-203 modulates epithelial differentiation of human embryonic stem cells towards epidermal stratification. Dev Biol. 2011;356:506–515. doi: 10.1016/j.ydbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Pennartz S, Belvindrah R, Tomiuk S, Zimmer C, Hofmann K, Conradt M, Bosio A, Cremer H. Purification of neuronal precursors from the adult mouse brain: comprehensive gene expression analysis provides new insights into the control of cell migration, differentiation, and homeostasis. Mol Cell Neurosci. 2004;25:692–706. doi: 10.1016/j.mcn.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Saini S, Majid S, Yamamura S, Tabatabai L, Suh SO, Shahryari V, Chen Y, Deng G, Tanaka Y, Dahiya R. Regulatory role of mir-203 in prostate cancer progression and metastasis. Clin Cancer Res. 2011;17:5287–5298. doi: 10.1158/1078-0432.CCR-10-2619. [DOI] [PubMed] [Google Scholar]
- Tang L, Ling X, Liu W, Das GM, Li F. Transcriptional inhibition of p21WAF1/CIP1 gene (CDKN1) expression by survivin is at least partially p53-dependent: evidence for survivin acting as a transcription factor or co-factor. Biochem Biophys Res Commun. 2012;421:249–254. doi: 10.1016/j.bbrc.2012.03.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JH, Malouf GG, Lu E, Sphyris N, Vijay V, Ramachandran PP, Ueno KR, Gaur S, Nicoloso MS, Rossi S, Herschkowitz JI, Rosen JM, Issa JP, Calin GA, Chang JT, Mani SA. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci Rep. 2013;3:2687. doi: 10.1038/srep02687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Li M, Ge J, Guo Y, Sun Y, Liu M, Xiao H. MiR–203 is downregulated in laryngeal squamous cell carcinoma and can suppress proliferation and induce apoptosis of tumours. Tumour Biol. 2014 Apr 1; doi: 10.1007/s13277-014-1790-7. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–1328. doi: 10.1016/s0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene. 2007;26:2678–2684. doi: 10.1038/sj.onc.1210094. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–297. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- Wang C, Zheng X, Shen C, Shi Y. MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells. J Exp Clin Cancer Res. 2012;31:58. doi: 10.1186/1756-9966-31-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Wang Q, An Y, Xu L. MiR203 regulates the proliferation, apoptosis and cell cycle progression of pancreatic cancer cells by targeting Survivin. Mol Med Rep. 2013;8:379–384. doi: 10.3892/mmr.2013.1504. [DOI] [PubMed] [Google Scholar]
- Yoon S, Choi YC, Lee S, Jeong Y, Yoon J, Baek K. Induction of growth arrest by miR-542-3p that targets survivin. FEBS Lett. 2010;584:4048–4052. doi: 10.1016/j.febslet.2010.08.025. [DOI] [PubMed] [Google Scholar]
- Zhao S, Yao DS, Chen JY, Ding N. Aberrant expression of miR-20a and miR-203 in cervical cancer. Asian Pac J Cancer Prev. 2013;14:2289–2293. doi: 10.7314/apjcp.2013.14.4.2289. [DOI] [PubMed] [Google Scholar]
- Zhu N, Gu L, Findley HW, Li F, Zhou M. An alternatively spliced survivin variant is positively regulated by p53 and sensitizes leukemia cells to chemotherapy. Oncogene. 2004;23:7545–7551. doi: 10.1038/sj.onc.1208038. [DOI] [PubMed] [Google Scholar]
- Zhu X, Er K, Mao C, Yan Q, Xu H, Zhang Y, Zhu J, Cui F, Zhao W, Shi H. miR-203 suppresses tumor growth and angiogenesis by targeting VEGFA in cervical cancer. Cell Physiol Biochem. 2013;32:64–73. doi: 10.1159/000350125. [DOI] [PubMed] [Google Scholar]