

Abstract
Despite significant advances in the physician’s ability to initiate myocardial reperfusion and salvage heart tissue, ischemic heart disease remains one of the leading causes of death in the United States. Consequently, alternative therapeutic strategies have been intensively investigated, especially methods of enhancing the heart’s resistance to ischemic cell death - so called “cardioprotective” interventions. However, although a great deal has been learned regarding the methods and mechanisms of cardioprotective interventions, an efficacious therapy has yet to be successfully implemented in the clinical arena. This review discusses the current understanding of cardioprotection in the context of ischemic heart disease pathophysiology, highlighting those elements of ischemic heart disease pathophysiology that have received less attention as potential targets of cardioprotective intervention.
1. Impact of Ischemic Heart Disease
Heart disease is the leading cause of death in the United States, and ischemia/reperfusion-induced cell death (IR), such as seen during myocardial infarction (MI), is a major cause of morbidity and mortality, as about 1.5 million Americans suffer from an MI annually [1-3]. Because terminally differentiated myocytes do not regenerate, the loss of myocardial tissue due to MI forces the remaining viable myocytes to work harder to maintain sufficient cardiac output. To accomplish this, these remaining myocytes hypertrophy to increase their strength of contraction. While this remodeling is initially adaptive, in severe cases this remodeling can decompensate and become pathological, ultimately leading to heart failure [3]. Thus, the best strategy to improve both survival and quality of life in patients suffering from MI is to minimize myocardial death that occurs due to IR. Clinically, this is achieved through arterial reperfusion of the ischemic myocardium which, in most cases, is accomplished through active dissolution and/or physical obliteration of an occlusive intracoronary lesion. Since the therapeutic importance of prompt myocardial reperfusion has been emphasized and implemented into today’s standard of care, the morbidity and mortality of MI has decreased. Aiding that decrease has been refinement in the techniques physicians employ to re-establish coronary blood flow, including the development of percutaneous coronary angioplasty and coronary stenting, as well as the administration of pharmacological adjuvants, such as antiplatelet therapeutics, that help maintain vessel patency [4]. However, as the prevalence of major risk factors for ischemic heart disease, most notably diabetes, hyperlipidemia and hypertension, continues to be substantial, the burden of disease for MI will remain significant.
Because further advances in methods to provide prompt myocardial reperfusion in patients suffering from MI are unlikely to yield significant benefits in morbidity and mortality, there is a great need for the development of novel ischemic heart disease therapies [4]. Consequently, many researchers have investigated strategies that can make the heart more resistant to ischemic death – so called “cardioprotective” interventions. While many cardioprotective strategies have been identified in the laboratory setting, attempts to translate these protective laboratory interventions into a successful clinical therapy have been largely unsuccessful. While reasons for this lack of success may be due to the inherent difficulty of translating results generated in tightly controlled animal models into a heterogeneous patient population [5], the lack of success may also be attributed, in part, to an incomplete understanding of how cardioprotective signaling may be initiated at the level of the cardiac myocyte in response to myocardial stress. Thus, there is great interest in elucidating the mechanisms by which IR induces lethal cellular injury and how cardioprotection may be elicited in the myocardium to allow for the identification of novel targets for ischemic heart disease therapy. Accordingly, the goal of this review is to highlight these potential avenues of cardioprotection in the context of MI pathophysiology.
2. Pathology of Myocardial Infarction
2.1 Ischemic Cell Death
Myocardial ischemia results in numerous deleterious consequences at the level of the cardiac myocyte that, if left uncorrected, culminate in necrotic cell death. A major consequence of myocardial ischemia is the depletion of adenosine triphosphate (ATP) and other high energy phosphates due to cessation of aerobic metabolism and oxidative phosphorylation. Because the continually contracting myocardium is highly dependent on aerobic metabolism, ATP depletion occurs rapidly in the ischemic heart and contractility is halted within 60 seconds. ATP depletion has numerous detrimental effects on myocyte biochemistry and metabolism, including relaxation of myofilaments, glycogen depletion, disruption of ionic equilibrium and cell swelling. Nevertheless, these effects can be reversed and normal myocyte contractile function restored if the duration of ischemia is sufficiently brief (generally considered to be less than 20 minutes of severe ischemia). However, if the ischemia is prolonged, irreversible injury will develop, which is characterized by damage and/or disruption of the myocyte sarcolemmal membrane. Plasma membrane damage leads to loss of osmotic balance and the leakage of cellular metabolites into the extracellular space. Damage to the mitochondrial membranes compromises the cell’s ability to generate ATP upon reperfusion, as well as results in release of mitochondrial proteins that can directly stimulate the apoptotic cell death pathway. Disruption of lysosomal membranes is especially dire, as this can lead to the release of degradative enzymes capable of digesting essentially all cellular constituents, invariably leading to cellular necrosis [6].
In addition to necrosis, apoptosis also contributes significantly to myocyte death during IR, although the exact contributions of each during the sustained ischemic episode and during myocardial reperfusion remain unclear. While apoptotic myocyte death is most pronounced in the reperfused myocardium [7, 8], apoptosis has also been shown to contribute to cell death in ischemic-only hearts [9]. Some studies suggest there may be overlap between the early signaling events that lead to either necrosis or apoptosis, as interventions known to inhibit apoptosis, such as Bcl-2 overexpression, have also been demonstrated to reduce cellular necrosis [10, 11]. Other investigators propose that, during myocardial IR, the distinction between apoptosis and necrosis becomes blurred, as injured cells attempting to undergo apoptosis may be unable to maintain plasma membrane integrity, resulting in necrosis instead [12]. In addition, a more recently defined form of cell death known as necroptosis or “programmed necrosis,” a form of cell death with characteristics of both necrosis and apoptosis, has been suggested to contribute to myocyte death during IR [13-15].
Although the determining factors by which IR will lead to either necrotic or apoptotic cell death are not completely understood, it is known that both ischemia and reperfusion result in substantial ionic changes capable of predisposing the cardiac myocyte to both forms of cell death. Ischemia causes a cessation of aerobic respiration and a shift to of anaerobic respiration, resulting in depletion of glycogen stores and a resulting accumulation of hydrogen ions and tissue acidosis [5, 16-18]. To counterbalance this acidosis, Na+/H+ exchange occurs at the sarcolemmal membrane, with the efflux of H+ balanced by the influx of Na+ and resulting in an accumulation of intracellular ions relative to the extracellular environment. As a result of the accumulation of Na+ ions, reverse action of the Na+/Ca2+ exchanger (NCX) is stimulated, which leads to an accumulation of intracellular Ca2+ as the accumulated Na+ is extruded from the cell in exchange for extracellular Ca2+. Compounding these ionic disturbances is the depletion of high energy phosphates that occurs rapidly in the ischemic myocardium, preventing the normal activity of the Na+/K+ ATPase, ATP-dependent reuptake of calcium at the sarcoplasmic reticulum and active Ca2+ excretion [16, 19], resulting in further exacerbation of intracellular Na+ and Ca2+ accumulation. As the calcium concentration within the cytosol continues to rise, mitochondria begin to passively uptake calcium into the mitochondrial matrix via the mitochondrial calcium uniporter [18]. If the ischemia is severe and these ionic imbalances are sustained, this cytosolic and mitochondrial calcium overload will ultimately lead to irreversible cellular injury as described below.
Loss of myocyte calcium homeostasis results in a number of cellular changes that predispose the myocyte to irreversible injury. Mitochondrial calcium overload is the primary stimulus for mitochondrial permeability transition (MPT), a stress response mediated by the opening of a high conductance pore located on the inner mitochondrial membrane (discussed further in section 2.2.2). While enhanced intracellular calcium concentration is capable of directly stimulating apoptosis [20], elevated calcium levels can also stimulate the activation of numerous intracellular degradative enzymes with the potential to damage several different cellular structures and precipitate cell death, including phospholipases, proteases and endonucleases. Activation of phospholipases can lead to the damage of cellular membranes which, as described above, can lead to necrotic cell death as a consequence of disruption of cellular osmotic balance and the release of lysosomal enzymes in the cytoplasm [6]. The ionic imbalances engendered by ischemia and the consequent causes of ischemic myocyte death are summarized in Figure 1.
Figure 1. Causes and consequences of ionic imbalances during ischemia.
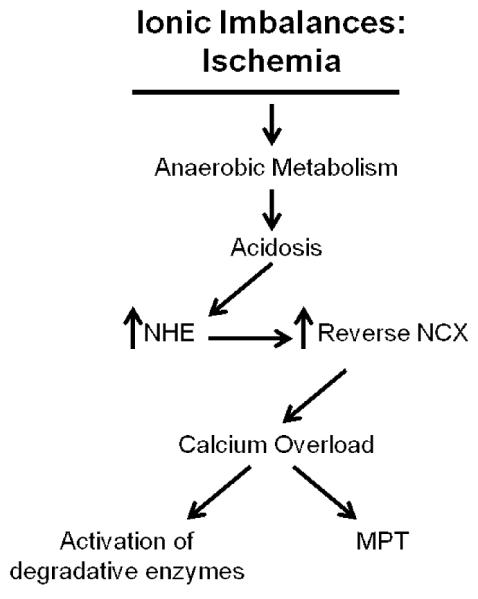
Ischemia results in a cessation of aerobic metabolism and a reliance on anaerobic metabolism, resulting in cellular and tissue acidosis. Accumulation of intracellular H+ stimulates NHE, resulting in accumulation of intracellular Na+. Accumulation of intracellular NA+, in turn, stimulates reverse activity of the NCX, resulting in intracellular Ca+2 accumulation. If ischemia is sustained, cellular Ca2+ overload may develop, resulting in activation of degradative enzymes (e.g., proteases, phosphatases and endonucleases), and stimulation of MPT, culminating in myocyte death. NHE, Na+/H+ exchanger; NCX, Na+/Ca2+ exchanger; MPT, mitochondrial permeability transition.
2.2 Reperfusion Injury
Studies conducted by Jennings and Reimer in the late 1960s and 1970s were pivotal for demonstrating the time-dependent progression of myocardial necrosis as the index ischemia was extended and the critical importance of prompt reperfusion for salvaging myocardium [21, 22]. Interestingly, in an even earlier study conducted by Jennings et al. in 1960, it was observed that reperfused myocardium displayed a more advanced injury pattern than myocardium subjected to the same ischemic duration that had not been reperfused [23]. Specifically, it was observed that reperfused myocardium displayed myocyte swelling, plasma membrane disruption and dense mitochondrial bodies – histological features that do not manifest in the absence of reperfusion unless ischemia is extended for a much longer duration. This observation led others to later hypothesize that reperfusion could result in lethal myocardial injury above and beyond that due to the ischemic insult alone – a concept known as lethal reperfusion injury.
The mechanisms of lethal reperfusion injury have been well studied in an effort to discern if the conditions of myocardial reperfusion could be modified to maximize the salvage of ischemic myocytes. Chief among the primary mediators of reperfusion injury are the rapid correction of intracellular acidosis, myocyte calcium overload and oxidative stress [5, 24]. The primary mediators of lethal reperfusion injury are depicted in Figure 2.
Figure 2. Primary mediators of lethal reperfusion injury.

Oxidative stress results in sensitization of mitochondria to MPT as well as direct damage to membrane phospholipids. The abrupt correction of acidosis relieves an inhibitory influence on MPT and degradative enzymes and serves as an important impetus for calcium overload. Calcium overload can culminate in cell death through varying mechanisms, including activation of calpain proteases, MPT and hypercontracture of fragile myocytes. Activation of calpains can result in additional damage to the myocyte cytoskeleton, predisposing myocytes to rupture of the sarcolemma following imposed mechanical stress, such as cell swelling or hypercontracture.
2.2.1 Reperfusion Injury: Correction of Intracellular pH
While the acidotic conditions of the ischemic myocyte precipitate ionic imbalance and cell swelling, intracellular myocyte acidosis also inhibits several subcellular processes which can potentially culminate in irreversible injury. Acidosis reduces the reverse activity of the NCX, thereby attenuating calcium overload [19]. Furthermore, H+ ion accumulation directly inhibits MPT [25], ostensibly by interfering with the binding of calcium to the MPT trigger site. The acidotic cytosolic environment of the ischemic myocyte also inhibits the activation of proteases called calpains - calcium-dependent enzymes which are capable of digesting and weakening the myocyte’s cytoskeletal support [26, 27]. However, upon reperfusion there is rapid washout of accumulated H+ ions, alleviating the acidotic inhibition and predisposing reperfused myocytes to cellular injury.
2.2.2 Reperfusion Injury: Calcium Overload
As discussed above, the accumulation of calcium during ischemia occurs as a result of the reverse mode of the NCX. In addition, ATP depletion and subsequent cessation of active calcium extrusion and ATP-dependent reuptake into the sarcoplasmic reticulum further contribute to myocyte calcium overload. Unfortunately, upon reperfusion, this myocyte calcium overload is not corrected, but rather is further exacerbated. At reperfusion, extracellular washout of accumulated H+ ions establishes a large gradient greatly favoring the influx of sodium via the Na+/H+ exchanger and leading to the rapid correction of cellular pH. This sudden and robust influx of sodium strongly stimulates the reverse action of the NCX, especially now that the acidotic inhibition of the NCX has been relieved, resulting in even greater elevations of intracellular calcium concentration [16]. Enhanced cytosolic calcium levels favor the electrophoretic uptake of calcium into mitochondria via the mitochondrial calcium uniporter [20]. The end result is cytosolic and mitochondrial calcium overload, culminating in irreversible myocyte injury through many different potential mechanisms including induction of MPT, activation of calpains and myocyte hypercontracture. Each of these mechanisms is discussed further below. Figure 3 summarizes the establishment and consequences of calcium overload during reperfusion.
Figure 3. Causes and consequences of ionic imbalances during reperfusion.
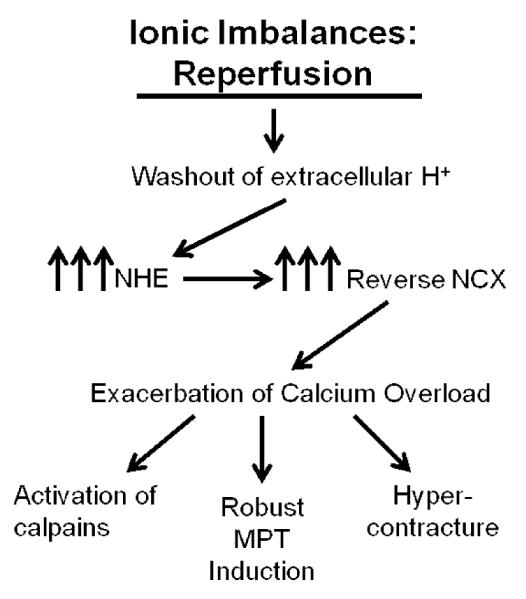
Reperfusion results in an abrupt alleviation of tissue acidosis and a washout of extracellular H+, establishing a highly favorable gradient for Na+ influx via the NHE. The resulting Na+ accumulation potently stimulates reverse NCX activity, resulting in Ca2+ overload. Ca2+ overload in the setting of reperfusion results in activation of the calcium-dependent proteases calpains, the activity of which is attenuated in the acidotic conditions of ischemia. The cellular milieu during reperfusion (e.g., Ca2+ overload, oxidative stress and alleviation of acidosis) is particularly well-suited for the induction of MPT. Finally, resumption of contractile activity in the presence of Ca2+ overload results in hypercontracture, imposing a significant physical strain on a weakened myocyte cellular architecture. NHE, Na+/H+ exchanger; NCX, Na+/Ca2+ exchanger; MPT, mitochondrial permeability transition.
MPT is a prominent cause of irreversible cell injury elicited by IR and is characterized by the opening of a high conductance, non-specific pore located within the inner mitochondrial membrane. While calcium overload is the primary determinant of MPT, there are other cellular conditions that can influence the calcium concentration threshold necessary to induce MPT. For example, both oxidative stress and high energy phosphate depletion sensitize mitochondria to permeability transition, lowering the calcium concentration threshold necessary to induce MPT [28]. Importantly, in addition to the robust influx of calcium that may occur upon reperfusion, oxidative stress and high energy phosphate depletion are both present at reperfusion, highlighting the fact that the cellular milieu is particularly well-suited for mitochondria to undergo MPT following reperfusion. Indeed, many studies have demonstrated that MPT occurs upon reperfusion, primarily in ex vivo models of myocardial IR [29, 30].
MPT results in the uncoupling of the mitochondrial membrane potential, as the enhanced inner membrane permeability dissipates the proton gradient necessary for oxidative phosphorylation. Furthermore, because the mitochondrial matrix exerts a positive colloidal osmotic pressure due to its higher protein concentration than either the cytosol or intermembranous space, MPT results in marked mitochondrial swelling [6, 28]. While the inner mitochondrial membrane can withstand this swelling without rupturing by virtue of “unfolding” of the cristae of the inner membrane, mitochondrial swelling is capable of rupturing the outer mitochondrial membrane, which leads to the release of cytochrome c and the stimulation of the intrinsic apoptotic pathway [31]. As mitochondria undergo MPT, swelling, and outer membrane rupture, more calcium is released from these “open” mitochondria back into the cytosol which, in turn, predisposes mitochondria that have so far remained “closed” to undergo MPT. If a critical number of mitochondria undergo MPT, the most likely outcome is bioenergetic failure of the myocyte and necrotic cell death [28].
The presence of myocyte calcium overload upon reperfusion can also lead to the activation of members of the calcium-dependent family of cysteine proteases known as calpains. μ-calpain and m-calpain are the most abundant calpain isoforms, both of which are expressed in the heart [27]. Calpains can become activated as a result of the calcium accumulation that occurs during ischemia as well as the more robust increase in calcium influx that occurs during reperfusion [5]. Calpain activation results in the hydrolysis of a number of proteins that predispose the myocyte to lethal reperfusion injury. Chief among these calpain substrates are cytoskeletal proteins, the enzymatic cleavage of which damages myocyte contractile machinery and weakens myocyte structural support [32, 33]. This latter consequence is particularly troublesome as it can potentially lead to the rupture of the sarcolemmal membrane upon the resumption of contractile activity [26].
Studies have supported a role for cytoskeletal lesions as important mediators of irreversible IR injury. Seminal studies conducted by Vander Heide and Ganote demonstrated using a Langendorff perfused rat heart model that anoxic perfusion results in enhanced myocardial fragility and decreased resistance to applied mechanical force. Electron microscopy of hearts subjected to anoxic perfusion prior to mechanical stress displayed rupture of the sarcolemmal membrane and the formation of large subsarcolemmal blebs caused by detachment of the sarcolemmal membrane from the Z-disc [34]. A similar study conducted by Steenbergen et al. at nearly the same time correlated the appearance of subsarcolemmal blebs and plasma membrane rupture induced by ischemia with the breakdown of vinculin, a focal adhesion cytoskeletal scaffolding protein, at the plasma membrane [35]. Thus, while the exact series of events that underlie the transition from reversible to irreversible injury during ischemia are not completely understood, studies support a role of cytoskeletal lesions as critical determinants of irreversible ischemic injury that manifests upon reperfusion.
Further compounding the problem of enhanced myocyte fragility during IR is the development of hypercontracture, a state of nonphysiologic myocyte sarcomere shortening due to ATP generation in a setting of intracellular calcium overload [24, 36, 37]. The concept that myocyte contracture can lead to irreversible injury and sarcolemmal membrane rupture in the reperfused ischemic myocyte by imposing a great physical stress on an already fragile sarcolemmal membrane was originally postulated by Ganote and Kaltenbach in 1979 [38]. Subsequent studies supported this hypothesis by demonstrating that the calcium paradox, a phenomenon by which the readmission of calcium to calcium depleted hearts results in contraction band necrosis, was reproduced simply with caffeine-stimulated sarcoplasmic reticulum calcium release and resulting myocyte contracture and occurred independently of extracellular calcium repletion [39, 40]. Furthermore, a study by Garcia-Dorado et al. demonstrated that attenuation of myocyte contractility at reperfusion significantly reduced infarct size in an in vivo porcine model of MI [41].
2.2.3 Reperfusion Injury: Oxidative Stress
While reactive oxygen species (ROS) are generated during normal cellular metabolism, the reperfusion of ischemic myocytes results in a sudden burst of ROS which overwhelms the cell’s capacity to scavenge these radicals, especially since the activity of scavenging enzymes is attenuated as a result of the ischemic insult [42]. Consequently, the reperfused myocyte enters a state of oxidative stress, resulting in a number of deleterious consequences that can culminate in lethal reperfusion injury. First, in addition to mitochondrial calcium overload, this sudden onset of oxidative stress sensitizes mitochondria to permeability transition [28]. Mitochondria that undergo permeability transition can, in turn, release additional free oxygen radicals that can sensitize other mitochondria to MPT; this phenomenon is termed “ROS-induced ROS release” [43]. Furthermore, ROS can directly damage calcium handling proteins, thereby contributing to calcium overload at reperfusion [5, 42]. Importantly, ROS are capable of peroxidizing membrane lipids, contributing to the disruption of cellular membranes that occurs during IR [42]. Finally, ROS are also capable of inflicting further cellular damage by cross-linking proteins and creating DNA breaks [6].
There are multiple sources of increased ROS in the reperfused myocyte. Xanthine oxidase is one prominent contributor of oxygen radicals at reperfusion [16, 44]. Normally, this enzyme exists in its dehydrogenase form, which oxidizes hypoxanthine while using NAD+ as its electron acceptor. However, during ischemia, a calcium-dependent protease converts xanthine dehydrogenase to xanthine oxidase, an enzyme which oxidizes hypoxanthine while reducing O2 to O2− [44, 45]. During ischemia, the degradation of adenine nucleotides results in a buildup of hypoxanthine, the primary substrate for xanthine oxidase. Thus, the sudden reintroduction of oxygen at reperfusion results in an abundance of xanthine oxidase substrates, hypoxanthine and O2, largely contributing to the observed oxidative burst at reperfusion [44]. Damage to mitochondria during the ischemic episode may elicit increased ROS generation directly from the electron transport chain as a result of incomplete reduction of oxygen [6, 42]. Finally, ischemic myocardial injury results in the influx of leukocytes into the ischemic risk region upon reperfusion. These inflammatory cells are subsequently activated and become another important source of ROS generation through the action of the enzyme NADPH oxidase [42].
3. Mechanisms of Cardioprotection
Because the best strategy for improving the morbidity and mortality of MI is to minimize the ischemic death of myocardial tissue, there has been great research interest in learning how the heart can be protected from ischemic death. Various laboratory interventions have been shown to be capable of reducing infarct size including hypothermia [46], heat stress [47] and ischemic preconditioning (IP) [48], as well as a myriad of pharmacologic agents, including calcium channel blockers [49], adenosine [50], alpha 1 adrenergic agonists [51] and delta 1 opioid agonists [52]. However, among these interventions, IP has demonstrated the most consistent ability to confer robust cardioprotection in several different mammalian models, including rats, rabbits, dogs, pigs, sheep and monkeys [53]. Importantly, the ability of IP to confer protection has also been supported in humans undergoing coronary artery bypass surgery, as patients subjected to an IP protocol displayed reduced ATP depletion following a controlled ischemic stress produced during the normal course of the surgery [54]. Although the clinical value of IP is limited by the fact that patients suffering from MI do not present until after the onset of ischemia, the discovery of IP provided definitive proof that the heart can be protected from ischemic death, spawning intensive research focused on elucidating these protective mechanisms.
3.1 Ischemic Preconditioning and Cardioprotective Signaling
The phenomenon of IP was originally described by Murry et al. in 1986 [48]. Following the original description of IP, it was discovered that the protection afforded by IP is not monophasic, but rather IP produces two temporally distinct windows of cardioprotection. The first window of protection begins within minutes following the cycles of preconditioning. This early window of protection is lost if the time between the cycles of preconditioning and the sustained myocardial ischemia is extended beyond 1-2 hours. The second window of protection develops 6-12 hours following IP and lasts for 3-4 days. The mechanisms mediating early and late preconditioning are distinct: the protection within the early window is associated with post-translational modification of pre-existing proteins, while the late window of protection is thought to be produced through changes in gene expression and the synthesis of new proteins, notably inducible nitric oxide synthase (iNOS) and cyclo-oxygenase 2 (COX-2) [55]. This review focuses on cardioprotective signaling cascades elicited by acute myocardial stress and therefore will discuss IP in context of the early window of cardioprotection as originally described by Murry et al.
The mechanism of IP’s protective effect has been extensively investigated, and the research leading up to the present understanding of IP’s protective mechanism is briefly summarized below. IP’s cardioprotection is currently thought to be triggered through the release of ischemic metabolites during the index ischemia and the subsequent binding to their respective transmembrane receptors. Adenosine was the first such metabolite to be recognized for having cardioprotective potential [50]. Later, other metabolite triggers were described, including bradykinin [56] and opioids [57]. This ability of multiple triggers to mimic the protective effect of IP led to the conclusion that the respective signaling pathways converge downstream at a common point. Protein kinase C (PKC) was one of the first proposed points of convergence, as PKC inhibitors are not only capable of abolishing the protective effect of IP [58], but also the protection afforded by pharmacological mimickers of IP [59]. PKC has been postulated to elicit cardioprotection, in part, by activating mitochondrial ATP-sensitive K+ channels (mitoKATP) [60]. The cardioprotection associated with mitoKATP is not completely understood, but activation of mitoKATP has been suggested to enhance the resistance of mitochondria to MPT. mitoKATP channel opening results in an influx of K+ into the mitochondrial matrix, thereby depolarizing the mitochondrial membrane potential. This depolarization, in turn, mitigates the driving force of calcium into the mitochondrial matrix during the sustained ischemic episode, thereby attenuating mitochondrial calcium overload and decreasing the likelihood of MPT occurring upon reperfusion [61].
In addition to PKC, several other downstream effector proteins have been implicated as mediators of cardioprotective signaling elicited by IP. Because phosphatidylinositol-3-kinase (PI3K), a lipid kinase implicated in cell survival signaling [62], had been reported to activate PKC, Tong et al. investigated the role of PI3K in IP-elicited cardioprotection. Using a Langendorff perfused adult rat heart model, they found that IP induced the activation of the cardioprotective proteins Akt and PKC-ε and that the activation of these proteins was blocked by pharmacological inhibition of PI3K. PI3K inhibition also abolished the protective effect of IP on ventricular functional recovery, further implicating PI3K as an important mediator of IP-elicited cardioprotection [63].
Concurrent to the research investigating the mechanism by which IP induces cardioprotection, other animal research emerged that demonstrated that various interventions initiated at the onset of reperfusion, notably the administration of insulin [64], insulin-like growth factor-1 (IGF-1) [65], bradykinin [66] and the hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor atorvastatin [67], also protected the myocardium from lethal IR injury. Importantly, research into the mechanism of these protective interventions revealed that in each case either PI3K/Akt and/or ERK 1/2 signaling was involved in mediating the conferred cardioprotection. Because PI3K/Akt signaling had been previously implicated in mediating the protective effect of IP when stimulated during the preconditioning phase [63], Hausenloy et al. postulated that these survival kinases could stimulate cardioprotection not only when activated during the cycles of preconditioning, but also when activated at the onset of reperfusion [68]. Indeed, a subsequent study by Hausenloy et al. found that activated Akt and ERK 1/2 expression displayed a biphasic pattern with expression significantly enhanced both immediately following preconditioning and again at the onset of reperfusion [69]. Because activation of both PI3K/Akt and ERK 1/2 at reperfusion was demonstrated to reduce infarct size, these signaling kinases were termed “reperfusion injury salvage kinases” or RISKs. Importantly, this study demonstrated for the first time that IP may elicit cardioprotection by mitigating reperfusion injury. This conclusion led to the formulation of the hypothesis that the robust protective effect of IP can be triggered at the onset of reperfusion, a viable target for clinical intervention.
The mechanisms responsible for the cardioprotection associated with RISK activation have been well studied. Although strong evidence consistently supports a cardioprotective role of PI3K/Akt signaling, the evidence surrounding ERK signaling is somewhat conflicting. While most data indicate that ERK is important in cardioprotective signaling [69, 70], other studies have not supported a causal role of ERK signaling in mediating IP-elicited cardioprotection [71, 72]. The cardioprotective phenotype associated with ERK signaling is not completely understood, but thought to be due to its ability to inhibit pro-apoptotic proteins such as BAD [68, 73]. The protective effect of PI3K is largely attributed to its ability to stimulate the downstream activation of the serine/threonine protein kinase Akt, also known as protein kinase B, a well-established cardioprotective signaling molecule [74, 75]. Akt is thought to provide protection from IR injury through multiple different mechanisms, including inhibition of apoptosis [74], stimulation of nitric oxide synthesis [76] and prevention of MPT through phosphorylation and inhibition of glycogen synthase kinase 3ß (GSK-3ß) [77, 78].
In addition to the well characterized cardioprotective role of RISK signaling, recent evidence has implicated reversible cysteine redox-based mechanisms of post-translational modifications, such as S-nitrosylation (SNO) and S-glutathionylation, in mediating cardioprotection. Strong evidence supports SNO in particular as an important contributor to preconditioning-elicited cardioprotection. SNO, a process in which NO forms a covalent bond with the thiol groups of cysteine residues, is thought to mediate protection from IR injury by preventing the irreversible oxidation of protein thiols during periods of oxidative stress, such as during the respiratory burst of early myocardial reperfusion [79]. Furthermore, SNO of the L-type calcium channel [80] and SR/ER calcium-ATPase [81] results in decreased cytosolic influx of calcium and increased uptake of calcium into the sarcoplasmic reticulum respectively, thereby attenuating cellular calcium overload during IR [79]. Importantly, studies have shown that SNO is enhanced in the preconditioned heart and that induction of SNO during IR results in reduced ischemic cell death [81, 82].
3.2 Targeting Ischemia-Reperfusion Injury-Clinical Trials
The discovery by Hausenloy and Yellon that IP-elicited cardioprotection could be aborted by inhibition of RISK signaling at reperfusion, along with numerous studies demonstrating that therapies initiated at reperfusion onset are protective, strongly renewed interest in investigating the pathology of lethal reperfusion injury and how it could be prevented or attenuated. Consequently, the therapeutic potential of methods and therapies capable of mimicking IP and eliciting cardioprotective signaling at reperfusion has been intensively evaluated by numerous animal studies and clinical trials. The findings of these studies have been thoroughly discussed by several excellent reviews to which the reader is directed [4, 5, 73, 83], and these studies will not be reviewed here. Instead, this review will focus on studies examining the cardioprotective potential of interventions capable of interfering with mediators of reperfusion injury, including correction of acidosis, calcium overload, oxidative stress and MPT. The results of these studies are summarized in Table 1.
Table 1. Summary of animal studies and clinical trials assessing cardioprotective potential of interventions against known mediators of lethal reperfusion injury.
IR, ischemia-reperfusion; NHE, Na+-H+ exchanger; KO, knockout; CABG, coronary artery bypass graft; CVA, cerebrovascular accident; NCX, Na+-Ca2+ exchanger; MI, myocardial infarction; SOD, superoxide dismutase; MPT; mitochondrial permeability transition.
| IR Injury Mediator |
Study | Design | Results |
|---|---|---|---|
| Rapid correction of acidosis |
Karmazyn89 | Cardioprotective potential of NHE inhibitor amiloride assessed in isolated-perfused rat heart |
Enhanced ventricular recovery in amiloridetreated hearts |
| Wang et al90 | Resistance to IR injury assessed in NHE1 KO mice using isolated-perfused heart model |
Improved ventricular function in NHE1 KO mice |
|
| ESCAMI91 | Clinical trial examining therapeutic potential of NHE1 inhibitor eniporide when administered 10 min before reperfusion |
No reduction in infarct size or adverse outcomes |
|
| EXPEDITION92 | Clinical trial examining therapeutic potential of NHE1 inhibitor cariporide in high risk patients undergoing CABG |
Reduced MI mortality, but increased CVA mortality |
|
|
| |||
| Calcium overload |
Imahashi et al93 | Resistance to ischemic death assessed in isolated-perfused hearts of NCX KO mice |
Reduced infarct size in NCX KO hearts |
| Kawasumi et al94 | Cardioprotective potential of NCX inhibitor caldaret assessed in in vivo canine model of MI |
Reduced infarct size in caldaret-infused hearts |
|
| CASTEMI95 | Clinical trial examining therapeutic potential of NCX inhibitor caldaret in patients presenting with ST-elevation MI |
No reduction in infarct size | |
|
| |||
| Oxidative stress |
Ambrosio et al96 | Ability of SOD to protect against IR injury assessed in isolated-perfused rabbit hearts |
Improved post-ischemic function in SOD hearts |
| Guan et al97 | Small clinical trial assessing therapeutic potential of xanthine oxidase inhibitor allopurinol |
Enhanced ventricular recovery in patients receiving allopurinol |
|
| Ye et al98 | Meta-analysis of clinical trials assessing effect of antioxidant vitamin supplementation on cardiac outcomes |
No effect on incidence of adverse cardiovascular events |
|
|
| |||
| MPT | Hausenloy et al101 | Cardioprotective potential of MPT inhibitor cyclosporin A assessed in isolated-perfused rat heart |
Infarct size reduction when cyclosporin A administered at reperfusion |
| Piot et al102 | Small clinical trial assessing therapeutic potential of cyclosporin A when administered at reperfusion |
Infarct size reduction | |
Perhaps the best demonstration that modification of the conditions of reperfusion can result in significant cardioprotection is a phenomenon known as ischemic postconditioning (PostC). In an effort to enhance the clinical value of IP, Zhao et al. investigated whether subjecting the heart to cycles of preconditioning following the sustained coronary occlusion could result in infarct size reduction. Using an open chest canine model of MI, they found that PostC, performed by subjecting the heart to three cycles of 30 seconds of reperfusion and 30 seconds of coronary re-occlusion immediately following a 60 minute coronary occlusion, resulted in significant reduction in infarct size, comparable to the protection afforded by IP [84].
Shortly after Zhao and colleague’s description of PostC, clinical trials were underway to determine the clinical efficacy of PostC. The results of these clinical trials have been variable, with some trials showing improved patient outcomes [85, 86] and others showing no difference in patient outcomes between the PostC and placebo groups [87, 88]. Reasons for the variability of results between trials may be due to the small number of patients enrolled in each of the trials and/or differences in inclusion criteria and comorbidities of the enrolled patient populations. Regardless, PostC may offer a therapeutic benefit in specific patient populations undergoing angioplasty for the treatment of acute MI. However, it should be noted that the therapeutic utility of PostC is limited in the clinical arena, as patients must be undergoing angioplasty in a catheterization lab to receive the therapy. Patients who require thrombolytic therapy for restoration of coronary blood flow cannot benefit from PostC. Thus, a therapeutic strategy that could be employed in all patients regardless of how blood flow is restored to the ischemic myocardium would be preferable to PostC [4].
As discussed above, the rapid correction of ischemic acidosis at reperfusion can result in irreversible injury to myocytes by stimulating calcium overload and cellular swelling while concomitantly alleviating the acidotic inhibition of MPT and calpains. Thus, investigators hypothesized that by curbing the effects of ischemic acidosis through inhibition of Na+-H+ exchange myocytes could be protected from reperfusion injury. An early study by Karmazyn demonstrated that amiloride, an inhibitor of the Na+-H+ exchanger (NHE), administered at reperfusion improved post-ischemic ventricular recovery [89]. Later, Wang et al. demonstrated that NHE-1 knockout (KO) mice were resistant to lethal IR injury [90].
Based on these salutary animal studies, clinical trials were conducted to assess NHE inhibition as a potential therapy for IR injury. Overall, these trials did not demonstrate a benefit of NHE inhibition in patients suffering from acute MI. The ESCAMI (Evaluation of the Safety and Cardioprotective Effects of Eniporide in Acute Myocardial Infarction) trial found no differences in clinical outcomes between patients receiving eniporide, a specific inhibitor of the NHE-1 isoform (the predominant NHE expressed in the heart) or placebo [91]. The EXPEDITION trial examined the therapeutic potential of cariporide, a specific NHE-1 inhibitor, in high risk patients undergoing coronary artery bypass graft (CABG) surgery [92]. While the EXPEDITION trial found that NHE inhibition reduced mortality associated with MI, all-cause mortality was paradoxically increased in the NHE inhibition group due to an increased incidence of cerebrovascular events. Therefore, the EXPEDITION investigators concluded that the trial provided proof of concept for the cardioprotective potential of NHE inhibition, but that cariporide was likely not suitable for clinical use due to unacceptable adverse effects.
As myocyte calcium overload is responsible for much of the irreversible injury that occurs during both ischemia and at reperfusion, inhibition of the Na+-Ca2+ exchanger (NCX), has been proposed to be cardioprotective. Imahashi et al. found that Langendorff-perfused hearts of NCX KO mice sustained less necrosis following IR than control hearts [93]. Furthermore, a study conducted by Kawasumi et al. found that a 30 minute intravenous infusion of caldaret, an NCX inhibitor, at reperfusion significantly attenuated ischemic death in the canine heart [94]. Unfortunately, a clinical trial examining the therapeutic efficacy of caldaret, the CASTEMI trial, provided no evidence of cardioprotection in patients presenting with large acute ST-segment elevation MI [95].
It has been well known for many years that scavengers of oxygen-derived free radicals could enhance contractile function and/or reduce infarct size in animal models of myocardial ischemia. Recombinant human superoxide dismutase administered at re-flow in the Langendorff perfused rabbit heart has been documented to improve post-ischemic contractile function [96]. Clinical trials examining the protective potential of attenuating oxidative stress as a treatment for MI have yielded conflicting results but, in general, have not supported a cardioprotective role. In a small trial enrolling 38 patients, allopurinol administered orally just after admission (approximately 60 minutes prior to reperfusion) assisted ventricular recovery in patients undergoing angioplasty for acute MI [97]. However, a meta-analysis of clinical trials examining the role of antioxidant vitamin supplementation in providing protection from cardiovascular disease found no benefit of antioxidant supplementation in improving patient outcomes [98].
Because MPT has been widely implicated as a central mediator of reperfusion injury, and because many known cardioprotective interventions, such as inhibition of NHE and NCX, have been shown to exert their protective effects partly through inhibition of MPT [28], many studies have focused on investigating methods by which MPT may be directly prevented. The immunosuppressant cyclosporin A (CsA) has been demonstrated to prevent MPT [99], and this prevention of MPT has been attributed to CsA’s ability to inhibit the MPT pore component cyclophilin D [28]. Animal models of myocardial IR have demonstrated that CsA administration is cardioprotective [100, 101]. In a small pilot clinical trial, CsA administered at time of re-flow to patients suffering from acute MI was demonstrated to reduce infarct size as assessed through release of creatine kinase and MRI, making CsA an attractive candidate for MI therapy [102]. However, concerns have been raised regarding the results of the trial, as, in some patients, reperfusion may not have been initiated up to 12 hours following onset of chest pain, a time point at which few myocytes within the risk region would remain salvageable [103]. Furthermore, there are some notable limitations in regards to CsA’s cardioprotective effect. First, although CsA administration was well-tolerated in the pilot clinical trial, CsA administration can potentially elicit some adverse effects at the level of the heart as a consequence of its interaction with calcineurin [31]. Secondly, the therapeutic range of CsA may be somewhat narrow, as a study conducted by Griffiths and Halestrap found that higher doses of CsA were not protective [104]. The protective effect of CsA can also be overcome by enhancing the stimulus of MPT, further limiting the therapeutic efficacy of CsA [28]. Thus, more work is required to establish the clinical utility of CsA as a treatment for acute MI.
Interestingly, a recent study by Pan et al. demonstrated that knockout of the mitochondrial calcium uniporter (MCU), which was demonstrated to abolish both mitochondrial calcium uptake and calcium-induced MPT, does not confer protection from IR injury [105]. This result is surprising in light of the many studies demonstrating that drugs and protocols that inhibit MPT, including administration of CsA at reperfusion, elicit cardioprotection [28]. One potential explanation as to why MCU knockout failed to confer cardioprotection may be due to the indirect effect that MCU knockout has on the cellular handling of calcium. Although calcium-induced MPT greatly predisposes myocytes to cell death during IR, the ability of mitochondria to buffer increases in cytosolic calcium concentration through passive uptake of calcium via the MCU may help protect cardiac myocytes from other calcium-mediated mechanisms of irreversible injury, such as the activation of calcium-dependent degradative enzymes and hypercontracture (discussed in section 2.2.2). However, MCU knockout eliminates the ability of mitochondria to uptake calcium, thereby abolishing the ability of mitochondria to buffer rising cytosolic calcium concentrations and potentially rendering MCU-deficient cardiomyocytes susceptible to these other forms of calcium overload-mediated irreversible injury. In contrast to MCU knockout, CsA, which reversibly inhibits MPT by binding to the permeability transition pore component cyclophilin D, does not absolutely abolish the ability of mitochondria to uptake calcium but rather increases the calcium load required for MPT to occur [28, 106]. This qualitative difference in the mechanism of MPT inhibition may partly explain why CsA administration has been shown to confer protection and MCU knockout has not.
While it is clear from the above discussion that a great many promising cardioprotective interventions have been described, the fact that none of these have become implemented into standard clinical practice underlines the importance of enhancing our understanding of how the heart can be protected from ischemic death, allowing for the identification of novel targets for MI therapy. Although the cytoskeleton has been demonstrated to play an important role in lethal reperfusion injury [26, 35], the role of the cytoskeleton in mediating cardioprotective signaling has received little attention and is largely unknown. However, it is known that cytoskeletal signaling has been shown to play a prominent role in maintaining cell viability in both non-muscle cells and in cardiac myocytes [107-109]. Furthermore, the cytosolic tyrosine kinase focal adhesion kinase (FAK), an essential mediator of cytoskeletal signaling [110], has been shown by numerous studies to play an important role in cell proliferation, survival and hypertrophy signaling cascades [111-113]. Based on this evidence, studies were conducted to assess the cardioprotective potential of cytoskeletal signaling; the results of these studies are reviewed below in section 4.
4. Cytoskeletal Signaling and Cardioprotection
Because previous studies had shown that cytoskeletal lesions are a critical determinant of irreversible cellular injury during IR [26, 41], it was hypothesized that interventions that protect the cytoskeleton from ischemic stress would be cardioprotective. Wei and Vander Heide demonstrated that heat stress (HS) significantly enhanced the expression of heat shock proteins (HSPs), proteins capable of associating with and stabilizing the actin cytoskeleton [114, 115], thereby enhancing the cell’s resistance to stress. Both HS and the dual expression of HSP27and HSP70 protected neonatal rat ventricular myocytes (NRVM) from cell death induced by metabolic inhibition (simulated IR). Furthermore, HS was associated with the assembly of an integrin-paxillin-FAK cytoskeletal signaling complex, which was abolished through targeted inhibition of FAK via overexpression of the endogenous FAK competitive inhibitor FRNK (FAK-related non-kinase). Interference with FAK activity also enhanced NRVM cell death in response to simulated IR [47].
In subsequent studies, Wei and Vander Heide hypothesized that FAK and cytoskeletal signaling may represent a unique cardioprotective signaling pathway elicited by myocardial stress. To test this hypothesis, the role of FAK was directly interrogated in HS and IP-elicited signaling pathways. In cultured NRVM, HS significantly enhanced the FAK activation. HS also enhanced the interaction between FAK and PI3K, and this interaction was associated with enhanced expression of activated Akt. Overexpression of FRNK in NRVM reduced the expression of activated Akt following HS both at baseline and following 10 minutes of simulated ischemia, implicating Akt as a mediator of cardioprotection downstream of FAK [116]. In Langendorff perfused adult mouse hearts, IP also significantly enhanced the expression of activated FAK and Akt and protected the hearts from ischemic death, suggesting that cytoskeletal signaling may mediate a common pathway by which myocardial stress leads to downstream cardioprotective signaling [117].
Whereas these earlier studies utilized in vitro and ex vivo model systems to demonstrate activation and protection resulting from a cytoskeletal-based cardioprotective signaling pathway, a later study conducted by Perricone et al. utilized a novel myocyte-restricted, inducible FAK KO mouse model to assess the importance of FAK and cytoskeletal signaling in an in vivo model of MI. This study found that, while IP elicited significant protection from lethal IR injury in control mice, the protective effect of IP was abrogated in FAK KO mice. Furthermore, the expression of activated PI3K and Akt was enhanced in preconditioned control hearts but not in preconditioned FAK KO hearts [118], in accordance with the findings of the results of the earlier studies conducted by Wei and Vander Heide discussed above [116, 117].
Because cytoskeletal lesions predispose cardiomyocytes to sarcolemmal membrane rupture and cell death, mechanisms that either protect cellular membranes during IR or enhance the repair of damaged membranes should be cardioprotective. Accordingly, a study by Wang et al. assessed the cardioprotective potential of the striated muscle-specific tripartite motif family protein MG53, a known mediator of skeletal muscle membrane repair. This study found that cardiomyocyte membrane damage stimulates the localization of MG53 exclusively to the damaged membrane, and that membrane healing is impaired in MG53 deficient cardiomyocytes. Importantly, genetic ablation of MG53 exacerbated IR injury in the Langendorff-perfused mouse heart, supporting a cardioprotective role of MG53 and membrane repair [119]. Interestingly, a study by Cao et al. found that MG53 knockout abolished the protective effect of IP in the Langendorff-perfused mouse heart and that overexpression of MG53 in cultured myocytes conferred protection from hypoxia and oxidative stress-induced cell death. This study determined that the protective mechanism of MG53 was due to its ability to stimulate downstream activation of PI3K-Akt and Erk1/2 signaling pathways [120]. Therefore, MG53 may exert its cardioprotective influence through multiple mechanisms.
5. Conclusion
Despite the great multitude of interventions implicated as having cardioprotective potential that have been identified over the past several years, an efficacious therapy based on what has been learned about cardioprotection is still lacking. The fact that so many different interventions are capable of reducing ischemic death underlines the fact that there are likely several different pathways within a robust cardiac signaling network that can ultimately elicit a cardioprotective state. The pathophysiology of ischemic heart disease is complex, and a therapeutic strategy capable of activating multiple arms of the cardioprotective signaling network and/or simultaneously counteracting multiple mediators of IR injury would likely have the greatest efficacy in mitigating ischemic death and improving the morbidity and mortality associated with MI. While the cardioprotective potential of therapies targeting many of the mediators of IR injury have been well studied in both animal models and clinical trials, the therapeutic potential of other mediators, especially cytoskeletal lesions and the role of cytoskeletal signaling, have received less attention and are not completely understood. Future studies will be important for enhancing both our knowledge of MI pathophysiology and mechanisms of cardioprotection, thereby providing additional avenues of cardioprotective signaling that may be potentially harnessed towards the ultimate goal of establishing a highly efficacious therapeutic strategy for enhancing the heart’s resistance to myocardial IR injury.
Glossary
- CsA
cyclosporin A
- ERK
extracellular regulated kinase
- FAK
focal adhesion kinase
- FRNK
FAK related non-kinase
- NCX
sodium-calcium exchanger
- NHE
sodium-hydrogen exchanger
- NRVM
neonatal rat ventricular myocytes
- PI3K
phosphatidylinositol-3-kinase
- PKC
protein kinase C
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Heron M. Deaths: leading causes for 2008. Natl Vital Stat Rep. 2012;60:1–94. [PubMed] [Google Scholar]
- [3].Schoen FJ, Mitchell RN. The Heart. In: Kumar V, Abbas AK, Fausto N, Aster JC, editors. Robbins and Cotran Pathologic Basis of Disease. Saunders; Philadelphia: 2010. pp. 529–87. [Google Scholar]
- [4].Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59:969–78. doi: 10.1016/j.jacc.2011.07.054. [DOI] [PubMed] [Google Scholar]
- [5].Vander Heide RS, Steenbergen C. Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res. 2013;113:464–77. doi: 10.1161/CIRCRESAHA.113.300765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kumar V, Abbas AK, Fausto N, Aster JC. Cellular Responses to Stress and Toxic Insults: Adaptation, Injury, and Death. In: Kumar V, Abbas AK, Fausto N, Aster JC, editors. Robbins and Cotran Pathologic Basis of Disease. Saunders; Philadelphia: pp. 3–42. [Google Scholar]
- [7].Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–8. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao ZQ, Velez DA, Wang NP, Hewan-Lowe KO, Nakamura M, Guyton RA, et al. Progressively developed myocardial apoptotic cell death during late phase of reperfusion. Apoptosis. 2001;6:279–90. doi: 10.1023/a:1011335525219. [DOI] [PubMed] [Google Scholar]
- [9].Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–56. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- [10].Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95:734–41. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- [11].Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, et al. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene. 1996;12:2045–50. [PubMed] [Google Scholar]
- [12].Gottlieb RA, Engler RL. Apoptosis in myocardial ischemia-reperfusion. Ann N Y Acad Sci. 1999;874:412–26. doi: 10.1111/j.1749-6632.1999.tb09255.x. [DOI] [PubMed] [Google Scholar]
- [13].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- [14].Smith CC, Yellon DM. Necroptosis, necrostatins and tissue injury. J Cell Mol Med. 2011;15:1797–806. doi: 10.1111/j.1582-4934.2011.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–33. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- [16].Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723–41. doi: 10.1152/ajpheart.00553.2011. [DOI] [PubMed] [Google Scholar]
- [17].Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106:360–8. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, et al. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011;2:e244. doi: 10.1038/cddis.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen S, Li S. The Na+/Ca(2)+ exchanger in cardiac ischemia/reperfusion injury. Med Sci Monit. 2012;18:RA161–5. doi: 10.12659/MSM.883533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium- apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- [21].Jennings RB, Sommers HM, Herdson PB, Kaltenbach JP. Ischemic injury of myocardium. Ann N Y Acad Sci. 1969;156:61–78. doi: 10.1111/j.1749-6632.1969.tb16718.x. [DOI] [PubMed] [Google Scholar]
- [22].Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab Invest. 1979;40:633–44. [PubMed] [Google Scholar]
- [23].Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol. 1960;70:68–78. [PubMed] [Google Scholar]
- [24].Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- [25].Szabo I, Bernardi P, Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem. 1992;267:2940–6. [PubMed] [Google Scholar]
- [26].Ganote CE, Vander Heide RS. Cytoskeletal lesions in anoxic myocardial injury. A conventional and high-voltage electron-microscopic and immunofluorescence study. Am J Pathol. 1987;129:327–44. [PMC free article] [PubMed] [Google Scholar]
- [27].Inserte J, Hernando V, Garcia-Dorado D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2012;96:23–31. doi: 10.1093/cvr/cvs232. [DOI] [PubMed] [Google Scholar]
- [28].Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- [29].Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–5. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- [30].Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307(Pt 1):93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- [32].Iizuka K, Kawaguchi H, Kitabatake A. Effects of thiol protease inhibitors on fodrin degradation during hypoxia in cultured myocytes. J Mol Cell Cardiol. 1993;25:1101–9. doi: 10.1006/jmcc.1993.1122. [DOI] [PubMed] [Google Scholar]
- [33].Liu X, Schnellmann RG. Calpain mediates progressive plasma membrane permeability and proteolysis of cytoskeleton-associated paxillin, talin, and vinculin during renal cell death. J Pharmacol Exp Ther. 2003;304:63–70. doi: 10.1124/jpet.102.043406. [DOI] [PubMed] [Google Scholar]
- [34].Vander Heide RS, Ganote CE. Increased myocyte fragility following anoxic injury. J Mol Cell Cardiol. 1987;19:1085–103. doi: 10.1016/s0022-2828(87)80353-x. [DOI] [PubMed] [Google Scholar]
- [35].Steenbergen C, Hill ML, Jennings RB. Cytoskeletal damage during myocardial ischemia: changes in vinculin immunofluorescence staining during total in vitro ischemia in canine heart. Circ Res. 1987;60:478–86. doi: 10.1161/01.res.60.4.478. [DOI] [PubMed] [Google Scholar]
- [36].Vander Heide RS, Angelo JP, Altschuld RA, Ganote CE. Energy dependence of contraction band formation in perfused hearts and isolated adult myocytes. Am J Pathol. 1986;125:55–68. [PMC free article] [PubMed] [Google Scholar]
- [37].Piper HM, Garcia-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300. doi: 10.1016/s0008-6363(98)00033-9. [DOI] [PubMed] [Google Scholar]
- [38].Ganote CE, Kaltenbach JP. Oxygen-induced enzyme release: early events and a proposed mechanism. J Mol Cell Cardiol. 1979;11:389–406. doi: 10.1016/0022-2828(79)90425-5. [DOI] [PubMed] [Google Scholar]
- [39].Vander Heide RS, Ganote CE. Caffeine-induced myocardial injury in calcium- free perfused rat hearts. Am J Pathol. 1985;118:55–65. [PMC free article] [PubMed] [Google Scholar]
- [40].Vander Heide RS, Altschuld RA, Lamka KG, Ganote CE. Modification of caffeine-induced injury in Ca2+-free perfused rat hearts. Relationship to the calcium paradox. Am J Pathol. 1986;123:351–64. [PMC free article] [PubMed] [Google Scholar]
- [41].Garcia-Dorado D, Theroux P, Duran JM, Solares J, Alonso J, Sanz E, et al. Selective inhibition of the contractile apparatus. A new approach to modification of infarct size, infarct composition, and infarct geometry during coronary artery occlusion and reperfusion. Circulation. 1992;85:1160–74. doi: 10.1161/01.cir.85.3.1160. [DOI] [PubMed] [Google Scholar]
- [42].Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–90. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- [43].Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Thompson-Gorman SL, Zweier JL. Evaluation of the role of xanthine oxidase in myocardial reperfusion injury. J Biol Chem. 1990;265:6656–63. [PubMed] [Google Scholar]
- [45].Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem Soc Symp. 1999;66:167–79. doi: 10.1042/bss0660167. [DOI] [PubMed] [Google Scholar]
- [46].Jones RN, Reimer KA, Hill ML, Jennings RB. Effect of hypothermia on changes in high-energy phosphate production and utilization in total ischemia. J Mol Cell Cardiol. 1982;14(Suppl 3):123–30. doi: 10.1016/0022-2828(82)90140-7. [DOI] [PubMed] [Google Scholar]
- [47].Wei H, Campbell W, Vander Heide RS. Heat shock-induced cardioprotection activates cytoskeletal-based cell survival pathways. Am J Physiol Heart Circ Physiol. 2006;291:H638–47. doi: 10.1152/ajpheart.00144.2006. [DOI] [PubMed] [Google Scholar]
- [48].Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- [49].Vander Heide RS, Schwartz LM, Reimer KA. The novel calcium antagonist Ro 40-5967 limits myocardial infarct size in the dog. Cardiovasc Res. 1994;28:1526–32. doi: 10.1093/cvr/28.10.1526. [DOI] [PubMed] [Google Scholar]
- [50].Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–6. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- [51].Bankwala Z, Hale SL, Kloner RA. Alpha-adrenoceptor stimulation with exogenous norepinephrine or release of endogenous catecholamines mimics ischemic preconditioning. Circulation. 1994;90:1023–8. doi: 10.1161/01.cir.90.2.1023. [DOI] [PubMed] [Google Scholar]
- [52].Schultz Je-J, Hsu AK, Nagase H, Gross GJ. TAN-67, a delta 1-opioid receptor agonist, reduces infarct size via activation of Gi/o proteins and KATP channels. Am J Physiol. 1998;274:H909–14. doi: 10.1152/ajpheart.1998.274.3.H909. [DOI] [PubMed] [Google Scholar]
- [53].Cohen MV, Downey JM. Myocardial preconditioning promises to be a novel approach to the treatment of ischemic heart disease. Annu Rev Med. 1996;47:21–9. doi: 10.1146/annurev.med.47.1.21. [DOI] [PubMed] [Google Scholar]
- [54].Yellon DM, Alkhulaifi AM, Pugsley WB. Preconditioning the human myocardium. Lancet. 1993;342:276–7. doi: 10.1016/0140-6736(93)91819-8. [DOI] [PubMed] [Google Scholar]
- [55].Bolli R, Li QH, Tang XL, Guo Y, Xuan YT, Rokosh G, et al. The late phase of preconditioning and its natural clinical application--gene therapy. Heart Fail Rev. 2007;12:189–99. doi: 10.1007/s10741-007-9031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Goto M, Liu Y, Yang XM, Ardell JL, Cohen MV, Downey JM. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ Res. 1995;77:611–21. doi: 10.1161/01.res.77.3.611. [DOI] [PubMed] [Google Scholar]
- [57].Schultz JE, Hsu AK, Gross GJ. Morphine mimics the cardioprotective effect of ischemic preconditioning via a glibenclamide-sensitive mechanism in the rat heart. Circ Res. 1996;78:1100–4. doi: 10.1161/01.res.78.6.1100. [DOI] [PubMed] [Google Scholar]
- [58].Ytrehus K, Liu Y, Downey JM. Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am J Physiol. 1994;266:H1145–52. doi: 10.1152/ajpheart.1994.266.3.H1145. [DOI] [PubMed] [Google Scholar]
- [59].Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24:225–34. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu Y, Gao WD, O’Rourke B, Marban E. Synergistic modulation of ATP- sensitive K+ currents by protein kinase C and adenosine. Implications for ischemic preconditioning. Circ Res. 1996;78:443–54. doi: 10.1161/01.res.78.3.443. [DOI] [PubMed] [Google Scholar]
- [61].Murata M, Akao M, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89:891–8. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- [62].Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ, et al. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373–9. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- [63].Tong H, Chen W, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res. 2000;87:309–15. doi: 10.1161/01.res.87.4.309. [DOI] [PubMed] [Google Scholar]
- [64].Baines CP, Wang L, Cohen MV, Downey JM. Myocardial protection by insulin is dependent on phospatidylinositol 3-kinase but not protein kinase C or KATP channels in the isolated rabbit heart. Basic Res Cardiol. 1999;94:188–98. doi: 10.1007/s003950050142. [DOI] [PubMed] [Google Scholar]
- [65].Otani H, Yamamura T, Nakao Y, Hattori R, Kawaguchi H, Osako M, et al. Insulin- like growth factor-I improves recovery of cardiac performance during reperfusion in isolated rat heart by a wortmannin-sensitive mechanism. J Cardiovasc Pharmacol. 2000;35:275–81. doi: 10.1097/00005344-200002000-00015. [DOI] [PubMed] [Google Scholar]
- [66].Bell RM, Yellon DM. Bradykinin limits infarction when administered as an adjunct to reperfusion in mouse heart: the role of PI3K, Akt and eNOS. J Mol Cell Cardiol. 2003;35:185–93. doi: 10.1016/s0022-2828(02)00310-3. [DOI] [PubMed] [Google Scholar]
- [67].Bell RM, Yellon DM. Atorvastatin, administered at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by up-regulating a pro- survival pathway. J Am Coll Cardiol. 2003;41:508–15. doi: 10.1016/s0735-1097(02)02816-4. [DOI] [PubMed] [Google Scholar]
- [68].Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- [69].Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H971–6. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- [70].Ping P, Zhang J, Cao X, Li RC, Kong D, Tang XL, et al. PKC-dependent activation of p44/p42 MAPKs during myocardial ischemia-reperfusion in conscious rabbits. Am J Physiol. 1999;276:H1468–81. doi: 10.1152/ajpheart.1999.276.5.H1468. [DOI] [PubMed] [Google Scholar]
- [71].Behrends M, Schulz R, Post H, Alexandrov A, Belosjorow S, Michel MC, et al. Inconsistent relation of MAPK activation to infarct size reduction by ischemic preconditioning in pigs. Am J Physiol Heart Circ Physiol. 2000;279:H1111–9. doi: 10.1152/ajpheart.2000.279.3.H1111. [DOI] [PubMed] [Google Scholar]
- [72].Mocanu MM, Bell RM, Yellon DM. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J Mol Cell Cardiol. 2002;34:661–8. doi: 10.1006/jmcc.2002.2006. [DOI] [PubMed] [Google Scholar]
- [73].Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–70. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–7. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Miura T, Tanno M. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res. 2012;94:181–9. doi: 10.1093/cvr/cvr302. [DOI] [PubMed] [Google Scholar]
- [78].Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage- dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–54. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- [79].Murphy E, Kohr M, Sun J, Nguyen T, Steenbergen C. S-nitrosylation: a radical way to protect the heart. J Mol Cell Cardiol. 2012;52:568–77. doi: 10.1016/j.yjmcc.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–93. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- [82].Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S- nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–25. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Minamino T. Cardioprotection from ischemia/reperfusion injury: basic and translational research. Circ J. 2012;76:1074–82. doi: 10.1253/circj.cj-12-0132. [DOI] [PubMed] [Google Scholar]
- [84].Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- [85].Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, et al. Postconditioning the human heart. Circulation. 2005;112:2143–8. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- [86].Laskey WK, Yoon S, Calzada N, Ricciardi MJ. Concordant improvements in coronary flow reserve and ST-segment resolution during percutaneous coronary intervention for acute myocardial infarction: a benefit of postconditioning. Catheter Cardiovasc Interv. 2008;72:212–20. doi: 10.1002/ccd.21583. [DOI] [PubMed] [Google Scholar]
- [87].Sorensson P, Saleh N, Bouvier F, Bohm F, Settergren M, Caidahl K, et al. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart. 2010;96:1710–5. doi: 10.1136/hrt.2010.199430. [DOI] [PubMed] [Google Scholar]
- [88].Freixa X, Bellera N, Ortiz-Perez JT, Jimenez M, Pare C, Bosch X, et al. Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J. 2012;33:103–12. doi: 10.1093/eurheartj/ehr297. [DOI] [PubMed] [Google Scholar]
- [89].Karmazyn M. Amiloride enhances postischemic ventricular recovery: possible role of Na+-H+ exchange. Am J Physiol. 1988;255:H608–15. doi: 10.1152/ajpheart.1988.255.3.H608. [DOI] [PubMed] [Google Scholar]
- [90].Wang Y, Meyer JW, Ashraf M, Shull GE. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. 2003;93:776–82. doi: 10.1161/01.RES.0000094746.24774.DC. [DOI] [PubMed] [Google Scholar]
- [91].Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, et al. The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol. 2001;38:1644–50. doi: 10.1016/s0735-1097(01)01608-4. [DOI] [PubMed] [Google Scholar]
- [92].Mentzer RM, Jr., Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg. 2008;85:1261–70. doi: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- [93].Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res. 2005;97:916–21. doi: 10.1161/01.RES.0000187456.06162.cb. [DOI] [PubMed] [Google Scholar]
- [94].Kawasumi H, Satoh N, Kitada Y. Caldaret, an intracellular Ca2+ handling modulator, limits infarct size of reperfused canine heart. J Pharmacol Sci. 2007;103:222–33. doi: 10.1254/jphs.fp0060765. [DOI] [PubMed] [Google Scholar]
- [95].Bar FW, Tzivoni D, Dirksen MT, Fernandez-Ortiz A, Heyndrickx GR, Brachmann J, et al. Results of the first clinical study of adjunctive CAldaret (MCC-135) in patients undergoing primary percutaneous coronary intervention for ST-Elevation Myocardial Infarction: the randomized multicentre CASTEMI study. Eur Heart J. 2006;27:2516–23. doi: 10.1093/eurheartj/ehl304. [DOI] [PubMed] [Google Scholar]
- [96].Ambrosio G, Weisfeldt ML, Jacobus WE, Flaherty JT. Evidence for a reversible oxygen radical-mediated component of reperfusion injury: reduction by recombinant human superoxide dismutase administered at the time of reflow. Circulation. 1987;75:282–91. doi: 10.1161/01.cir.75.1.282. [DOI] [PubMed] [Google Scholar]
- [97].Guan W, Osanai T, Kamada T, Hanada H, Ishizaka H, Onodera H, et al. Effect of allopurinol pretreatment on free radical generation after primary coronary angioplasty for acute myocardial infarction. J Cardiovasc Pharmacol. 2003;41:699–705. doi: 10.1097/00005344-200305000-00005. [DOI] [PubMed] [Google Scholar]
- [98].Ye Y, Li J, Yuan Z. Effect of antioxidant vitamin supplementation on cardiovascular outcomes: a meta-analysis of randomized controlled trials. PLoS One. 2013;8:e56803. doi: 10.1371/journal.pone.0056803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+- dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–60. [PMC free article] [PubMed] [Google Scholar]
- [100].Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–72. [PubMed] [Google Scholar]
- [101].Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–43. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- [102].Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- [103].Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113:428–38. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- [104].Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–9. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- [105].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–72. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- [107].van de Water B, Nagelkerke JF, Stevens JL. Dephosphorylation of focal adhesion kinase (FAK) and loss of focal contacts precede caspase-mediated cleavage of FAK during apoptosis in renal epithelial cells. J Biol Chem. 1999;274:13328–37. doi: 10.1074/jbc.274.19.13328. [DOI] [PubMed] [Google Scholar]
- [108].Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM. GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ Res. 2002;90:1282–9. doi: 10.1161/01.res.0000023201.41774.ea. [DOI] [PubMed] [Google Scholar]
- [109].Pfister R, Acksteiner C, Baumgarth J, Burst V, Geissler HJ, Margulies KB, et al. Loss of beta1D-integrin function in human ischemic cardiomyopathy. Basic Res Cardiol. 2007;102:257–64. doi: 10.1007/s00395-006-0640-1. [DOI] [PubMed] [Google Scholar]
- [110].Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- [111].Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;289:H2291–301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- [112].Daval M, Gurlo T, Costes S, Huang CJ, Butler PC. Cyclin-dependent kinase 5 promotes pancreatic beta-cell survival via Fak-Akt signaling pathways. Diabetes. 2011;60:1186–97. doi: 10.2337/db10-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Provenzano PP, Keely PJ. The role of focal adhesion kinase in tumor initiation and progression. Cell Adh Migr. 2009;3:347–50. doi: 10.4161/cam.3.4.9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Quinta HR, Galigniana NM, Erlejman AG, Lagadari M, Piwien-Pilipuk G, Galigniana MD. Management of cytoskeleton architecture by molecular chaperones and immunophilins. Cell Signal. 2011;23:1907–20. doi: 10.1016/j.cellsig.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Huot J, Lambert H, Lavoie JN, Guimond A, Houle F, Landry J. Characterization of 45-kDa/54-kDa HSP27 kinase, a stress-sensitive kinase which may activate the phosphorylation-dependent protective function of mammalian 27-kDa heat-shock protein HSP27. Eur J Biochem. 1995;227:416–27. doi: 10.1111/j.1432-1033.1995.tb20404.x. [DOI] [PubMed] [Google Scholar]
- [116].Wei H, Vander Heide RS. Heat stress activates AKT via focal adhesion kinase- mediated pathway in neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;295:H561–8. doi: 10.1152/ajpheart.00401.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wei H, Vander Heide RS. Ischemic preconditioning and heat shock activate Akt via a focal adhesion kinase-mediated pathway in Langendorff-perfused adult rat hearts. Am J Physiol Heart Circ Physiol. 2010;298:H152–7. doi: 10.1152/ajpheart.00613.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Perricone AJ, Bivona BJ, Jackson FR, Vander Heide RS. Conditional knockout of myocyte focal adhesion kinase abrogates ischemic preconditioning in adult murine hearts. J Am Heart Assoc. 2013;2:e000457. doi: 10.1161/JAHA.113.000457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wang X, Xie W, Zhang Y, Lin P, Han L, Han P, et al. Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ Res. 2010;107:76–83. doi: 10.1161/CIRCRESAHA.109.215822. [DOI] [PubMed] [Google Scholar]
- [120].Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. 2010;121:2565–74. doi: 10.1161/CIRCULATIONAHA.110.954628. [DOI] [PubMed] [Google Scholar]