

Abstract
Predicting protein domains is essential for understanding a protein’s function at the molecular level. However, up till now, there has been no direct and straightforward method for predicting protein domains in species without a reference genome sequence. In this study, we developed a functionality with a set of programs that can predict protein domains directly from genomic sequence data without a reference genome. Using whole genome sequence data, the programming functionality mainly comprised DNA assembly in combination with next-generation sequencing (NGS) assembly methods and traditional methods, peptide prediction and protein domain prediction. The proposed new functionality avoids problems associated with de novo assembly due to micro reads and small single repeats. Furthermore, we applied our functionality for the prediction of leucine rich repeat (LRR) domains in four species of Ficus with no reference genome, based on NGS genomic data. We found that the LRRNT_2 and LRR_8 domains are related to plant transpiration efficiency, as indicated by the stomata index, in the four species of Ficus. The programming functionality established in this study provides new insights for protein domain prediction, which is particularly timely in the current age of NGS data expansion.
Introduction
With the advent of next-generation sequencing (NGS) technology, a massive amount of DNA data is currently being produced in both model and non-model species. However, there are many problems associated with de novo assembly, i.e., when there is no reference genome on which to map reads, especially when the genome structure is complex with large parts of repetitive elements, as it is often the case in plant species [1]. In such cases, the DNA reads can only be assembled to scaffold or contig level [2]. Thus, methods based on an analysis of the fragments are needed.
A protein domain is a conserved part of a protein sequence which has a specific structure and function. The typical length of a protein domain is from about 25 to 500 amino acids. For some protein domain analysis, the whole protein sequence is not required [3]. Hence, some of the problems associated with full-length assembly without a reference genome can be avoided by protein domain analysis.
In the present study, fig trees belonging to the Ficus genus of the Moraceae family were examined to verify the above hypothesis. The Ficus genus has been found to have great diversity in tropical and subtropical areas, which is linked to geographical evolution within the genus [4], [5]. Ficus altissima Blume, Ficus tinctoria G. Forst, Ficus langkokensis Drake and Ficus fistulosa Reinw. ex Blume usually have overlapping distributions. However, their ecological niches are different due to their physiology. F. altissima and F. tinctoria are semi-epiphytic and their leaves are coriaceous. As a result, they can tolerate environments with drought episodes [6]. In contrast, F. langkokensis and F. fistulosa grow in relatively humid habitats, such as waterside rocks, and their leaves are thin coriaceous [7]. The ecological differences in the growing areas of these different Ficus species might thus exert different types of drought stress pressures, leading to different responses in stomatal development and morphology [8]. Hence, it would be valuable to develop a model that predicts the peptide domains of proteins for genes potentially involved in responses to drought stress, using genomic data.
One of the strategies used by plants to respond to drought stress events is plant transpiration efficiency. In the model plant Arabidopsis, plant transpiration efficiency is a quantitative trait, which has been shown to be controlled by several genes based on quantitative trait loci (QTLs) mapping studies [9]. To date, only a few contributing genes have been identified, one of which is the ERECTA gene, which explains 21–46% of the total phenotypic variation in Δ(leaf carbon isotopic discrimination) [9]. In Arabidopsis, ERECTA is one of the best studied receptor like kinases (RLKs) with leucine rich repeat (LRR) domains, which not only participates in plant transpiration efficiency but also regulates aerial architecture, stomatal patterning and confers resistance to the pathogenic bacteria Ralstonia solanacearum, the necrotrophic fungi Plectosphaerella cucumerina and Pythium irregulare [10], [11]. Structurally, the protein encoded by the ERECTA gene in Arabidopsis has one LRRNT_2 protein domain at the N-terminal, two LRR_8 protein domains in the middle part, and one Pkinase domain at the C-terminal (Fig. 1A). The LRR_8 domains form the hydrophobic core of the proteins, and they are frequently involved in the formation of protein-protein interactions [11], [12]. The LRRNT_2 domain of the protein encoded by ERECTA in Arabidopsis has LRRs flanked by cysteine rich sequences (Fig. 1B).
Figure 1. Protein domain structure of the protein encoded by the ERECTA gene in Arabidopsis thaliana.

A. From the N- to C-terminal, the protein is composed of one LRRNT_2 domain, two LRR_8 domains and one Pkinase domain. B. Amino acids of the protein. The LRRNT_2 domain and two LRR_8 domains are underlined. Leucine repeats can be found in the latter domains.
In contrast to model species, the molecular mechanism of plant transpiration efficiency still remains unclear in many plant and tree species, especially those without reference genomes. Improving functional annotation of assembled data obtained from NGS technology may provide new insights into genes potentially involved in this important trait. In this study, our first objective was to develop a method for obtaining high quality contigs from low coverage NGS data. Secondly, we attempted to predict protein domains from contigs obtained via the above method. Finally, we utilized our programming functionality to predict LRR domains homologous to those from the Arabidopsis ERECTA gene in four Ficus species that respond differently to drought environments and examined the relationship between LRR domain numbers and plant transpiration efficiency.
Materials and Methods
DNA extraction and genome sequence
Leaf material of four species, F. altissima, F. tinctoria, F. langkokensis and F. fistulosa, was collected from the Xishuangbanna Tropical Botanical Garden, Yunnan Province, P. R. China (101°25′E, 21°41′N) in April 2013 and stored in a paper bag with silica gel until DNA extraction. The four species had been transplanted in 1990 from the natural Xishuangbanna Tropical Forest, Yunna Province, P. R. China (101°57′E, 21°48′N). Genomic DNA of each individual was extracted from dried leaves using the DNeasy Plant Kit (Qiagen). DNA quality was checked on 2% agarose gels stained with ethidium bromide using a UV-Vis spectrometer (Bio-Rad Molecular Imager ChemiDoc XRS+ Imaging System) coupled with a Qubit fluorometer (ds DNA BR, Invitrogen). 40 ug RNA-free genomic DNA were used for the library construction. Library preparation (400-bp and 150-bp paired-end reads) and sequencing on an Illumina HiSeq2000 were performed by the Beijing Genomics Institute.
Quality control methods
The raw data from the Illumina Hiseq2000 were trimmed with two programs for performing quality control written in the Practical Extraction and Report Language (PERL). The first program was used to remove nucleotides with a Phred score lower than 20 (Script S1). The second program was used to delete fastq reads with length less than 20 base pairs as well as “orphanage” reads (single reads not in a pair) created by the first program (Script S2).
Sequence assembly
To generate a better genome assembly, we used a combination of four popular assembly software packages: ABySS, SOAPdenovo, Velvet and Phrap. ABySS, SOAPdenovo and Velvet were used to align the trimmed Illumina fastq reads to obtain contigs. These contigs were then aligned again with Phrap to improve the alignment.
First of all, ABySS (http://www.bcgsc.ca/platform/bioinfo/software/abyss) which allows de novo, parallel, paired-end sequence assembly for short sequence reads was used to construct alignments [13]–[15] on our Ficus genome data. We employed 25 as the k-mer length and 10 as the minimum number of pairs needed for two joined contigs.
Secondly, SOAPdenovo (http://soap.genomics.org.cn/soapdenovo.html) [16], which is particularly designed to assemble Illumina GA short reads, was used for building the contigs. The detailed parameter set was as follows: k-mer length 25; average insert size 250; cutoff value of pair number for a reliable connection between two contigs of pre-scaffolds 3; and minimum alignment length between a read and contig required for a reliable read location 32.
Thirdly, Velvet (http://www.ebi.ac.uk/~zerbino/velvet/) [17], which is a sequence assembler for very short sequence reads, was also applied for the sequence alignment. We set the k-mer length as 25 and the average insert size as 250.
Finally, Phrap (http://www.phrap.org/) [18], which is a program for assembling shotgun DNA sequence data was further applied on the sequence to increase the maximum length and remove redundancy. We analyzed the results of ABySS, SOAPdenovo and Velvet by Phrap (for parameters see Table S1 and some connection Script S3).
Gene structure identification
GENSCAN (http://genes.mit.edu/GENSCANinfo.html) was used to identify complete gene structures in genomic DNA. It is a GHMM-based program that can be used to predict the location of genes and their exon-intron boundaries in genomic sequences are from a variety of organisms. The “Arabidopsis.smat” file was downloaded and used as parameter file for the Ficus genome data [19].
Protein domain prediction
HMMER 3.0 (http://hmmer.janelia.org/) was used for searching sequence databases for homologs of protein sequences and making protein sequence alignments [20]. It employs methods using probabilistic models called profile hidden Markov models (profile HMMs). We used hmmscan to predict protein domains in the gene ERECTA, which were then predicted in Ficus and Populus by hmmsearch.
Experimental analysis
Stomata index evaluation
The study was conducted in the Xishuangbanna Tropical Botanical Garden in Yunnan Province, P. R. China (101°25′E, 21°41′N) in August 2013. Four to six trees of each species showing good growth performance were sampled. We collected three mature and well-exposed leaves from each tree. To obtain a better view of the stomata, we removed the main vein of leaves and then boiled them in hot alkaline buffer to remove the mesophyll. Treated leaves were examined under a light microscope (DM2500, Leica, Germany). The numbers of stomata and epidermal cells were counted using ImageJ (National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/index.html). We used the formula (100×stomatal density)/(stomatal density + epidermal cell density) to calculate the stomata index as in Hara et al., (2009) [21]. We repeated the experiments six times.
Results and Discussion
Genomic contigs assembled from Illumina genomic sequence data
The Illumina Hiseq2000 genomic sequence data in fastq format for each of F. altissima, F. tinctoria, F. langkokensis and F. fistulosa was about 15 Gigabytes, and thus the coverage was about 15x [22]. The quality control methods improved the quality of the Illumina genomic sequence data for all four species, where the number of reads was about 135 million for each species. For the gene and protein domain prediction, it was most appropriate to use the contigs that had a base pair number >250 as the input for Phrap as the typical number of nucleotides in a DNA fragment encoding a protein domain of the LRR family is about 250 (Fig. 2). The maximum lengths of contigs predicted by Phrap in F. altissima, F. tinctoria, F. langkokensis and F. fistulosa were 6914 base pairs, 10755 base pairs, 6665 base pairs and 9203 base pairs, respectively. These numbers showed that the genomic contigs finally assembled could be used for the gene prediction (Table 1).
Figure 2. The proposed programming functionality for predicting protein domains directly from genomic sequence data without a reference genome.
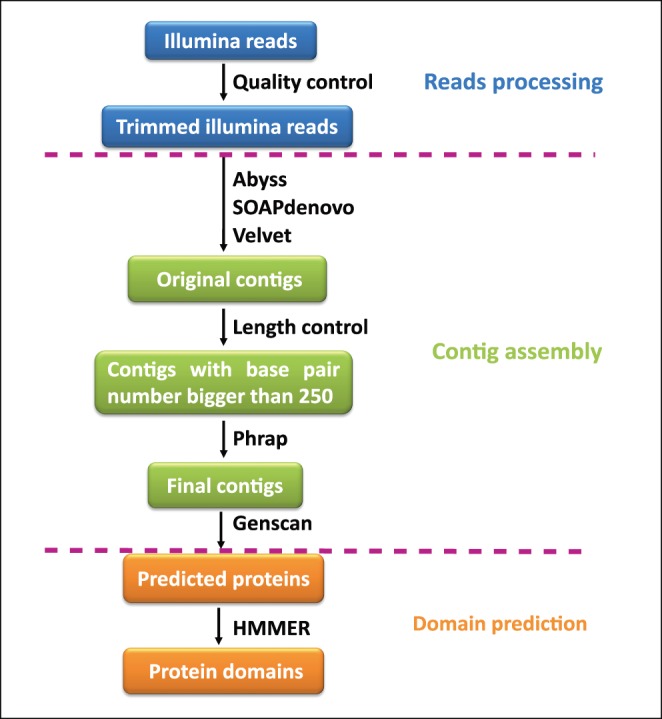
The Illumina reads were first trimmed with quality control methods. Then, assembly software ABySS, SOAPdenovo and Velvet were used separately to obtain original contigs. Next, length control methods were used to select contigs larger than 250 base pairs. Afterwards, the assembly software Phrap was used to obtain final contigs and Genscan was used to predict peptides from these contigs. Finally, Hmmsearch was used to predict protein domains.
Table 1. Results from the assembly software.
| Species | #fastqreads | Coverage | Software | #contig_250 | max_len(bp) | #pep | max_len(aa) | #LRRNT_2 | #LRR_8 |
| FA | 2,185,253,886 | 4.86 | Abyss | 26,816 | 1,968 | 10,846 | 606 | 5 | 72 |
| SOAP | 26,898 | 1,906 | 10,735 | 578 | 11 | 71 | |||
| Velvet | 123,763 | 6,407 | 23,086 | 702 | 22 | 120 | |||
| Phrap | 114,596 | 6,914 | 21,901 | 880 | 19 | 132 | |||
| FT | 2,197,543,362 | 4.88 | Abyss | 54,144 | 2,739 | 8,595 | 436 | 3 | 40 |
| SOAP | 59,831 | 2,524 | 8,419 | 306 | 4 | 33 | |||
| Velvet | 170,753 | 9,251 | 15,319 | 418 | 6 | 59 | |||
| Phrap | 154,710 | 10,755 | 14,807 | 467 | 9 | 62 | |||
| FL | 1,993,136,266 | 4.43 | Abyss | 7,679 | 2,002 | 2,426 | 506 | 1 | 24 |
| SOAP | 8,321 | 3,430 | 2,611 | 506 | 3 | 23 | |||
| Velvet | 86,717 | 6,718 | 6,479 | 534 | 3 | 32 | |||
| Phrap | 84,287 | 6,665 | 6,822 | 550 | 3 | 45 | |||
| FF | 869,615,244 | 1.93 | Abyss | 7,087 | 5,558 | 2,669 | 772 | 2 | 14 |
| SOAP | 7,049 | 7,064 | 2,609 | 772 | 0 | 12 | |||
| Velvet | 12,129 | 7,511 | 3,092 | 772 | 0 | 17 | |||
| Phrap | 13,972 | 9,203 | 3,827 | 1,536 | 2 | 19 |
FA, FT, FL and FF stands for Ficus altissima, Ficus tinctoria, Ficus langkokensis and Ficus fistulosa, respectively.
#fastq reads: number of fastq reads from Illumina Hiseq2000.
#contig_250: number of predicted contigs longer than 250 base pairs.
max_len (bp): number of base pairs (bp) of the contigs predicted with maximum length.
#pep: number of peptides predicted.
max_len (aa): number of amino acids (aa) of the peptides predicted with maximum length.
#LRRNT_2: number of LRRNT_2 domains predicted.
#LRR_8: number of LRR_8 domains predicted.
Peptides and protein domains predicted from genomic contig data
Genscan was used to predict peptide sequences from the genomic contigs assembled by Phrap for F. altissima, F. tinctoria, F. langkokensis and F. fistulosa (Fig. 2). The maximum lengths of peptides predicted by Genscan for the four species (880, 467, 550 and 1576 amino acids, respectively) were employed since longer peptides enable better protein domain prediction. These numbers indicated that the predicted peptides could be used to predict protein domains (Table 1).
HMMER was used to predict protein domains from the peptides predicted from Genscan for the four species (Fig. 2). The numbers of LRRNT_2 domains predicted from the contigs assembled by Phrap were 19, 9, 3, and 2 in F. altissima, F. tinctoria, F. langkokensis and F. fistulosa, respectively, whereas the numbers of LRR_8 domains were 132, 62, 45, and 19, respectively (Table 1).
Phrap can improve assembly of contigs and remove identical segments of genomic sequences
Phrap cannot directly work on Illumina fastq reads. However, it can increase the maximum length of the contigs assembled through ABySS, SOAPdenovo and Velvet, which can directly work on Illumina fastq reads (Table 1). Thus, the maximum length of the peptides predicted by Genscan may also be increased (Fig. 3).
Figure 3. Maximum length (number of amino acids) of peptides predicted by the programming functionality.
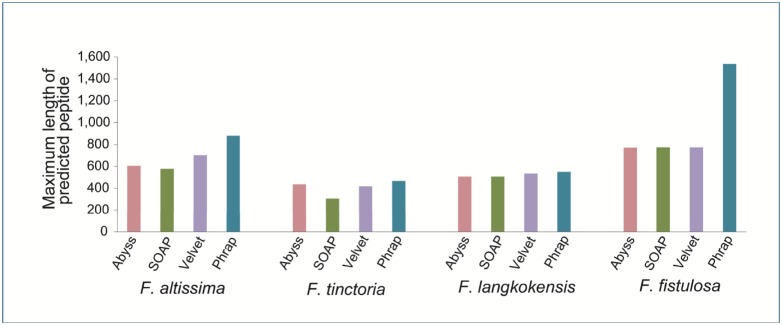
The Illumina reads for F. altissima (FA), F. tinctoria (FT), F. langkokensis (FL) and F. fistulosa (FF) were assembled by ABySS, SOAPdenovo and Velvet. Phrap was used to assemble the contigs from ABySS, SOAPdenovo and Velvet, and then Genscan was used to predict peptides from these contigs. The maximum length of the peptides could be increased by Phrap in FA, FT, FL and FF.
Assembly through Phrap can remove redundancy of the contigs, including the identical genomic sequence segments predicted by ABySS, SOAPdenovo and Velvet. The percentage of redundancy removed from F. altissima, F. tinctoria, F. langkokensis and F. fistulosa was 36.51, 45.91, 18.64 and 46.91 respectively (Table 2). The Phrap software can be used to combine identical DNA fragments into one sequence, thus avoid the effect of gene expression difference produced by NGS methods. Programs like CAP3 and TIGR Assembler may also offer similar functions. However, it is important to choose correct parameters for different species. When applying on the contigs which are assembled by NGS assembly methods, we found that Phrap has more suitable parameters to be adjusted comparing to other programs by testing different combinations of parameters values. The raw data sequences used here were submitted to NCBI under accession number SRP041276.
Table 2. Redundancy removed by Phrap.
| Species | #contig_250from Abyss,SOAP andVelvet | #basepairs | #contig_250not usedby Phrap | #basepairs | #contig_250used byPhrap | #basepairs | #contig_250after Phrap | #basepairs | Percent ofredundancy removedby Phrap |
| FA | 177477 | 79652086 | 86943 | 37241030 | 90534 | 42411056 | 27653 | 13333742 | 36.51 |
| FT | 284698 | 124099037 | 95706 | 40299597 | 188992 | 83799440 | 59004 | 26821922 | 45.91 |
| FL | 102717 | 40680387 | 75107 | 29210626 | 27610 | 11469761 | 9180 | 3886983 | 18.64 |
| FF | 26265 | 9447858 | 7416 | 2344792 | 18849 | 7103066 | 6556 | 2670964 | 46.91 |
FA, FT, FL and FF stands for Ficus altissima, Ficus tinctoria, Ficus langkokensis and Ficus fistulosa, respectively.
#contig_250: number of contigs longer than 250 base pairs.
#base pairs: number of base pairs.
The numbers of LRRNT_2 and LRR_8 domains in Ficus correlate with stomata index values
To test whether the LRRNT_2 domains and LRR_8 domains are related to transpiration efficiency, we used our programming functionality to predict their numbers in the four species of Ficus. The mean values of the stomata index for F. altissima, F. tinctoria, F. langkokensis and F. fistulosa were 5.3, 10.9, 13.6 and 15.9, respectively (Table 3). As the stomata index values increased in these species the numbers of LRRNT_2 and LRR_8 domains decreased accordingly (Fig. 4). To eliminate the contingency in protein domain selection, we used the actin domain from actin1 protein in Arabidopsis thaliana (NCBI accession number NP_850284.1) for control analysis. Actin is a house-keeping protein expressed in every plant cell as a component of the cytoskeleton [23], and thus provides a good control. Among all the peptides predicted for the four Ficus species, one actin domain was found to be longer than 100 amino acids and another was shorter than 50 amino acids (Fig. 4). These results suggest that the transpiration efficiency could be related to the LRRNT_2 and LRR_8 domains in Ficus.
Table 3. Physiological, anatomical and stomata response data in Ficus.
| Species | #stomata | #epidermalcells | Stomataldensity | Epidermalcell density | Stomatalindex | |
| FA | M | 12.91667 | 231.1944 | 326.5458 | 5844.819 | 5.301273 |
| SD | 2.061553 | 20.15769 | 52.11805 | 509.606 | 0.79305 | |
| SE | 0.343592 | 3.359615 | 8.686342 | 84.93433 | 0.132175 | |
| FT | M | 20.84848 | 169.0303 | 527.0699 | 4273.25 | 10.90198 |
| SD | 4.016538 | 13.41754 | 101.542 | 339.2083 | 1.331365 | |
| SE | 0.699189 | 2.335693 | 17.67619 | 59.04859 | 0.231761 | |
| FL | M | 15.66667 | 99.47619 | 396.0685 | 2514.854 | 13.61349 |
| SD | 1.932184 | 7.35268 | 48.84747 | 185.8829 | 1.467321 | |
| SE | 0.421637 | 1.604486 | 10.65939 | 40.56297 | 0.320196 | |
| FF | M | 19.125 | 99.2 | 483.4985 | 2507.872 | 15.90947 |
| SD | 3.879433 | 10.0584 | 98.07582 | 254.2861 | 1.932021 | |
| SE | 0.969858 | 2.597068 | 24.51895 | 65.65639 | 0.498846 |
FA, FT, FL and FF stands for Ficus altissima, Ficus tinctoria, Ficus langkokensis and Ficus fistulosa, respectively.
M, SD, and SE: mean, standard deviation and standard error, respectively.
#stomata: number of stomata.
#epideman cells: number of epidermal cells.
Figure 4. Number of LRRNT_2, LRR_8 and actin domains predicted in F. altissima (FA), F. tinctoria (FT), F. langkokensis (FL) and F. fistulosa (FF) (A); and stomata index in FA, FT, FL and FF (B).

As the number of LRRNT_2 and LRR_8 domains decreased for FA, FT, FL and FF, the stomata index increased.
The ERECTA gene has not only a positive regulatory role on respiration in drought conditions but also benefits plants in the absence of water shortage [9]. Therefore, the protein domains in the ERECTA gene might show a cumulative positive evolution. The LRR_8 domain has more LRRs than the LRRNT_2 domain, and thus may have a more important role in protein-protein interactions (Fig. 1). Hence, this could explain why the number of LRR_8 domains was more than that of LRRNT_2 domains (Fig. 4).
Conclusion
The programming functionality in this study was proved to be a useful tool in biological studies by showing that the LRRNT_2 and LRR_8 domains were potentially related to plant transpiration efficiency, as we can see from the stomata index in F. altissima, F. tinctoria, F. langkokensis, and F. fistulosa. The main benefit of the functionality is that it overcomes many of the complex problems associated with de novo assembly. However, with the increasing read lengths produced by NGS and improvements in third-generation sequencing, such problems may also be solved with the rapid developments of de novo assembly methods. The main limitation of the functionality is GENSCAN prediction step, which requires a suitable model. In addition, it is hard for some species to choose a perfect model to predict the gene structure. Confronting with this situation, researchers normally prefer to pick a widely used model which turns out to have more or less shortage. Nevertheless, methods of whole genome protein domain analysis will still help researchers to better understand some mechanisms of biological function from the perspective of genetic sequence, if combined with a large amount of NGS data.
Supporting Information
Table for Phrap parameters.
(DOCX)
Perl program used to remove the nucleotides which have Phred score lower than a specific value.
(DOCX)
Perl program used to delete the fastq reads which have length less than a specific value as well as to erase the “orphange” reads (single reads without pair).
(DOCX)
Perl programs used for dealing with the assembly files which were created by Phrap as well as for making statistic analysis.
(DOCX)
Acknowledgments
We would like to thank the following colleagues from the Xishuangbanna Tropical Botanical Garden (XTBG), Chinese Academy of Sciences (CAS): Bo Pan for collecting samples and Jun Yang for providing experimental equipment.
Funding Statement
This work was funded by National Natural Science Foundation of China (grant number 61271447) to TL; National Natural Science Foundation of China (grant number 41201051), 111 Project (grant number B13007) and Program for Changjiang Scholars and Innovative Research Team in University (grant number IRT13047) to FKD. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, et al. (2008) ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res 18: 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corpet F, Servant F, Gouzy J, Kahn D (2000) ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res 28: 267–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg CC (1989) Classification and distribution of Ficus. Experientia 45: 605–611. [Google Scholar]
- 5. Harrison RD (2005) Figs and the diversity of tropical rainforests. Bioscience 55: 1053–1064. [Google Scholar]
- 6. Hao GY, Sack L, Wang AY, Cao KF, Goldstein G (2010) Differentiation of leaf water flux and drought tolerance traits in hemiepiphytic and non-hemiepiphytic Ficus tree species. Funct Ecol 24: 731–740. [Google Scholar]
- 7.Wu ZY, Zhou ZK, Gilbert MG (2004) Flora of China. Beijing: Science Press. Vol. 5: 21 p. [Google Scholar]
- 8. Hamanishi ET, Thomas BR, Campbell MM (2012) Drought induces alterations in the stomatal development program in Populus. J Exp Bot 63: 4959–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masle J, Gilmore SR, Farquhar GD (2005) The ERECTA gene regulates plant transpiration efficiency in Arabidopsis. Nature 436: 866–870. [DOI] [PubMed] [Google Scholar]
- 10. Torii KU, Mitsukawa N, Oosumi T, Matsuura Y, Yokoyama R, et al. (1996) The arabidopsis ERECTA gene encodes a putative receptor protein kinase with extracellular leucine-rich repeats. Plant Cell 8: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobe B, Kajava AV (2001) The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol 11: 725–732. [DOI] [PubMed] [Google Scholar]
- 12. Wei T, Gong J, Jamitzky F, Heckl WM, Stark RW, et al. (2008) LRRML: a conformational database and an XML description of leucine-rich repeats (LRRs). BMC Struct Biol 8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robertson G, Schein J, Chiu R, Corbett R, Field M, et al. (2010) De novo assembly and analysis of RNA-seq data. Nat Methods 7: 909–912. [DOI] [PubMed] [Google Scholar]
- 15. Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, et al. (2009) De novo transcriptome assembly with ABySS. Bioinformatics 25: 2872–2877. [DOI] [PubMed] [Google Scholar]
- 16. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 17. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Machado M, Magalhães WC, Sene A, Araújo B, Faria-Campos AC, et al. (2011) Phred-Phrap package to analyses tools: a pipeline to facilitate population genetics re-sequencing studies. Investig Genet 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burge C, Karlin S (1997) Prediction of complete gene structures in human genomic DNA. J Mol Biol 268: 78–94. [DOI] [PubMed] [Google Scholar]
- 20. Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hara K, Yokoo T, Kajita R, Onishi T, Yahata S, et al. (2009) Epidermal cell density is autoregulated via a secretory peptide, EPIDERMAL PATTERNING FACTOR 2 in Arabidopsis leaves. Plant Cell Physiol 50: 1019–1031. [DOI] [PubMed] [Google Scholar]
- 22. Deepak O, Genome Size Variation, Plant Systematics (1998) Annals of Botany. 82 (Supplement A)75–83. [Google Scholar]
- 23. Shah DM, Hightower RC, Meagher RB (1983) Genes encoding actin in higher plants: intron positions are highly conserved but the coding sequences are not. J Mol Appl Genet 2: 111–126. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table for Phrap parameters.
(DOCX)
Perl program used to remove the nucleotides which have Phred score lower than a specific value.
(DOCX)
Perl program used to delete the fastq reads which have length less than a specific value as well as to erase the “orphange” reads (single reads without pair).
(DOCX)
Perl programs used for dealing with the assembly files which were created by Phrap as well as for making statistic analysis.
(DOCX)