

Abstract
Congenital insensitivity to pain (CIP) is a rare autosomal recessive genetic disease caused by mutations in the SCN9A gene. We report a patient with the clinical features consistent with CIP in whom we detected a novel homozygous G2755T mutation in exon 15 of this gene. Routine electrophysiological studies are typically normal in patients with CIP. In our patient, these studies were abnormal and could represent the consequences of secondary complications of cervical and lumbosacral spine disease and associated severe Charcot's joints.
1. Introduction
Autosomal recessive congenital insensitivity to pain (CIP) is a rare condition, affecting very few individuals, but with a worldwide distribution. CIP is clinically characterized by the ability to feel a given stimulus but also the inability to perceive pain. This is in contrast to congenital “indifference” to pain which implies a lack of concern to a painful stimulus that is received through normal sensory pathways and may be associated with central nervous system disorders such as schizophrenia or pervasive development disorder [1].
CIP is genetically and clinically heterogeneous caused by mutations in several different genes. For example, mutations in the neurotrophic tyrosine kinase receptor type 1 gene (NTRK1) and nerve growth factor-β (NGFB) result in CIP with an anhidrosis phenotype [2, 3]. In contrast, homozygous loss of function mutations in sodium channel voltage-gated type IX, alpha subunit (SCN9A) gene has been reported to result in the CIP with an anosmia phenotype [4]. Although this condition is rare, genotype phenotype studies of such patients are important.
We report the results of our analysis of a patient who we encountered in our neurology clinic with a history of insensitivity to pain.
2. Case Report
This 58-year-old woman presented with a long history of insensitivity to pain since childhood and increased numbness in her legs for several years. As a child, she recalled developing cuts on her feet that she could not feel. She could distinguish between hot and cold temperature although there was no uncomfortable sensation associated with extremes of either one. Since the age of 15 years she started to develop frequent fractures involving multiple bones which were also painless. In addition, she has two children and suffered no pain during childbirth. She also had anosmia. Over the ten years prior to evaluation, she had started to develop sensory loss in her legs. She had previously been diagnosed with cervical and lumbar spine disease and had undergone surgical treatment of both of these regions of her spine. She is of Caucasian English descent and the product of a nonconsanguineous marriage. She has a healthy brother and two healthy children. There is no indication that either her parents or any other relative was affected by symptoms suggestive of CIP suggesting an autosomal recessive form of inheritance. The remainder of the general medical history was significant for absence of diabetes, cancer, or rheumatologic disease. Neurological examination revealed normal mental status and cranial nerve examination except for anosmia. She was diffusely areflexic with flexor plantar responses. She had multiple joint deformities involving both ankles, elbows, and knees (Charcot's joints) which limited the testing of power. When she could provide a good effort, she had good strength. She had decreased sensation to pin prick, proprioception, and vibration distally in her feet. She could not perform a tandem walk and had a positive Romberg's test.
An electromyogram (EMG) was performed; the motor nerve conduction parameters were normal in the right tibial nerve but showed a markedly reduced response amplitude in the right peroneal nerve recording the extensor digitorum brevis muscle (this was severely atrophied). No evoked response could be elicited with stimulation of the peroneal nerve at the fibular head. No evoked sensory nerve action potentials were obtained in the right ulnar, sural, and superficial peroneal nerves. Needle electromyogram showed no abnormal spontaneous activity in any muscle sampled and the presence of high amplitude polyphasic units in the distal muscles of the right arm and legs associated with a mildly reduced interference pattern with maximal effort. Overall the study was interpreted as showing chronic neurogenic changes with a superimposed entrapment neuropathy of the right ulnar nerve. The other abnormalities noted in the nerve conductions were interpreted as partly secondary to Charcot's joints and technical factors such as increased subcutaneous tissues.
3. Genetic Analysis
Following IRB approved policies and procedures, a blood sample was obtained and DNA was extracted. Whole exome sequencing was performed by commercial sequencing company. Exome capture was performed by HiSeq2000 using a paired-end (2 × 100) protocol, Illumina raw data processing, and Agilent SureSelect exome kit for exome enrichment. The sequences were aligned to human genome reference (UCSC version hg 19). Nucleotide-level variation analysis of the exome sequence data was performed using the DNA nexus platform (https://dnanexus.com/). The variants obtained with this platform were further annotated using Ensembl variant effect predictor tool (Ensembl release 75, February 2014) (http://useast.ensembl.org/info/docs/tools/vep/index.html) [5]. Since CIP is a rare disorder, the minor allele frequency was assigned at less than 1%. These results were further filtered for homozygous, nonsynonymous variants with deleterious, possible damaging and unknown effect using SIFT and Polyphen analysis. This narrowed down the list of variants to 584.
Those single nucleotide polymorphisms (SNPs) involving genes known to cause insensitivity to pain were then analyzed. A potentially significant variant was identified on chromosome 2 at position 167133579, a homozygous A/A variant (Figure 1(a)). This homozygous c. G2755T mutation in exon 15 of SCN9A gene results in a stop mutation, causing premature truncation of the protein p. E919X. This SNP was reconfirmed by amplification and Sanger's sequencing (Figure 1(b)).
Figure 1.
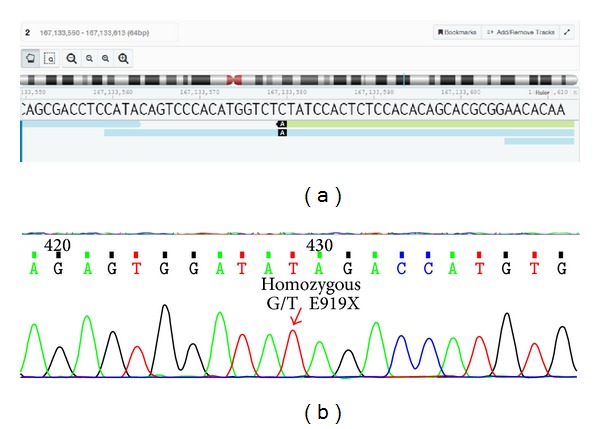
Image showing the homozygous variant on chromosome 2. (a) Image identifying the homozygous A/A mutation on chromosome 2 at position 167133579 using the DNA nexus platform. (b) Image showing the homozygous c. G2755T mutation in exon 15 of SCN9A gene following amplification and Sanger's sequencing.
4. Discussion
The SCN9A gene is expressed in all sensory neurons and is a key molecule in the processing of peripheral pain. This gene encodes a voltage-gated sodium channel (Nav 1.7) which plays a significant role in nociceptive signaling and both gain and loss of function mutations have been reported. Interestingly, depending upon the specific mutation, there is a marked diversity of resulting phenotype. For example, gain of function mutations causes inherited erythromelalgia and paroxysmal extreme pain disorder which follow an autosomal dominant pattern of inheritance [6, 7]. More recently there have been reports of mutations causing seizures or a small fiber neuropathy [8, 9].
Studies in individuals with CIP from seven different populations identified homozygous mutations in SCN9A gene [10]. Loss of function mutations in SCN9A gene causes truncation of the encoded sodium channel Nav 1.7 protein, resulting in channelopathy-associated autosomal recessive congenital insensitivity to pain. Twenty-seven different SCN9A gene mutations have been reported in CIP patients so far (Table 1). Given the predicted consequences of the novel change in the SCN9A gene in our patient, it is likely to be a disease producing mutation and brings the total number of mutations to twenty-eight.
Table 1.
SCN9A mutations causing congenital indifference to pain.
| SCN9A mutations | Reference |
|---|---|
| c.1376C>G, p.Ser459Ter | Cox et al., 2006 [4] |
| c.2298delT, pIle767Ter | |
| c.2691G>A, p.Trp897Ter | |
|
| |
| c.828delGT | Nilsen et al., 2009 [9] |
| c.2575C>T | |
|
| |
| c.829C>T, p.Arg277Ter | Goldberg et al., 2007 [10] |
| c.984C>A, p.Tyr328Ter | |
| c.2455C>T, p.Arg630X | |
| c.3600delT, p.Phe1200LeufsX33 | |
| c.4462C>T, p.Arg1488X | |
| c5067G>A, p.Trp1689X | |
| c.2076_2077InsT, p.Glu693X | |
| c.4366-7_10delGTTT, del 4 bp, splice junction mutation | |
| c.3703_3713del, del 11 bp, pIle1235LeufsX2 | |
| c.4975A>T, p.Lys1450X | |
|
| |
| c.1126A>C, p.K376Q | Shorer et al., 2014 [14] |
| c.1124delG, p.G375AfsX5 | |
|
| |
| c.984C>A, p.Y328X | Ahmad et al., 2007 [15] |
|
| |
| c.2687G>A, p.R896Q | Cox et al., 2010 [16] |
| c.4108_4122delCGATGGAAAAACCTG, p.R1370-L1374 del | |
| c.4474delA, p.I1493SfsX8 | |
|
| |
| c.1567C>T, p.Arg523Ter | Kurban et al., 2010 [17] |
|
| |
| c.2697G>A, p.Met899Ile | Yuan et al., 2011 [18] |
| c.2796A→C, p.Met932Leu | |
|
| |
| c.5155T>C; C1719R | Staud et al., 2011 [19] |
| c.3467+3delA, or IVS17+3 delA | |
|
| |
| c.1567C>T, p.Arg523Ter | Klein et al., 2013 [20] |
| IVS8-2A>G | |
Although the primary consequence of the homozygous SCN9A mutation is the absence of pain sensation, there are associated conditions including anosmia, self-mutilation resulting in oral and digit lesions, multiple injuries due to repeated trauma, burn-related injuries, orthopedic complications that include bone deformities from untreated fractures, osteomyelitis, and neuropathic joints later in life [4, 8, 9, 11–14]. Although Charcot's joints are commonly reported in patients with CIP, bony involvement of the spine as seen in our patient is rare; however, anosmia and Charcot's joints noted in our patient are comorbidities that were previously reported associated with CIP [4, 8, 9, 14].
Routine EMG studies of patients with CIP are typically normal. In our patient, it is likely that the abnormalities detected on both the nerve conduction studies and needle examination are secondary to cervical and lumbosacral spine disease, joint deformities, and muscle wasting associated with Charcot's joints. However, a sensory motor peripheral neuropathy is not excluded by this examination. It is possible that the patient has an associated large fiber neuropathy which may be related to the G2755T mutation or alternatively to another unrelated etiology. A possible relationship between mutations in the SCN9A and a large fiber neuropathy could be supported by genotype/phenotype analysis in further patients with CIP.
The study of our patient expands the spectrum of mutations that have been reported to cause this disorder. In addition, our analysis demonstrates the power of next generation sequencing that can enable genetic confirmation of a suspected diagnosis of a rare disorder.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Dyck PJ, Mellinger JF, Reagan TJ, et al. Not “indifference to pain” but varieties of hereditary sensory and autonomic neuropathy. Brain. 1983;106(2):373–390. doi: 10.1093/brain/106.2.373. [DOI] [PubMed] [Google Scholar]
- 2.Miura Y, Mardy S, Awaya Y, et al. Mutation and polymorphism analysis of the TRKA (NTRK1) gene encoding a high-affinity receptor for nerve growth factor in congenital insensitivity to pain with anhidrosis (CIPA) families. Human Genetics. 2000;106(1):116–124. doi: 10.1007/s004390051018. [DOI] [PubMed] [Google Scholar]
- 3.Carvalho OP, Thornton GK, Hertecant J, et al. A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5 neuropathy. Journal of Medical Genetics. 2011;48(2):131–135. doi: 10.1136/jmg.2010.081455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flicek P, Amode MR, Barrell D, et al. Ensembl 2014. Nucleic Acids Research. 2014;42(1):D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of Medical Genetics. 2004;41(3):171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michiels JJ, Te Morsche RHM, Jansen JBMJ, Drenth JPH. Autosomal dominant erythermalgia associated with a novel mutation in the voltage-gated sodium channel α subunit Nav1.7. Archives of Neurology. 2005;62(10):1587–1590. doi: 10.1001/archneur.62.10.1587. [DOI] [PubMed] [Google Scholar]
- 8.Piazza MR, Bassett GS, Bunnell WP. Neuropathic spinal arthropathy in congenital insensitivity to pain. Clinical Orthopaedics and Related Research. 1988;(236):175–179. [PubMed] [Google Scholar]
- 9.Nilsen KB, Nicholas AK, Woods CG, Mellgren SI, Nebuchennykh M, Aasly J. Two novel SCN9A mutations causing insensitivity to pain. Pain. 2009;143(1-2):155–158. doi: 10.1016/j.pain.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg YP, Macfarlane J, Macdonald ML, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clinical Genetics. 2007;71(4):311–319. doi: 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 11.Weiss J, Pyrski M, Jacobi E, et al. Loss-of-function mutations in sodium channel Na v 1.7 cause anosmia. Nature. 2011;472(7342):186–190. doi: 10.1038/nature09975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirsch E, Moye D, Dimon JH., III Congenital indifference to pain: Long-term follow-up of two cases. Southern Medical Journal. 1995;88(8):851–857. doi: 10.1097/00007611-199508000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Cassidy RC, Shaffer WO. Charcot arthropathy because of congenital insensitivity to pain in an adult. Spine Journal. 2008;8(4):691–695. doi: 10.1016/j.spinee.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Shorer Z, Wajsbrot E, Liran T-H, Levy J, Parvari R. A novel mutation in SCN9A in a child with congenital insensitivity to pain. Pediatric Neurology. 2014;50(1):73–76. doi: 10.1016/j.pediatrneurol.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad S, Dahllund L, Eriksson AB, et al. A stop codon mutation in SCN9A causes lack of pain sensation. Human Molecular Genetics. 2007;16(17):2114–2121. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- 16.Cox JJ, Sheynin J, Shorer Z, et al. Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations. Human Mutation. 2010;31(9):E1670–E1686. doi: 10.1002/humu.21325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurban M, Wajid M, Shimomura Y, Christiano AM. A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology. 2010;221(2):179–183. doi: 10.1159/000314692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan R, Zhang X, Deng Q, et al. Two novel SCN9A gene heterozygous mutations may cause partial deletion of pain perception. Pain Medicine. 2011;12(10):1510–1514. doi: 10.1111/j.1526-4637.2011.01237.x. [DOI] [PubMed] [Google Scholar]
- 19.Staud R, Price DD, Janicke D, et al. Two novel mutations of SCN9A (Nav1.7) are associated with partial congenital insensitivity to pain. European Journal of Pain. 2011;15(3):223–230. doi: 10.1016/j.ejpain.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein CJ, Wu Y, Kilfoyle DH, et al. Infrequent SCN9A mutations in congenital insensitivity to pain and erythromelalgia. Journal of Neurology, Neurosurgery and Psychiatry. 2013;84(4):386–391. doi: 10.1136/jnnp-2012-303719. [DOI] [PMC free article] [PubMed] [Google Scholar]