

Abstract
Manipulating biochemical reactions in living cells to synthesize nanomaterials is an attractive strategy to realize their synthesis that cannot take place in nature. Yeast cells have been skillfully utilized to produce desired nanoparticles through spatiotemporal coupling of intracellular nonrelated biochemical reaction pathways for formation of fluorescent CdSe quantum dots. Here, we have successfully transformed Staphylococcus aureus cells into cellular beacons (fluorescing cells), all of which are highly fluorescent and photostable with perfect uniformity. Importantly, on the basis of such cells, we efficiently fabricated fluorescent nanobioprobes by a specific interaction between the protein A expressed on the S. aureus surface and the Fc fragment domain of antibodies, avoiding the use of other common methods for cell surface modifications, such as molecular covalent connection or more difficult genetic and metabolic engineering. Coupled with immunomagnetic beads, the resulting fluorescent-biotargeting bifunctional cells, i.e., biotargeting cellular beacons, can be employed as nanobioprobes for detection of viruses, bacteria, and tumor cells. With this method, H9N2 AIV can be detected specifically with a limit of 8.94 ng/mL (based on protein content). Furthermore, diverse probes for detection of different pathogens or for other biomedical applications can be easily obtained by simply changing the antibody conjugated to the cell surface.
Keywords: anobioprobe, beacon, quantum dot, pathogen, detection
In recent years, the biosynthesis of nano-materials by using organisms has attracted a lot of attention due to the need to develop clean, nontoxic, and eco-friendly sustainable “green chemistry” engineering. Organisms are highly organized systems with a sophisticated level of molecular control over the biosynthesis of inorganic materials. Numerous organisms have been employed to synthesize inorganic nanomaterials.1–5 Despite tremendous progress made in this area, it is still challenging to produce high-quality luminescent nano-particles by using organisms, and it is even more difficult to conveniently utilize the intracellular fluorescence function for biomedical applications.3,6,7 It is laborious and time-consuming to isolate the luminescent nanoparticles from the organism.8 In addition, even after successful isolation, such nanocrystals need to be further engineered with targeting ligands through complicated processes for further applications. Often, these processes can reduce the targeting capabilities of the ligand,9 cause nonspecific binding,10,11 and even induce the aggregation of nanoparticles.11–13 Moreover, the activity of biomolecules may be affected after coupling to the nanoparticles.14 Thus, there is an urgent need for a simple and efficient method to generate and use the intracellular fluorescence from nanomaterials.
In our previous work, by the “space–time coupling strategy”, i.e., by temporally and spatially collaborative coupling of intracellular glutathione reductase-involved selenium-(IV) reduction reactions with nonrelated intracellular Cd(II) detoxification, the cadmium precursor formed by Cd(II) detoxification can react with low-valence organoselenium resulting from selenium(IV) reductions to yield color-tunable fluorescent CdSe quantum dots in living yeast cells, which can never occur in live cells in nature.1
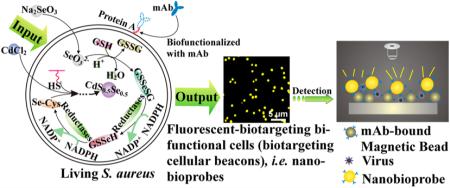
Here, our aim is to transform Staphylococcus aureus into cellular beacons by using a “space–time coupling strategy”, which can then be used to fabricate fluorescent nanobioprobes via a specific interaction between protein A expressed on the S. aureus surface and the Fc region of antibodies against specific targets of interest (Figure 1a).15–17 The space–time coupling strategy involves first the co-incubation of Na2SeO3 and S. aureus cells at the stationary phase to reduce Na2SeO3 to organoselenium compounds such as selenocysteine and then the addition of CdCl2 into the seleniumized cell culture at an appropriate time, such that intracellular detoxification of a Cd2+-forming Cd precursor can react precisely with the intracellularly reduced selenium and endogenous biomolecules containing mercapto groups to create CdS0.5Se0.5 at the right place. By using this strategy, cells that are internally labeled with fluores-cent quantum dots (QDs) can be obtained via facile chemical handling and rational utilization of intracellular biochemical processes. We refer to such cells as cellular beacons due to their fluorescence-enabled easy detection. The resulting cellular beacons have outstanding photostability, high luminance, good monodispersity, and perfect uniformity. Furthermore, protein A endogenously expressed on the surface of S. aureus can be utilized as a natural capture probe capable of binding the Fc region of antibodies. Thus, just by mixing the cellular beacons with monoclonal antibodies (mAbs) and simply centrifuging to remove excess antibodies, fluorescent S. aureus can effectively conjugate with mAbs to produce biotargeting fluores-cent cells without any additional fabrications. Coupled with immunomagnetic beads,18–22 the resulting bio-targeting fluorescent cells, namely, biotargeting cellular beacons (here referred to as nanobioprobes), can be used for detection of viruses, bacteria, tumor cells, etc. (Figure 1b). Such bioprobes can potentially be used generally for detection of different kinds of pathogens just by changing the antibody conjugated to the cell surface.
Figure 1.

Schematic illustration for the generation and use of nanobioprobes. (a) Fabrication of bioprobes. Space–time coupling strategy coupled intercellular biochemical processes in the natural systems including reduction of Na2SeO3 and detoxification of Cd2+ inside the cell. The first step is the cellular reduction of Na2SeO3 leading to the accumulation of selenocysteine, which is the precursor for CdS0.5Se0.5 QDs. The S element for the formation of CdS0.5Se0.5 QDs comes from endogenous biomolecules. The second step is treatment of seleniumized cells with CdCl2 for 12 h, which allows the synthesis of the fluorescent CdS0.5Se0.5 QDs to form cellular beacons. As protein A is a cell-wall-associated protein expressed on the S. aureus surface, which can specifically bind to the Fc fragment domain of immunolglobulins, just by mixing such cellular beacons with mAbs, the cells could effectively conjugate with mAbs to form biotargeting cellular beacons without any additional modification. (b) Using biotargeting cellular beacons (fluorescent-biotargeting bifunctional cells) as bioprobes for pathogen detection. The bioprobes and magnetic beads coupled with the same mAbs for a specific virus as described in the text can be used together for the detection of viruses with the mAb-bound magnetic beads used for isolation to enrich the virus and the probes used for detection as diagrammed.
RESULTS AND DISCUSSION
Optical Properties of the Cellular Beacons
When we co-incubated seleniumized cells with CdCl2 for 12 h, the cells displayed unique yellow fluorescence internally, while no fluorescence appeared from the control cells (Figure 2a–d). The cellular beacons showed good dispersibility and strong intracellular fluorescence (Figure 2b,e). The average hydrodynamic diameter of the cellular beacons was measured to be about 800.7 nm with a PDI (polydispersity index) of 0.082, suggesting that the cellular beacons were well-mono-dispersed with perfectly uniform sizes (Figure 2f).
Figure 2.

Characterization of cellular beacons. (a, b) Bright and fluorescent field images of cellular beacons (i.e., cultured with Na2SeO3 and CdCl2), respectively. (c, d) Corresponding images for the control of cells cultured neither with Na2SeO3 nor CdCl2. (e) Enlarged panel of the boxed region in (b), showing perfectly uniform size and strong fluorescence of the individual cells. (f) Histogram showing the size distribution of cellular beacons, with the average hydrodynamic diameter of the cellular beacons at about 800.7 nm with a PDI of 0.082. (g) Histogram showing the fluorescence intensity distribution of the cellular beacons based on the measurement of the intensity of 500 individual cells in a fluorescence image.
To determine the intracellular fluorescence intensity distribution pattern, the fluorescence intensity of 500 individual cells was analyzed by using IPP software. The results (Figure 2g) showed that the fluorescence intensity of the cellular beacons was strong and perfectly uniform. Importantly, essentially 100% of the cells were fluorescent (Figure S1), indicating the high efficiency to transform cells into cellular beacons under our conditions. Furthermore, we investigated the average fluorescence intensity of the cellular beacons under different conditions: without treatment, stored at 4 °C for two months, incubated in a 65 °C water bath for 30 min, or incubated in a 65 °C water bath for 30 min after storing at 4 °C for two months, respectively. The fluorescence images, e.g., Figure 3a–d, were used to determine the mean fluorescence intensity value by using IPP software. The results showed that there were no obvious differences among the fluorescing cells under different conditions (Figure 3e). These results demonstrated that the cellular beacons had the proper size and shape, uniformity of fluorescence intensity, outstanding photostability, and high luminance, making them good photoluminescence reporters for potential biomedical applications.
Figure 3.

Photostability of the cellular beacons under different conditions. The fluorescing cells without treatment (a), stored at 4 °C for two months (b), incubated in a 65 °C water bath for 30 min (c), or incubated in a 65 °C water bath for 30 min after placing at 4 °C for two months (d). (e) Mean fluorescence intensity values of the images analyzed by IPP software.
Characterization of the Intracellularly Fluorescent QDs
The presence of strong fluorescence in the cells above was consistent with our intention to biosynthesize CdS0.5Se0.5 QDs intracellularly. To characterize the QDs formed intracellularly, high-performance liquid chromatography coupled with inductively coupled plasma mass spectrometry (HPLC-ICP-MS) was used to determine the intermediate of the formation of the QDs in the cells. We found that the component of organoselenium compound was selenocystine ((Se-Cys)2) (Figure 4), which was similar to our previous report.1,8
Figure 4.

Characterization of intracellular reduced selenium species. HPLC-ICP-MS chromatograms of selenium species standards containing l-selenocystine (Se-Cys)2, Se-methylseleno-l-cysteine (SeMC), and d,l-selenomethionine (Se-Met) (a) and selenoamino acids isolated from the seleniumized cells (b) and the control cells untreated with Na2SeO3 (c).
The photoluminescence emission peak of fluores-cent nanocrystals isolated from the cells was at 520 nm with excellent photostability (Supplementary Note 2 and Figures S2, S3). TEM images (Figure 5a) of the isolated QDs revealed well-monodispersed nanocrystals with a diameter of 1.8 ± 0.5 nm (Figure 5b). These particles remained monodisperse, uniform in size, and very stable for more than two months without aggregation and precipitation. The long-term stability of the nanoparticle solution could be due to the presence of the capping protein/peptide in the cells that were bound to the surface of the nanoparticles prior to isolation and prevented their flocculation.
Figure 5.

In situ HRTEM for intracellular CdS0.5Se0.5 QDs and the characterization of the isolated CdS0.5Se0.5 QDs from the cellular beacons. (a) TEM image of monodispersed isolated CdS0.5Se0.5 QDs. (b) Size distribution histogram of the isolated CdS0.5Se0.5 QDs, with the median diameter of 1.8 ± 0.5 nm. (c) HRTEM image of the isolated CdS0.5Se0.5 QDs. (d) In situ HRTEM image of intracellular CdS0.5Se0.5 QDs. (e) XRD patterns of CdS0.5Se0.5 QDs. The lines under the spectrum represent the JCPDS pattern for CdS0.52Se0.48.
Energy-dispersive X-ray (EDX) analysis of the QDs isolated from the cells (Figure S4b) showed that the products consisted of elements Cd, S, and Se. The HRTEM image of the isolated QDs (Figure 5c) and the in situ HRTEM image of intracellular QDs (Figure 5d) showed that the lattice spacing (ca. 0.19 nm) agreed with the distance of the adjacent facet (103) of CdS0.52Se0.48. XRD results (Figure 5e) of the nanocrystals showed that all characteristic peaks matched well with the main peaks of standard CdS0.52Se0.48 (JCPDS Card No. 49-1460), although the diffraction peaks were wide, likely due to the low crystallinity of small-sized particles. Furthermore, when the isolated nanoparticles were subjected to atomic composition determination by using ICP-AES, the results showed that the molar ratio of elements Cd to S to Se was about 1:0.5:0.5 (Supplementary Note 2 and Table S1). These results thus suggested that the nanocrystals synthesized in live S. aureus were CdS0.5Se0.5.
Fluorescent-Biotargeting Cells as Probes for Virus Detection Coupled with Immunomagnetic Beads
Given the superior properties of the fluorescing cells as described above, we reasoned that such cells might be easily used as cellular beacons in various biomedical applications. It is well known that protein A is a cell-wall-associated protein endogenously expressed on the surface of S. aureus and can bind with strong specificity and affinity to the Fc fragment through the consensus sequence (Asn-Gln-Phe-Asn-Lys-Glu).17 Thus, monoclonal antibodies can be directly immobilized on the protein A endogenously expressed on the S. aureus surface. To test this possibility, the cellular beacons were treated with a 65 °C water bath for 30 min to inactivate the cells and then incubated with rabbit anti-mouse IgG conjugated with Cy3. The fluorescent microscopic images of the resulting cells (Figure S5, Supplementary Note 3) showed that no red fluorescence was detected in the negative control samples (i.e., adding Cy3 alone), while the IgG conjugated with Cy3-treated samples had strong fluorescence, indicating that the rabbit anti-mouse IgG was bound to the bacteria. Thus, S. aureus can be effectively conjugated with mAbs at their surfaces to form biotargeting cells without any other modifications, avoiding the use of other common methods for cell surface modifications, such as molecular covalent connection or more difficult genetic and metabolic engineering.23–25
To generate the bioprobes with our cellular beacons, we thus can simply mix S. aureus cells with the mAbs against specific biotargets and remove the excess mAbs by simply using centrifugation. Early studies have estimated that there are ~80 000 binding sites, i.e., protein A units, on the cell wall of an S. aureus upon which IgG can anchor,26 making it easy to label the cellular beacons with mAbs. To investigate the possible use of our cellular beacons as bioprobes for detection, we attached mAbs against a specific virus to the cells as the nanobioprobes and the magnetic beads coupled with the same mAbs for immunomagnetic capture of the virus.
Type A influenza virus from avian influenza virus (AIV) has caused several worldwide outbreaks of influenza-like illness in the recent decade.27 With the increasing concerns of an influenza-like illness caused by avian influenza A viruses, there is an urgent need for easy detection of the virus. We first used H9N2 AIV as a model virus. There are two viral proteins, hemagglutinin (HA) and neuraminidase (NA), expressed on the virus surface. Thus, we first conjugated superparamagnetic beads with monoclonal anti-HA antibody to recognize the surface protein HA of H9N2 AIV for the immunomagnetic capture of viruses. To determine whether the mAbs were effectively conjugated to the beads, we monitored the average hydrodynamic diameters of the beads before and after modification with the mAbs. As shown in Figure S6 (Supplementary Note 4), after the addition of the mAbs, the average hydrodynamic diameters increased from 503.8 nm to 657.3 nm, consistent with efficient coupling of the mAbs to the beads. Moreover, we used FITC-labeled goat anti-mouse IgG to demonstrate that the anti-HA antibody was conjugated to the beads. As shown in Figure S7b, the green fluorescence of FITC on the anti-HA beads was obvious compared to the control beads with no green fluorescence due to the binding of anti-HA antibody to the goat anti-mouse IgG-FITC (Supplementary Note 4 and Figure S7). In addition, when the conjugated beads were mixed with viruses and analyzed microscopically by TEM (Figure 6k), the virions were found to be intact and an immunomagnetic bead had more than one specific binding site for the virions. Thus, mAb-bound beads could be easily generated to capture the H9N2 AIV. Similarly, we generated the mAb-bound fluorescent bioprobes by adding mAbs to the cellular beacons.
Figure 6.

Fluorescent-biotargeting cells are very specific for the desired target. TEM images of fluorescent-biotargeting cells as probes for virus detection and bead–virus composites. The binding of the bioprobe to the immunomagnetic beads is proportional to H9N2 AIV concentrations. The concentration of H9N2 AIV is 1.75 × 104 ng/mL, while the concentrations of the negative control virus were 100 times as high as that of H9N2 AIV. The fluorescence images of H9N2 AIV sample (b), control sample H1N1 (e), and blank sample (h). Bright fields from the corresponding areas of H9N2 sample (a), H1N1 sample (d), and buffer blank (g), respectively. The merging of a and b (c), d and e (f), and g and h (i). (j) Histogram showing the amount of fluorescent probes bound to the positive sample (H9N2 AIV) and controls (inactive H1N1, NDV, baculovirus, PRV, and blank). (k) The bead–virus composites were negatively stained with phosphotungstic acid for TEM observation. The blackish round spots are the beads, while the smaller spots are characteristic of virion morphology around the beads, which showed the virons. (l) TEM image of beads–virus–bioprobe composites. (m, n, o, p, q) Fluorescence images of the detection of H9N2 AIV at different concentrations (1.75 × 104, 1.75 × 103, 89.4, 8.94, and 0 ng/mL, respectively). (r) Histogram showing the number of bioprobes captured on the beads in the presence of different concentrations of H9N2 virus.
For virus detection, the viruses captured by the immunomagnetic beads were mixed with nanobiop-robes. The resulting complex was carefully collected with a magnetic separator and washed thoroughly, transferred to a glass slide, and imaged with a microscope. As shown in Figure 6, when beads–H9N2 virus complexes were mixed with the bioprobes, the apparent formation of the beads–virus–bioprobes complexes was observed (Figure 6a,b). The merged image of the bright and fluorescence fields (Figure 6c) showed that the virus acted as the bridge between the beads and bioprobes (note that no fluorescence was observed around some magnetic beads, because the excess of the beads without capture of H9N2 virus cannot bind to biotargeting cellular beacons). In contrast, the negative control samples (inactive H1N1 virus, NDV, PRV, baculovirus), whose concentrations were 100 times as high as the concentration of H9N2 AIV, or a blank solution had no fluorescent complex (Figure 6d,e,g,h; images of the negative control with NDV, PRV, and baculovirus are shown in Figure S8, where no fluorescence was observed), supporting the high specificity of this method.22 Furthermore, based on the fact that more than 99% of the cellular beacons uniformly shine (see above), the number of fluorescence dots in the complexes could be used as a measure of the number of biotargeting cellular beacons bound to the magnetic beads. Using 20 randomly chosen fields, we determined the number of fluorescent dots in the virus sample and the controls and found that the binding of the bioprobe to the virus was highly specific (Figure 6j). TEM analysis showed that the biotargeting cellular beacons were attached onto the virus conjugated to the immunomagnetic beads, forming a sandwich immunocomplex (Figure 6l), again supporting the specific capture of the virus by the beads and specific detection of the virus by the bioprobes.
To determine whether our approach allows quantitative detection of the virus, we used decreasing concentrations of the target virus for the assay and found that the number of fluorescent dots and the degree of formation of sandwich immunocomplexes decreased gradually (Figure 6m–q), indicating that the probe binding was dependent upon the concentrations of the virus. Detection limit is an assessment of the sensitivity of the method and is defined as virus concentration at which the average number of fluorescent dots in each field was equivalent to the average number of the blank control plus three times the standard deviations. Quantitative analysis showed that there was a small quantity of fluorescent dots in the blank control sample, which was likely due to the nonspecific adsorption caused by the electrostatic interaction between capping molecules on the surface of cells and biomolecules in this polydispersity system (Figure 6r).21 Importantly, we found that the detection limit of this method was as low as 8.94 ng/mL (based on protein content), which was similar to the human enterovirus 71 (EV71) detection.28 Thus, not only is our method of using biotargeting cellular beacons as nanobioprobes applied to virus detection as sensitive as the available detection method, but also the approach is much simpler and more convenient.
A major breakthrough in our approach is the direct use of a whole microorganism self-synthesizing fluorescent QDs as a fluorescence tracking unit of bioprobes for biodetection. Compared with common QD-based fluorescent probes, our fluorescent nanobioprobes have the following advantages. First, our fluorescent probes are monodisperse with perfectly uniform sizes, high luminance, and photostability. Second, our bioprobes are highly accurate, reliable, and repeatable. Common fluorescent probe construction involves multiple process steps such as synthesis of colloidal QDs, water-solubilization of QDs, and their functionlization with biotargeting molecules, which are easily affected by the proficiency and subjectivity of researchers. Our method, based on a new strategy for one-step preparation of fluorescent bioprobes, is time- and labor-saving and cost-effective, with high sensitivity and good specificity. As known, nanocrystals cannot be applied in biology and medicine until they are further conjugated to biotargeting ligands through complicated process steps. However, it is usually difficult to control such conjugation, which often leads to an excess of ligands on the surface of the nanoparticles, causing nonspecific binding and aggregation of the nanoparticles. Thus, the targeting capabilities of the ligands will also decrease. Nevertheless, our new approach is versatile and can be even easily adapted to construct nanobioprobes for detection of a variety of species. For example, we demonstrated that, using different antibodies attached to the cellular beacons, pseudorabies virus (PRV), baculovirus, Salmonella typhimurium bacteria, and SK-BR-3 cells can be successfully detected (more details in Supplementary Note 6 and Figures S9 – S13). Thus, this strategy can be easily used as a general method for detection of different pathogens by simply and easily changing the conjugated antibodies.
CONCLUSIONS
In summary, we have successfully transformed S. aureus into cellular beacons by a space–time coupling strategy. The resulting cellular beacons have outstanding photostability, high luminance efficiency, and perfect uniformity and can be directly and easily conjugated to mAbs based on specific interaction between the protein A expressed on the cell surface and the Fc region of antibodies to generate fluores-cent-biotargeting bifunctional probes. Such probes can be used for virus detection in an easy user-friendly approach when used in combination with immuno-magnetic beads for virus capture. With this method, H9N2 AIV can be detected specifically with a limit of 8.94 ng/mL. Such new bioprobes can be extended to detect other pathogens, such as pseudorabies virus, baculovirus, Salmonella typhimurium bacteria, and SKBR-3 cells, just by changing the antibodies conjugated to the cells. Our cellular beacons offer several distinct advantages over other QD-based detection systems. These include easy attachment of biotargeting molecules due to the presence of surface protein A, high efficiency (essentially 100%) in transforming the cells into fluorescent beacons, perfectly uniform QD levels in individual beacons, and superior properties (highly stable and monodispersible). Just with the help of a magnet and a fluorescence microscope, such a simple, sensitive, but specific approach can become a general platform for detection of pathogens and other biotargets.
METHODS
Transforming Cells into Fluorescent Nanobioprobes
S. aureus (CCTCC AB91093) was provided by the Chinese Center for Type Culture Collections of Wuhan University (Wuhan, PR China). The S. aureus was cultured for 12 h with LB broth (yeast extract 5 g/L, tryptone 10 g/L, NaCl 5 g/L) at 37 °C to ensure that the cells were in the stationary phase. The cells were incubated with Na2SeO3 (final concentration: 5 mM) for 12 h and then harvested by centrifugation at 4000 rpm for 5 min, followed by transferring them back into fresh LB broth. CdCl2 (final concentration: 1 mM) was added to the medium, and the cells were cultured for 12 h to obtain the cellular beacons due to the formation of intracellular fluorescent CdS0.5Se0.5 QDs.
The cellular beacons were harvested by centrifuging at 4000 rpm for 5 min, washed three times with Tris-HCl buffer (pH 8.0, 0.1 M), and then resuspended in the same Tris-HCl buffer. The cellular beacons with an OD of 1.5 were heated in a 65 °C water bath for 30 min, a condition that allows the entrance of the cells into a nonculturable state (no colony formation when such treated cells were plated in the LB agar plate).29–31 The cellular beacons were then bound with Abs as follows: The cellular beacons with an OD of 1.5 (a final volume of 100 μL) were incubated with 2 μL of mouse monoclonal anti-HA antibody (0.59 mg/mL) for 3 h at 37 °C with shaking at 150 rpm. The mAb-bound cells were harvested by centrifugation at 4000 rpm for 5 min, washed three times with 100 μL of Tris-HCl buffer, blocked with 1% BSA (w/v) for 30 min at 37 °C, and washed with Tris-HCl buffer again. Finally, the cells were dispersed in 100 μL of Tris-HCl buffer. The resulting S. aureus thus became fluorescent-biotargeting bifunctional probes for HA targets.
Separation of Seleno Compounds and Characterization of Intracellular Reduced Selenium Species
A single S. aureus colony was picked from the agar plate and precultured in 100 mL of LB broth for 12 h at 37 °C. Then cells (1 mL) were diluted 100 times into 99 mL of fresh media and cultured for another 12 h at 37 °C. After that, the cells were incubated with Na2SeO3 (final concentration: 5 mM) for 12 h to obtain the seleniumized cells. The cells were harvested, washed three times with ultrapure water to exclude the culture medium, and wet weighed prior to enzymatic extraction. After 2 mL of ultrapure water was added into 1 g of the wet weighed sample, the sample was ultrasonicated at 500 W, 75 times a circle, 2 s per time, 2 s intervals each time. Finally the extracted samples were centrifuged at 12 000 rpm for 30 min, and the upper aqueous phase was collected. Trypsin (250 μg/mL) from bovine pancreas (Sigma) and 0.1% (w/v) SDS (final concentration) were added to the samples for digestion. After digestion for 8 h at 37 °C, the samples were filtered with MILLEX GP filters (unit 0.22 μm).
A 100 μL amount of each sample was analyzed on an HPLC system equipped with LC-6AD high-pressure pumps and an SPD-20A UV spectrometry detector (Shimadzu, Japan) for chromatographic separation with a Superdex 200 10/300 GL gel filtration column (13 μm, 10 × 300 mm, GE Healthcare, Amersham Biosciences), operating at a mobile phase flow rate of 0.5 mL/min and column temperature of 30 °C. The mobile phase consisted of 0.05 M phosphate buffer (NaH2PO4 Na2HPO4, pH 7.2). The detection wavelengths were 254 and 280 nm. The fraction from 35 to 47 min and that from 47.01 to 55 min were collected separately and were injected into HPLC-ICP-MS for further chromatographic separation by using an HPLC system (Shimadzu, Japan) equipped with a high-pressure liquid chromatographic pump (LC-10AD, Japan) and C18 chromatographic column (Shim-pack CLC, 5 μm, 4.6 mm × 150 mm, Japan) (Figure S3). An Agilent 7500a ICP-MS (Agilent, USA) was used for online element-specific detection. The mobile phase consisted of 0.02 M COONH4 (pH 3.0). Selenium standards, d,l-selenomethionine (>99%), Se-methylseleno-l-cysteine (98%), and l-selenocystine (97%), were purchased from Acros Organics, USA, J&K Scientific Ltd., and Sigma-Aldrich, respectively. Standard stock solutions (100 μg/mL as Se) were prepared by dissolving each of them in ultrapure water, except for l-selenocystine, with 0.1 M HCl.
Characterization of the Cellular Beacons
The fluorescing cells were harvested by centrifugation at 4000 rpm for 5 min, then washed three times with Tris-HCl (pH 8.0, 0.1 M) and redispersed in the Tris-HCl buffer (pH 8.0, 0.1 M). The fluorescence images were obtained with a Nikon microscope (TE 2000-U, Nikon Corp, 100× objective, Canada) equipped with a charge-coupled device (RETIGA 2000R, Qimaging Corp, Canada). The images were analyzed with the Image-Pro Plus 6.0 software (Media Cybernetics, USA).
In situ high-resolution transmission electron microscopy (HRTEM) images were obtained by using a JEM-2010 FEE (UHR) microscope with an acceleration voltage of 200 kV. The cellular beacons were harvested by centrifugation at 4000 rpm for 5 min, washed three times with ultrapure water (18.2 MΩ 3 cm, Millipore, Molsheim, France), and then resuspended in ultra-pure water. The HRTEM samples were prepared by drying the ultrapure water dispersion of the fluorescing cells on a copper grid coated with ultrathin carbon film. In situ energy-dispersive X-ray data of cellular beacons were obtained by using a JEM-2010 FEE (UHR) microscope equipped with EDX spectrometry (EDAX Inc.).
One gram (wet weight) of the cellular beacons was resus-pended in 20 mL of Tris-HCl buffer (pH 8.0, 0.01 M). The fluorescing cells were lysed with a Vibra-Cell ultrasonic processor (model no. VC 505, Sonics & Materials. Inc., CT, USA). The parameters of operation were designed as follows: TIMER, 20 min; PULSER ON, 2 s; PULSER OFF, 2 s; AMPL, 50%. The lysed cells were centrifuged at 12 000 rpm for 5 min, and the super-natant was collected. The supernatant was centrifuged at 12 000 rpm for 15 min and collected again, and the procedure was repeated once more. Finally, the samples were purified with the Amicon centrifugal filter unit (MWCO 10 kDa) to remove free small substances and resuspended in 400 μL of ultrapure water. The samples were then filtered with MILLEX GP filters (unit 0.22 μm).
The transmission electron microscopy (TEM) and high-resolution transmission electron microscopy images of isolated CdS0.5Se0.5 QDs were obtained with the same microscope as above-mentioned. The samples were purified with a gel electrophoresis apparatus (Beijing Liu Yi instrument factory, DYY-III-88) by using a 1% (w/v) agarose gel in Na2CO3–NaHCO3 buffer (5 × 10 −3 M, pH 10.0). For each well, 70 μL of a sample was mixed with 20 μL of loading buffer (Na2CO3–NaHCO3, 20% (v/v) glycerol at pH 10.0) before loading into the gel. The electrophoresis was run at 100 V, 35 mA for 40 min. Then fluorescent Gershgorin band of the gel was cut out and placed into a dialysis bag with a molecular weight cutoff of 3 kDa (MWCO 3 kDa). The bag was then placed into the electrophoresis chamber for 20 min at 100 V, 35 mA, to extract the product from the gel. The sample was collected and ultrafiltered as above. The purified samples for TEM and HRTEM were prepared by dropping 10 μL of each sample onto ultrathin-carbon-coated copper grids and allowing it to dry. The X-ray powder diffraction (XRD) pattern was obtained with a Bruker AXS D8 Advance X-ray diffractometer. Samples of isolated fluorescent quantum dots from cells were dropped onto the slide and allowed to dry as a uniform thin film.
Preparation of Immunomagnetic Beads with mAb-HA
The Super-paramagnetic Carboxyl-Adembeads (500 nm diameter) were obtained from Ademtech SA (Pessac, France); N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma. The monoclonal anti-HA antibody was obtained from Sino Biological Inc. The superparamagnetic beads with carboxyl groups were functionalized with Abs as follows: First, 10 μL of 50 mg/mL superparamagnetic beads was washed three times with a phosphate buffer (PB, 0.1 M, pH = 7.2). Then, 50 mM EDC and 50 mM NHS dissolved in MES buffer (0.1 M MES, 0.15 M NaCl, pH = 6.5) were added into the superparamagnetic beads (a final volume of 800 μL) to activate carboxyl groups for 30 min at 37 °C with shaking at 150 rpm. After that, the activated superpara-magnetic beads were washed three times with PB buffer (pH 7.2, 0.1 M). Second, 16 μL of monoclonal anti-HA antibody (0.59 mg/mL) was added onto the activated superparamagnetic beads and adjusted with PB buffer (pH 7.2, 0.1 M) to 400 μL. Then, the mAbs and activated superparamagnetic beads were incubated for 4 h at 37 °C with shaking at 150 rpm. Unbound mAb was removed by washing three times with PB buffer (pH 7.2, 0.1 M), and the immunomagnetic beads were kept in 400 μL of PB buffer (pH 7.2, 0.1 M) containing 1% (w/v) BSA and 0.05% (w/v) NaN3 at 4 °C for further use.
Protocol for Target Virus Detection
H9N2 AIV, the inactivated H1N1 AIV, the inactivated NDV, pseudorabies virus, and baculovirus were obtained from Wuhan Institute of Virology, Chinese Academy of Sciences. The virus was quantitated by using a protein quantification method.28,32 A 50 μL amount of 1.25 mg/mL immunomagnetic beads (Superparamagnetic Carboxyl-Adembeads, 500 nm diameter, were attached to anti-HA antibody against a specific H9N2 virus; the conjugation process of the immunomagnetic beads is described in detail in Preparation of Immunomagnetic Beads with mAb-HA) was washed three times with 0.01 M PBS (pH 7.2); then a H9N2 sample of 1.75 × 104 ng/mL was added into the immunomagnetic beads in 0.01 M PBS (pH 7.2, a final volume of 100 μL), and the mixture were incubated for 30 min at 37 °C with shaking at 150 rpm. The bead–virus composites were collected with a magnetic separator (Invitrogen), and washed three times with the same PBS buffer. The mAb-bound cells with an OD of 1.5 resuspended in Tris-HCl (pH 8.0, 0.1 M) were added into the bead–virus complex (a final volume of 100 μL) and then incubated for 30 min at 37 °C with shaking at 150 rpm. After washing five times with the same Tris-HCl solution by using the magnetic separator, the bead–virus–cell complexes were resuspended in 10 μL of the same Tris-HCl solution. Five control experiments were also performed: the four control experiments by using several other viruses (inactive H1N1, NDV, PRV, and baculovirus) were used as negative controls, and another control experiment without any virus was used as the blank control. The concentrations of the negative control virus were 100 times as high as that of H9N2 AIV. In this experiment, 20 fields were also chosen randomly to count the total number of fluorescent dots, and then the average number of fluorescence dots in each field was calculated as the result of this experiment. For quantitative detection of the virus, experiments at each concentration were repeated three times, while the control experiment was repeated seven times. Transmission electron microscopy images were obtained by using a FEI Tecnai G2 20 TWIN. The TEM images of bead–virus composites were obtained with a Hitachi H-7000FA electron microscope. The TEM images of the samples were negatively stained in the phosphotungstic acid for 2 min.
Supplementary Material
Acknowledgment
This work was supported by the National Basic Research Program of China (973 Program, Grant No. 2011CB933600), the National Natural Science Foundation of China (Grant No. 21105075), and the 111 Project (111-2-10). We thank Xue-Qin Guo for HPLC-ICP-MS experiments and Yong Li for his assistance with the experiments.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information Available: Thirteen additional figures and one additional table (Figures S1 S13 and Table S1) and information. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.Cui R, Liu H-H, Xie H-Y, Zhang Z-L, Yang Y-R, Pang D-W, Xie Z-X, Chen B-B, Hu B, Shen P. Living Yeast Cells as a Controllable Biosynthesizer for Fluorescent Quantum Dots. Adv. Funct. Mater. 2009;19:2359–2364. [Google Scholar]
- 2.Dameron CT, Reese RN, Mehra RK, Kortan AR, Carroll PJ, Steigerwald ML, Brus LE, Winge DR. Biosynthesis of Cadmium Sulphide Quantum Semiconductor Crystallites. Nature. 1989;338:596–597. [Google Scholar]
- 3.Sweeney RY, Mao C, Gao X, Burt JL, Belcher AM, Georgiou G, Iverson BL. Bacterial Biosynthesis of Cadmium Sulfide Nanocrystals. Chem. Biol. 2004;11:1553–1559. doi: 10.1016/j.chembiol.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Klaus T, Joerger R, Olsson E, Granqvist C-G. Silver-Based Crystalline Nanoparticles, Microbially Fabricated. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13611–13614. doi: 10.1073/pnas.96.24.13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siponen MI, Legrand P, Widdrat M, Jones SR, Zhang W-J, Chang MCY, Faivre D, Arnoux P, Pignol D. Structural Insight into Magnetochrome-Mediated Magnetite Biomineralization. Nature. 2013;502:681–684. doi: 10.1038/nature12573. [DOI] [PubMed] [Google Scholar]
- 6.Kowshik M, Vogel W, Urban J, Kulkarni SK, Paknikar KM. Microbial Synthesis of Semiconductor PbS Nanocrystallites. Adv. Mater. 2002;14:815–818. doi: 10.1002/bit.10233. [DOI] [PubMed] [Google Scholar]
- 7.Tilley RD, Cheong S. Earthworms Lit with Quantum Dots. Nat. Nanotechnol. 2013;8:6–7. doi: 10.1038/nnano.2012.241. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Cui R, Zhang P, Chen B-B, Tian Z-Q, Li L, Hu B, Pang D-W, Xie Z-X. Mechanism-Oriented Controllability of Intracellular Quantum Dots Formation: The Role of Glutathione Metabolic Pathway. ACS Nano. 2013;7:2240–2248. doi: 10.1021/nn305346a. [DOI] [PubMed] [Google Scholar]
- 9.Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004;10:7063–7070. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Wang L, Yeh J, Wu X, Cao Z, Wang YA, Zhang M, Yang L, Mao H. Reducing Non-Specific Binding and Uptake of Nanoparticles and Improving Cell Targeting with an Antifouling PEO-b-PγMPS Copolymer Coating. Biomaterials. 2010;31:5397–5407. doi: 10.1016/j.biomaterials.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doshi N, Mitragotri S. Designer Biomaterials for Nano-medicine. Adv. Funct. Mater. 2009;19:3843–3854. [Google Scholar]
- 12.Bagwe RP, Hilliard LR, Tan W-H. Surface Modification of Silica Nanoparticles to Reduce Aggregation and Nonspecific Binding. Langmuir. 2006;22:4357–4362. doi: 10.1021/la052797j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grubbs RB. Roles of Polymer Ligands in Nanoparticle Stabilization. Polym. Rev. 2007;47:197–215. [Google Scholar]
- 14.Chan WCW, Nie S-M. Quantum Dot Bioconjugates for Ultrasensitive Nonisotopic Detection. Science. 1998;281:2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 15.Löfdahl S, Guss B, Uhlén M, Philipson L, Lindberg M. Gene for Staphylococcal Protein A. Proc. Natl. Acad. Sci. U.S.A. 1983;80:697–701. doi: 10.1073/pnas.80.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moks T, Abrahmsén L, Nilsson B, Hellman U, Sjöquist J, Uhlén M. Staphylococcal Protein A Consists of Five IgG-Binding Domains. Eur. J. Biochem. 1986;156:637–643. doi: 10.1111/j.1432-1033.1986.tb09625.x. [DOI] [PubMed] [Google Scholar]
- 17.Abrahmsén L, Moks T, Nilsson B, Hellman U, Uhlén M. Analysis of Signals for Secretion in the Staphylococcal Protein A Gene. EMBO J. 1985;4:3901–3906. doi: 10.1002/j.1460-2075.1985.tb04164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y, Du X-W, Zhao F, Xu B. Magnetic Nanoparticles for the Manipulation of Proteins and Cells. Chem. Soc. Rev. 2012;41:2912–2942. doi: 10.1039/c2cs15315g. [DOI] [PubMed] [Google Scholar]
- 19.Xie H-Y, Zuo C, Liu Y, Zhang Z-L, Pang D-W, Li X-L, Gong J-P, Dickinson C, Zhou W. Cell-Targeting Multi-functional Nanospheres with Both Fluorescence and Magnetism. Small. 2005;1:506–509. doi: 10.1002/smll.200400136. [DOI] [PubMed] [Google Scholar]
- 20.Song E-Q, Hu J, Wen C-Y, Tian Z-Q, Yu X, Zhang Z-L, Shi Y-B, Pang D-W. Fluorescent-Magnetic-Biotargeting Multifunctional Nanobioprobes for Detecting and Isolating Multiple Types of Tumor Cells. ACS Nano. 2011;5:761–770. doi: 10.1021/nn1011336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao W, Zhang W-P, Zhang Z-L, He R-L, Lin Y, Xie M, Wang H-Z, Pang D-W. Robust and Highly Sensitive Fluorescence Approach for Point-of-Care Virus Detection Based on Immunomagnetic Separation. Anal. Chem. 2012;84:2358–2365. doi: 10.1021/ac203102u. [DOI] [PubMed] [Google Scholar]
- 22.Wen C-Y, Hu J, Zhang Z-L, Tian Z-Q, Ou G-P, Liao Y-L, Xie M, Sun Z-Y, Pang D-W. One-Step Sensitive Detection of Salmonella typhimurium by Coupling Magnetic Capture and Fluorescence Identification with Functional Nanospheres. Anal. Chem. 2013;85:1223–1230. doi: 10.1021/ac303204q. [DOI] [PubMed] [Google Scholar]
- 23.Kellam B, Bank PAD, Shakesheff KM. Chemical Modification of Mammalian Cell Surfaces. Chem. Soc. Rev. 2003;32:327–337. doi: 10.1039/b211643j. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Brock A, Chen S, Chen S-B, Schultz PG. Genetic Incorporation of Unnatural Amino Acids into Proteins in Mammalian Cells. Nat. Methods. 2007;4:239–244. doi: 10.1038/nmeth1016. [DOI] [PubMed] [Google Scholar]
- 25.Mahal LK, Yarema KJ, Bertozzi CR. Engineering Chemical Reactivity on Cell Surfaces through Oligosaccharide Biosynthesis. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 26.Langone JJ. Protein A of Staphylococcus aureus and Related Immunoglobulin Receptors Produced by Streptococci and Pneumonococci. Adv. Immunol. 1982;32:157–252. [PubMed] [Google Scholar]
- 27.Schnitzler SU, Schnitzler P. An Update on Swine-Origin Influenza Virus A/H1N1: A Review. Virus Genes. 2009;39:279–292. doi: 10.1007/s11262-009-0404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, Zhang X, Zhang C, Zhou G, Zhang W, Xiang D, He Z, Wang H. Dual-Color Fluorescence and Homogeneous Immunoassay for the Determination of Human Enterovirus 71. Anal. Chem. 2011;83:7316–7322. doi: 10.1021/ac201129d. [DOI] [PubMed] [Google Scholar]
- 29.Oliver JD. The Viable but Nonculturable State in Bacteria. J. Microbiol. 2005;43:93–100. [PubMed] [Google Scholar]
- 30.Oliver JD. Recent Findings on the Viable but Nonculturable State in Pathogenic Bacteria. FEMS Microbiol. Rev. 2010;34:415–425. doi: 10.1111/j.1574-6976.2009.00200.x. [DOI] [PubMed] [Google Scholar]
- 31.Kell DB, Kaprelyants AS, Weichart DH, Harwood CR, Barer MR. Viability and Activity in Readily Culturable Bacteria: A Review and Discussion of the Practical Issues. Antonie van Leeuwenhoek. 1998;73:169–187. doi: 10.1023/a:1000664013047. [DOI] [PubMed] [Google Scholar]
- 32.Liu S-L, Zhang Z-L, Tian Z-Q, Zhao H-S, Liu H, Sun E-Z, Xiao G-F, Zhang W, Wang H-Z, Pang D-W. Effectively and Efficiently Dissecting the Infection of Influenza Virus by Quantum-Dot-Based Single-Particle Tracking. ACS Nano. 2012;6:141–150. doi: 10.1021/nn2031353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.