

Abstract
Multiple processes are involved in gene expression including transcription, translation and stability of mRNAs and proteins. Each of these steps are tightly regulated, affecting the final dynamics of protein abundance. Various regulatory mechanisms exist at the translation step, rendering mRNA levels alone an unreliable indicator of gene expression. In addition, local regulation of mRNA translation has been particularly implicated in neuronal functions, shifting 'translatomics' to the focus of attention in neurobiology. The presented method can be used to bridge transcriptomics and proteomics.
Here we describe essential modifications to the technique of polyribosome fractionation, which interrogates the translatome based on the association of actively translated mRNAs to multiple ribosomes and their differential sedimentation in sucrose gradients. Traditionally, working with in vivo samples, particularly of the central nervous system (CNS), has proven challenging due to the restricted amounts of material and the presence of fatty tissue components. In order to address this, the described protocol is specifically optimized for use with minimal amount of CNS material, as demonstrated by the use of single mouse spinal cord and brain. Briefly, CNS tissues are extracted and translating ribosomes are immobilized on mRNAs with cycloheximide. Myelin flotation is then performed to remove lipid rich components. Fractionation is performed on a sucrose gradient where mRNAs are separated according to their ribosomal loading. Isolated fractions are suitable for a range of downstream assays, including new genome wide assay technologies.
Keywords: Neuroscience, Issue 86, central nervous system, CNS, translation, polyribosome fractionation, RNA, Brain, spinal cord, microarray, next-generation sequencing, gradient, translatome
Introduction
Gene expression is determined by the combined action of transcription, translation and stability of mRNA and proteins, with translation bearing the most predominant effect1. It is now evident that each of these steps is highly regulated. Micro-RNAs, formation of RNA granules, alternative and cytoplasmic polyadenylation are some examples of post-transcriptional regulation of gene expression2,3. Each of these mechanisms uncouples transcription from translation and influences the proteome in the biological system of interest. Thus, unsurprisingly, mRNA levels alone are an imperfect readout of protein levels4. Quantitative proteomics provides the most direct assessment of gene expression, however despite recent advances, there are still considerable limitations on sensitivity and protein sequence resolution. Therefore addressing the translatome, the repertoire of translating mRNAs, offers an excellent compromise between studying the transcriptome and proteome. It is more accurate than transcriptomics in assessing final gene expression, and provides both higher coverage and sequence resolution than proteomics.
In mammalian systems, the majority of translation events begin via cap-dependent initiation. A group of eukaryotic initiation factors together with the 40S small ribosomal subunit assemble at the 5' cap of an mRNA. The complex then scans the mRNA and upon reaching the start codon AUG, recruits the 60S large ribosomal subunit to form a complete 80S ribosome. The elongation stage proceeds with the ribosome moving along the mRNA, with elongation factors assisting the incorporation of amino acids from loaded tRNAs to the nascent peptide chain. Multiple ribosomes can proceed along a single mRNA simultaneously and the number of associated ribosomes has been shown to correlate with the rate of protein synthesis5,6. This makes ribosomal loading a reliable indication for translation and allows separation of actively translating mRNAs based on sedimentation rate. In addition to quantification of translating mRNAs, sequence information can be obtained to identify motifs involved in translation regulation. Also RNA binding proteins and other translation factors can be isolated from different fractions, facilitating the study and discovery of related regulatory proteins.
In the nervous system, translational control has been linked to processes such as mRNA storage, transport and local protein synthesis. Growth cones have been shown to harbor a specific localized pool of mRNAs distinct from the rest of the axon7. In addition, axons possess the ability to locally synthesize proteins8,9. As a result, local control of translation has become a crucial topic of study in neurobiology. The potential of polyribosome fractionation to address this has been illustrated in several studies, in which the technique was used to investigate axon guidance in spinal cord development, and demonstrated the activity-dependent translation of BDNF in the brain10,11.
Protocol
All animal experiments were performed in accordance with the institutional guidelines of the DKFZ. Caution: In order to prevent RNAse contamination of samples, take basic precautions for avoiding RNAse contamination and prepare all buffers with DEPC-treated water.
1. Preparation of Sucrose Gradients
Prepare 17.5, 25.6, 33.8, 41.9 and 50% sucrose solutions (see Table 1). Start preparing gradients by slowly adding 2 ml of 50% sucrose solution to the bottom of a polyallomer ultracentrifuge tube. Freeze the layer by placing tubes for 20 min at -80 °C.
Add the subsequent layers at decreasing sucrose percentages with freezing steps in between, ending up to 17.5% sucrose. Keep gradients on ice during addition of sucrose solutions in order to prevent thawing of underlying layers.
Prepare sucrose gradients freshly one day before use and keep at 4 °C. Alternatively, prepare gradients in advance, store at -80 °C and thaw at 4 °C the night before the experiment.
2. Tissue Preparation (Spinal Cord and/or Brain)
Anesthetize mouse by an overdose of Xylazin and Ketamin in NaCl and transcardially perfuse with 20 ml of Hank's balanced salts solution (HBSS) containing 200 µg/ml CHX which immobilizes ribosomes on associated transcripts. Confirm proper anesthetization by non-responsiveness to toe pinches.
Extract tissue rapidly. Open the cranium dorsally and extract the brain. Perform laminectomy of the entire thoracic and lumbar vertebral column and extract the spinal cord. Use whole brain (~400 mg) or a 4 cm piece of spinal cord (~90 mg), respectively.
Transfer tissue into ice cold homogenization buffer A (see Table 1) (spinal cord: 1 ml; brain: 4 ml) and dice into small pieces with a clean scalpel to allow increased uptake of cycloheximide. Perform all further steps on ice.
Incubate for 15 min and homogenize tissue mechanically using a Dounce homogenizer. Carefully controlled homogenization is necessary to ensure that nuclei remain intact. Treat all samples exactly the same way to ensure comparability. Note: for brain tissue: Disrupt tissue by 5 strokes with a tightly fitting pestle. For spinal cord tissue: disrupt by 5 strokes with a loosely fitting pestle, followed by 5 further strokes with a tightly fitting pestle.
Take aliquots (400 µl for brain and 200 µl for spinal cord), flash freeze and store at -80 °C for total RNA isolation.
Remove nuclei and undisrupted cell and tissue fragments by centrifugation at 500 x g for 10 min at 4 °C. Low speed centrifugation prevents loss of ribosomes.
Lyse membrane fragments in the nuclei free supernatant for 30 min by adding NP-40 and sodium deoxycholate detergents (each to a final concentration of 1%).
3. Myelin Flotation
This step removes fatty components from the sample which will otherwise mask the signal in the polyribosome profile. First, pre-chill ultracentrifuge buckets and rotor at 4 °C and prepare 2 M, 1.1 M and 0.9 M sucrose solutions (see Table 1).
Mix lysate with an 1.22 volumes of 2 M sucrose solution and transfer into a polyethylene ultracentrifuge tube. Fill up all tubes to equal volume of 10 ml with 1.1 M sucrose solution, thereafter overlay it carefully with 0.9 M sucrose solution.
Place the ultracentrifuge tubes into pre-chilled ultracentrifuge buckets. Centrifuge for 3 hr at 100,000 x g at 4 °C. During this process ribosomes become deposited in the pellet whereas fatty components float up and remain in the supernatant.
4. Sucrose Gradient Fractionation
Remove supernatant and dissolve pellet in homogenization buffer B (see Table 1). Measure the absorbance at 260 nm for each sample using a Nanodrop or equivalent apparatus and normalize amounts to load according to the absorbance value.
Place sucrose gradients (preparation is described in a previous section) into the pre-chilled ultracentrifuge buckets. Layer samples carefully on top of the gradient. Add one blank sucrose gradient as technical control.
Adjust the weight of each bucket with homogenization buffer. Centrifuge samples for 1.5 hr at 285,000 x g at 4 °C.
- Start preparing the Isco fractionator 30 min before the end of centrifugation. If another model of fractionator is used, follow the manufacturer's protocol.
- Set the appropriate sensitivity (0.2 AUFS for brain or 0.05 AUFS for spinal cord) for the UV lamp and switch it on for warming up. Assemble the tube piercer, connect it via the rolling pump to 60% sucrose solution through the tube piercer.
- Test the setup by pumping sucrose (flow rate: 1 ml/min), in particular make sure there is no leaks in the tubings which will introduce bubbles into the gradient.
- Blank background absorption by manually pumping gradient buffer into the UV detector and correcting the baseline.
Carefully remove the ultracentrifuge buckets containing the sucrose gradients with sedimented samples from the rotor and place them on ice. Avoid any bumping of gradients to prevent loss of resolution.
- Run gradients on the fractionator. Run empty gradients first to assess background absorption and ensure proper technical setup.
- Attach gradient to the UV detector and pierce the bottom of the tube with the tube piercer. Start the pumping of 60% sucrose, which will displace the gradient with sedimented samples upwards through the UV detector and into the drop dispenser.
- The absorbance at 260 nm is documented to generate the absorption profile for the sample, with peaks indicating the sedimentation of mRNAs associated with ribosomal subunits, monosomes, and subsequently increasing number of ribosomes.
Collect samples into 20 fractions via the drop dispenser (600 µl each, in 2 ml tubes).
5. RNA Isolation from Individual Fractions
Add 10% SDS to a final concentration of 1% to individual fractions and mix well in order to unfold proteins and dissociate ribosomes. At this point samples can be frozen at -80 °C. Depending on the question to be addressed, fractions can be pooled according to the absorbance profile.
- Isolate RNA with acidic phenol/chloroform extraction, which also removes contaminating traces of DNA.
- Add one volume of acidic phenol/chloroform (prewarmed to RT) to each sample, heat for 10 min at 65 °C and release pressure under the fume hood afterwards.
- Centrifuge samples for 20 min at 17,000 x g at RT. Carefully transfer aqueous phase to a new 1.5 ml tube. Be aware of phase inversion in denser sucrose fractions, where the aqueous phase might be at the bottom after centrifugation.
- Add one volume of isopropanol, 1/9 volume sodium acetate (pH 5.2) and 1 µl GlycoBlue to each sample in order to precipitate RNA. Keep samples at least 1 hr at -80 °C.
- Centrifuge for 30 min at 17,000 x g and 4 °C. GlycoBlue provides visualization of the pellet. Remove supernatant, wash pellet once with ice cold 80% ethanol and air dry pellet. Dissolve pellet in RNAse free water.
Quantify amount of RNA by Nanodrop and analyze RNA integrity by a Bioanalyzer chip. RNA which is obtained by the presented protocol is suitable for all state of the art high throughput assays including microarray and deep sequencing.
Representative Results
Figure 2 shows representative polyribosome profiles after fractionation. The profiles of brain and spinal cord show characteristic absorption curves as described before for cell lines. mRNAs bound to small ribosomal subunits (40S) sediment at lighter fractions and appear first as a peak on the profile, followed by the large ribosomal subunit (60S) and monosome (80S) bound mRNAs. mRNAs bound to multiple ribosomes sediment at heavier fractions, with the later peaks indicating increasing number of bound ribosomes. Global translation can be assessed from the profile. As an example, brain tissue has a higher polyribosome to monosome ratio than spinal cord tissue, indicating more active translation.
RNA extracted from individual fractions were assessed by Bioanalyzer, showing distribution of 18S and 28S rRNA. 18S rRNA appears earlier in the profile, in accordance with the small ribosomal subunits sedimenting in lighter sucrose fractions. Typical yields of total RNA are 10-20 µg for brain and 2-4 µg for spinal cord. RNA yields from individual fractions are up to 4 µg and 0.8 µg for brain and spinal cord respectively, depending on the fraction. A blank sucrose gradient already shows some background absorption at 260 nm, due to the presence of DTT. This background can be subtracted during data analysis in order to normalize sample values. EDTA treatment collapsed the polyribosome peaks, demonstrating the sedimentation profile is due to translation.

Figure 1. Workflow and potential applications of in vivo polyribosome fractionation. Please click here to view a larger version of this figure.
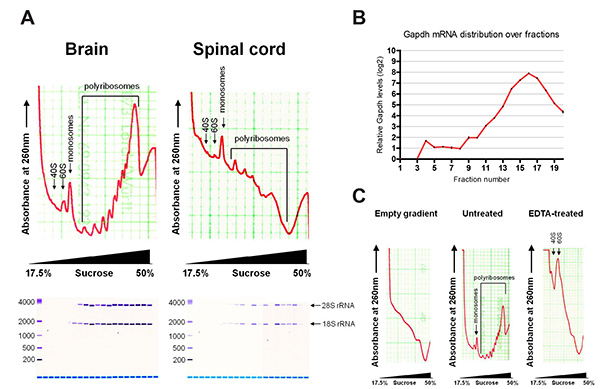 Figure 2. (A) Typical polyribosome profiles of brain and spinal cord, with assessment of extracted RNA by Bioanalyzer. (B) Distribution of Gapdh mRNA over fractions by qRT-PCR. (C) Polyribosome profiles of empty gradient and brain with and without EDTA treatment. Please click here to view a larger version of this figure.
Figure 2. (A) Typical polyribosome profiles of brain and spinal cord, with assessment of extracted RNA by Bioanalyzer. (B) Distribution of Gapdh mRNA over fractions by qRT-PCR. (C) Polyribosome profiles of empty gradient and brain with and without EDTA treatment. Please click here to view a larger version of this figure.
| 1.1 | 2x gradient buffer | 30 mM Tris-HCl pH7.4, 30 mM MgCl2, 600 mM NaCl, 200 µg/ml cycloheximide (CHX), 2mM dithiothreitol (DTT) | |
| 1.1 | sucrose solution 17.5% | 8.67 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 25.6% | 12.8 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 33.8% | 16.9 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 41.9% | 20.95 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 50% | 25 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 2.3 | homogenization buffer A | 0.25 M sucrose, 50 mM Tris/HCl, pH7.4), 5 mM MgCl2, 25 mM KCl | 200 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1mM phenylmethanesulfonylfluoride (PMSF), 100 U/ml RNAsin |
| 3.1 | 2M sucrose solution | 68.4% sucrose, 50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 25 mM KCl | 100 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1 mM PMSF |
| 3.1 | 1.1M sucrose solution | 38.5% sucrose, 50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 25 mM KCl | 100 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1 mM PMSF |
| 3.1 | 0.9M sucrose solution | 30.8% sucrose, 50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 25 mM KCl | 100 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1 mM PMSF |
| 4.1 | homogenization buffer B | 0.25 M sucrose, 50 mM Tris/HCl, pH7.4, 5 mM MgCl2, 25mM KCl, 1% NP-40, 1% sodium deoxycholate | 200 µg/ml CHX, 1x Roche complete protease inhibitor, 1mM DTT, 1 mM PMSF , 100 U/ml RNAsin |
Table 1. List of buffers and solutions.
Discussion
Although polyribosome fractionation is not a novel technique, it remains a particularly challenging one. Based on the input material, substantial optimization can be necessary. This is especially the case for in vivo CNS samples, where the amount of material is often a limitation and fatty tissue components hinder isolation of translating mRNAs. Most published fractionation protocols deal with yeast or mammalian cell lines, and there are established protocols for the brain12,13,14. In contrast, there are barely any publications describing fractionation of the spinal cords, and previous protocols require spinal cord from a large number of animals to be pooled15. For these reasons, several essential modifications were made to adapt the fractionation protocol for CNS tissues, including single mouse spinal cord. Ribosome immobilization with cycloheximide is performed during animal perfusion to avoid dissociation of ribosomes during the long process of tissue extraction. Subsequently, polysomal RNAs are extracted in a specific manner to maximize polyribosome yield. First, controlled homogenization of the tissue by douncing, keeps the nucleus intact and prevents DNA contamination. The nucleus is then removed reliably by centrifugation. The combination of the detergents NP-40 and sodium deoxycholate ensures lysis of the endoplasmic reticulum and release of its membrane associated ribosomes. In addition, UV-absorbing components within the lipid rich myelin obscure the polyribosome profiles. Myelin flotation is therefore necessary, where dense compounds such as polyribosomes are pelleted and lighter compounds such as myelin float and are removed16. Polyribosomes are then sedimented over a 17.5% to 50% sucrose gradient. Instead of manually layering and freezing each sucrose layer, gradients can also be prepared using a gradient mixer. Extraction of RNA from fractions using acidic phenol/chloroform allows DNA to be removed with minimal RNA loss. However, phase inversion may occur at dense sucrose fractions (above fraction 16).
This protocol provides advantages over other conventional methods that address the level of translation. For example, both the measurement of phosphorylated S6 (a prominent translation marker) as well as labeling of nascent chains with radioisotopes give information on global translation levels but reveal little on what specifically is being translated. Polyribosome fractionation, on the other hand, allows not only assessment of global translation, but also of the identity of translating mRNAs and the related regulatory proteins. Quantitative real time PCR can be performed on isolated RNAs for a quick read out of selected mRNAs, and microarrays and next generation RNA sequencing can be performed for genome wide studies. The optimizations presented here allow the technique to be used with minimal amounts of CNS tissues, therefore diminishing the number of animals needed and improving overall experimental quality by reducing tissue processing time. On the other hand, this technique cannot distinguish stalled ribosomes from translating ones. Transcripts carrying stalled ribosomes will sediment in the heavy fractions, and this should be kept in mind when interpreting the data.
There are recent reported techniques, namely RiboTaq and TRAP, in which ribosomes are labeled with epitope tags or reporters respectively in a cell type specific manner and associated mRNAs isolated by immunoprecipitation17,18. This technique can be coupled to polyribosome fractionation to offer high resolution read out for specific cell populations. Ribosome foot-printing is another novel technique that involves nuclease digestion to generate small fragments, or “footprints”, of mRNAs that are protected by ribosomes. Libraries are then generated from these footprints and sequenced. This method provides a codon-specific whole genome analysis on translation and is able to identify stalled ribosomes, non-AUG start codons and small upstream open reading frames, which cannot be achieved by polyribosome fractionation19. However, polyribosome fractionation can be coupled to ribosome profiling for isolation of digested fragments containing single ribosomes, thus enriching for the molecular species needed for library preparation in which high sample purity is often required. Taken together, polyribosome fractionation is a flexible method with lasting significance and can be coupled to various downstream applications including genome wide assays like next-generation sequencing.
Disclosures
No conflict of interest declared.
Acknowledgments
This work was supported by the German Federal Ministry of Education and Research (BMBF), the Systems Biology of Signaling in Cancer (Helmholtz Alliance on Systems Biology) and the German Cancer Research Center (DKFZ).
References
- Schwanhäusser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Lau AG, et al. Distinct 3'UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF) Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15945–15950. doi: 10.1073/pnas.1002929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqué M, López JM, Foissac S, Guigó R, Méndez R. A combinatorial code for CPE-mediated translational control. Cell. 2008;132(3):434–448. doi: 10.1016/j.cell.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grolleau A, et al. Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. The Journal of Biological Chemistry. 1074;277(25):22175–22184. doi: 10.1074/jbc.M202014200. [DOI] [PubMed] [Google Scholar]
- Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Molecular Cell. 2003;12(4):889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- Zivraj KH, et al. Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. The Journal of Neuroscience. 2010;30(46):15464–15478. doi: 10.1523/JNEUROSCI.1800-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittis Pa, Lu Q, Flanagan JG. Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell. 2002;110(2):223–235. doi: 10.1016/s0092-8674(02)00813-9. [DOI] [PubMed] [Google Scholar]
- Tcherkezian J, Brittis Pa, Thomas F, Roux PP, Flanagan JG. Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell. 2010;141(4):632–644. doi: 10.1016/j.cell.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Ji S-J, Porse BT, Jaffrey SR. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell. 2013;153(6):1252–1265. doi: 10.1016/j.cell.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AG, et al. Distinct 3'UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF) Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15945–15950. doi: 10.1073/pnas.1002929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete MJ, Vernal R, Dolznig H, Müllner EW, Garcia-Sanz JA. Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. RNA. 2007;13(3):414–421. doi: 10.1261/rna.79407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito AM, et al. Eukaryotic polyribosome profile analysis. J. Vis. Exp. 2010. [DOI] [PMC free article] [PubMed]
- Sampath P, Lee QY, Tanavde V, et al. Schlaeger T, editor. Identifying translationally regulated genes during stem cell differentiation. Current Protocols in Stem Cell Biology. 2011. [DOI] [PubMed]
- Chiu FC, Smith ME. Studies on rat spinal cord polysomes: postnatal development and experimental demyelination. Journal of Neurochemistry. 1978;31(4):835–844. doi: 10.1111/j.1471-4159.1978.tb00118.x. [DOI] [PubMed] [Google Scholar]
- Larocca JN, Norton WT, et al. Bonifacino JS, et al., editors. Isolation of myelin. Current Protocols in Cell Biology. 2007. [DOI] [PubMed]
- Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135(4):738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(33):13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar Ga, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nature Protocols. 2012;7(8):1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]