

Abstract
PALB2 was first identified as a partner of BRCA2 that mediates its recruitment to sites of DNA damage. PALB2 was subsequently found as a tumor suppressor gene. Inherited heterozygosity for this gene is associated with an increased risk of cancer of the breast and other sites. Additionally, biallelic mutation of PALB2 is linked to Fanconi anemia, which also has an increased risk of developing malignant disease. Recent work has identified numerous interactions of PALB2, suggesting that it functions in a network of proteins encoded by tumor suppressors. Notably, many of these tumor suppressors are related to the cellular response to DNA damage. The recruitment of PALB2 to DNA double-strand breaks at the head of this network is via a ubiquitin-dependent signaling pathway that involves the RAP80, Abraxas and BRCA1 tumor suppressors. Next, PALB2 interacts with BRCA2, which is a tumor suppressor, and with the RAD51 recombinase. These interactions promote DNA repair by homologous recombination (HR). More recently, PALB2 has been found to bind the RAD51 paralog, RAD51C, as well as the translesion polymerase pol η, both of which are tumor suppressors with functions in HR. Further, an interaction with MRG15, which is related to chromatin regulation, may facilitate DNA repair in damaged chromatin. Finally, PALB2 interacts with KEAP1, a regulator of the response to oxidative stress. The PALB2 network appears to mediate the maintenance of genome stability, may explain the association of many of the corresponding genes with similar spectra of tumors, and could present novel therapeutic opportunities.
Keywords: PALB2, tumor suppressor, DNA repair, homologous recombination, genome stability, breast cancer
1. Introduction
1.1. DNA double-strand break repair proteins as tumor suppressors
The identification of tumor suppressor genes has been highly important for understanding how tumors develop (reviewed in [1, 2]). Breast cancer is a good example, where numerous genes have been identified which can cause familial cancer when they are mutated [3–7]. These include the two major genes associated with familial, or inherited, breast cancer, BRCA1 and BRCA2 [8, 9]. Inherited germline mutations in BRCA1 or BRCA2, respectively, confer 57% and 49% lifetime risks of developing breast cancer by one estimate [10]. BRCA1 and BRCA2 are also major ovarian cancer genes [11]. Mutation of these genes has also been implicated in other cancers, such as those of the pancreas and prostate [12, 13].
The products of many of the identified breast cancer genes, including ATM, CHEK2, TP53, BRCA1, BRCA2, PALB2, BRIP1, ABRA1, NBS1, RAD50, RAD51C and XRCC2 [3–7], are involved in DNA damage signaling in response to DNA double-strand breaks (DSBs) and/or DSB repair by homologous recombination (HR). This includes roles for ATM, Chk2 (encoded by the CHEK2 gene), and RAP80 and Abraxas (encoded by the ABRA1 gene) in DNA damage signaling. Further, NBS1 and RAD50 are required for end resection during HR, and BRCA2, PALB2, RAD51C and XRCC2 are involved in strand invasion or resolution of Holliday junction intermediates during HR (reviewed in [2, 14–17]). BRCA1 appears to have roles in each of the above activities [18–21].
DSBs are among the most deleterious DNA lesions that occur in cells and are an important source of genome instability. Thus, understanding how DSBs are repaired is important for understanding the origins of many cancers. Further, the repair of DSBs modulates the therapeutic response to radiation and to a variety of chemotherapeutic agents [22, 23], many of which induce DSBs as a result of replication fork collapse or as an intermediate of repair.
1.2. Fanconi anemia, cancer susceptibility, and DNA interstrand crosslink repair
Sixteen Fanconi anemia (FA) genes have been identified (FANC- A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P and Q) [24–46]. FA is a chromosome instability syndrome that is characterized by progressive bone marrow failure, congenital anomalies, and a predisposition to cancer (reviewed in [47, 48]). The risk of acute myeloid leukemia, and of head and neck squamous cell carcinoma, are especially elevated. Cells from FA patients display spontaneous chromosome instability, a defect in DNA interstrand crosslink-induced HR, and hypersensitivity to mitomycin C (MMC) and other DNA interstrand crosslinking agents [47, 49, 50]. The phenotypes of FA cells emphasize the importance of DNA repair pathways as tumor suppressors which prevent genome instability.
A core complex containing eight of the corresponding proteins, FANC- A, B, C, E, F, G, L, and M, is required for monoubiquitination of FANCD2 and FANCI [40, 41, 51]. Four of the FA genes, BRCA2/FANCD1, BRIP1/FANCJ, PALB2/FANCN, and RAD51C/FANCO are also breast cancer susceptibility genes [6, 7, 9, 52–54], and the corresponding proteins are not required for monoubiquitination of FANCD2. This subset of FA genes/proteins is also critical for cellular resistance to ionizing radiation (IR) [55–59].
1.3. The PALB2 protein may scaffold, or coordinate, HR proteins and other DNA damage response proteins
PALB2 is among the more recently identified breast cancer genes [52, 53, 60] and is required for HR [24, 53, 58, 60–67]. The PALB2 protein directly binds to BRCA2 [58, 68] and is a functional partner of BRCA2. This issue is considered in more detail in Section 4.1.
Here, we will review how recent work on PALB2 has elucidated the cooperation of RAP80 and Abraxas, BRCA1, PALB2 and BRCA2, RAD51, and the RAD51 paralogs RAD51C and XRCC3, in a network of DNA DSB repair by HR [24, 53, 58, 60–64, 66]. Alterations of each of the corresponding genes, except for RAD51 and XRCC3, have been found to cause human cancer [4, 6, 7, 12, 69, 70]. Thus, understanding this DNA repair network, which functions to maintain genome stability, is important for understanding the etiology of cancer.
PALB2 also interacts with MRG15 [65, 71, 72], which is a component of various histone modifying complexes. This interaction represents a potential interface between chromatin regulation and DNA repair, as detailed in Section 3.2.
In addition, PALB2 also binds KEAP1 [73, 74], which is a regulator of Nrf2, a transcription factor for antioxidant genes. The interaction of PALB2 and KEAP1 will be examined in depth in Section 2.3. Mutations of Nrf2 and KEAP1 have been found associated with lung cancer [75]. This is of interest, given that reactive oxygen species (ROS) can lead to oxidative DNA damage that can yield DSBs. Further, ROS can either promote or suppress carcinogenesis, and can modulate the therapeutic response of tumors (reviewed in [76, 77]). Together, these various interactions, including those with HR proteins, a chromatin regulator, and a regulator of the response to oxidative stress, suggest that PALB2 potentially acts as a molecular scaffold that coordinates HR with other activities related to the DNA damage response [65, 66]. Ultimately, this network may prevent the development of cancerous cells by maintaining genome stability.
PALB2 has 1186 amino acids and a size of 131 kDa. Structural motifs which mediate protein interactions, including coiled-coil and WD40 domains, respectively, are present at the N- and C-termini of PALB2 (Fig. 1A). We will consider interactions with the N-terminus of PALB2, the middle region, and then the C-terminus, respectively, in Sections 2, 3, and 4. We will also examine how mutation of PALB2 in breast cancer affects its various protein interactions and may lead to cancer (Sections 5.1 and 5.2).
Figure 1.
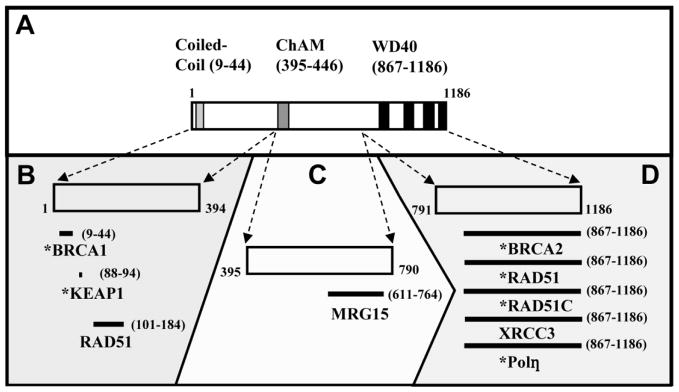
PALB2 has numerous interactions in its role in mediating DNA damage responses. (A) Three structural domains have been identified, one each in the N-terminal, central, and C-terminal thirds of the protein. (B) Protein interactions with the N-terminal portion of PALB2 (amino acids 1–394) which have been identified are displayed. (C) MRG15, the only known interaction of the central region of PALB2 (amino acids 395–790) with a specific protein, is shown. (D) Numerous proteins with functions in homologous recombination (HR) associate with the WD40 domain present in the C-terminus of PALB2. (B–D) Interacting proteins which have been identified as tumor suppressors are indicated (*).
2. The N-terminus of PALB2
The amino-terminal one-third of PALB2 (approximately amino acids 1–394) contains one prominent structural element, a coiled-coil domain. Coiled-coil motifs are known to mediate protein-protein interactions [78]. The coiled-coil motif in PALB2 mediates interactions with BRCA1, and may thereby confer proper localization of PALB2 [63, 64]. Further, this N-terminal region of PALB2 is required for interactions with KEAP1 and with the RAD51 recombinase. The N-terminus of PALB2 is shown in detail in Fig. 1B along with interactions that have been identified.
2.1 Binding to BRCA1 and role in PALB2 localization
BRCA1 and BRCA2 are both recruited to nuclear foci in response to DNA damage [79, 80]. Further, BRCA1- and BRCA2-deficient cells have similar cellular phenotypes, including defects in DSB-initiated HR, chromosome instability, and hypersensitivity to DNA damaging agents [21, 26, 81]. But a direct physical interaction had never been demonstrated between these proteins. As it turns out, they interact indirectly with PALB2 acting as a linker [63, 64, 66].
Different domains on PALB2 mediate direct interactions with BRCA1 and BRCA2 [58, 63, 64, 66, 68]. The interaction of PALB2 with BRCA1 is via coiled-coil motifs present on PALB2 (amino acids 9–44) and BRCA1 (1393–1424) [63, 64, 66]. This interaction promotes DSB-initiated HR [63, 64, 66] and resistance to MMC [63, 66], suggesting that BRCA1 and PALB2 function together in a DNA repair pathway.
The question arose as to whether the interaction of BRCA1 and PALB2 mediates the recruitment of one or the other to nuclear foci. Two initial reports suggested that this interaction mediates the assembly of PALB2 foci [63, 64]. Further, it was reported that this interaction also promotes the assembly of BRCA2 and RAD51 foci in a PALB2-dependent manner [63]. Another report did not support these conclusions, however [66].
Subsequently, it was demonstrated that defective localization of a mutant of the PALB2 coiled-coil domain, and of RAD51 downstream, can be rescued by fusion to the BRCT repeats that normally localize BRCA1 [62]. This lends strong support to the conclusion that BRCA1 localizes PALB2. Importantly, RNAi-mediated depletion of PALB2 [61], or a genetic deficiency for PALB2 in cells from a FA patient [63], does not affect BRCA1 foci. Together, these results suggest that BRCA1 acts upstream of PALB2, which in turn has a key role in a pathway that recruits BRCA2 and RAD51 to sites of DNA damage (Fig. 2).
Figure 2.

PALB2 links BRCA1 and BRCA2 into a pathway of products of breast cancer susceptibility genes. This pathway mediates HR, and as a result, the maintenance of genome stability. BRCA1 binds to PALB2 through a coiled-coil motif present on each protein and recruits it to sites of DNA damage. PALB2 then directly binds to the N-terminus of BRCA2 through its WD40 domain and recruits BRCA2. BRCA2 directly binds and recruits the RAD51 recombinase, which mediates strand invasion. PALB2 can also directly bind RAD51, but whether PALB2 can recruit RAD51 independent of BRCA2 is unknown. BRCA1, PALB2, and BRCA2 are all breast cancer susceptibility genes.
The recruitment of BRCA1 to DSBs is mediated by ubiquitin-dependent signaling that is centered around the RNF8 ubiquitin ligase (reviewed in [82]). Given that PALB2 is recruited in a BRCA1-dependent manner, the question arose as to whether ubiquitin-based signaling recruits PALB2 [62]. This possibility is supported by the finding that PALB2 colocalizes with ubiquitin, detected using the FK2 antibody, following exposure to IR [62]. Similar to the result for BRCA1 [83–85], RNAi-mediated depletion of RNF8 dramatically reduces IR-induced PALB2 foci [62]. Further, RNF8 binds to the MDC1 checkpoint mediator [83–85], and MDC1 is also required upstream of RNF8 in the recruitment of BRCA1 [86] and PALB2 [62] to sites of DNA damage.
Downstream of MDC1 and RNF8, RAP80 binds to K63-linked polyubiquitin chains and recruits its partner, Abraxas [87–90]. While less prominent than the roles of MDC1 and RNF8, RAP80 and Abraxas also function in recruiting PALB2 to sites of DNA damage that are induced by IR [62]. Since MDC1 binds to the histone variant, H2AX [86], which is phosphorylated after exposure to IR, the MDC1-RNF8-RAP80-Abraxas-BRCA1 network may connect PALB2-dependent HR to early signaling events that occur upon the generation of DSBs [62] (Figure 3).
Figure 3.

Ubiquitin-dependent signaling culminates in the recruitment of PALB2 to sites of DNA damage. (Left) MDC1 binds to the phosphorylated histone variant, H2AX, and is then recognized by the RNF8 E3 ubiquitin ligase. RNF8 mediates polyubiquitination of H2A and of H2AX. (Right) Polyubiquitinated H2A and H2AX are then recognized by RAP80, which has an ubiquitin-interaction motif. The partner of RAP80, Abraxas, is phosphorylated in a cell cycle-dependent manner. BRCA1 binds to phospho-Abraxas, which thereby results in the recruitment of PALB2.
2.2. Oligomerization of PALB2
In addition to interacting with BRCA1, the N-terminal coiled-coil of PALB2 can also self-associate [67, 91]. In this context, it should be noted that the coiled-coil domain of PALB2 mediates the assembly of PALB2 and RAD51 foci, as well as DSB-initiated HR and resistance to MMC [62–64, 66, 67, 91]. No separation of function mutant of the PALB2 coiled-coil domain has been identified that can directly compare the role of the BRCA1-PALB2 hetero-oligomer and the PALB2 homo-oligomer in vivo. Thus, the importance of the PALB2 homo-oligomer in these DNA damage response-related functions of PALB2, as compared to the role of the hetero-oligomer with BRCA1, has been unclear. An initial response to this question came from fusing the BRCT-repeats of BRCA1, which are required for its localization, to a mutant of the PALB2 coiled-coil domain that cannot interact with either BRCA1 or other PALB2 molecules [62]. Expression of this fusion protein in PALB2-deficient cells rescues various PALB2-dependent functions, including the assembly of RAD51 foci, DNA repair by HR, and resistance to MMC. On this basis, it appears that the interaction of PALB2 with BRCA1 may be sufficient to mediate each of these key functions of PALB2 and that the PALB2 homo-oligomer is not essential.
PALB2 promotes the assembly of RAD51 into a nucleoprotein filament with single-strand DNA in vitro [92, 93]. A PALB2 mutant lacking the coiled-coil domain (monomeric PALB2) shows increased DNA binding and increased assembly of the RAD51 filament in vitro, as compared to PALB2 which contains the coiled-coil [91]. Thus, based upon these results, the PALB2 homo-oligomer could have a regulatory role that is required for optimal maintenance of genome stability. However, whether the PALB2 homo-oligomer has such a regulatory role in intact cells is uncertain, especially since BRCA1 was not present in the in vitro experiments described above. It is also unknown how homo- versus hetero-oligomerization of PALB2 is regulated in vivo, and whether there are specific signals that induce one or the other at specific stages of DNA repair.
2.3. Interaction with KEAP1, a regulator of NRF2
Other interactions of PALB2, including that with KEAP1, have suggested functions for PALB2 that are not directly related to DNA repair but which may be complementary to its other characterized roles. KEAP1 is an E3 ubiquitin ligase which is a sensor of oxidative stress [94, 95]. KEAP1 can target NRF2, a master transcriptional factor for anti-oxidant genes, for cytoplasmic degradation. KEAP1 also has a role in nuclear export of NRF2 [73, 94]. But in response to oxidative stress, KEAP1-dependent down-regulation of NRF2 function is diminished, thereby promoting the anti-oxidant response. Importantly, PALB2 and NRF2 contain an identical LDEETGE sequence that can directly bind to KEAP1 [73]. As such, PALB2 can compete with NRF2 for binding to KEAP1 and can also disrupt pre-existing KEAP1-NRF2 complexes. Thus, PALB2 is a positive-regulator of NRF2 function and thereby has a role in cellular redox homeostasis [73]. In particular, by binding KEAP1, PALB2 can increase NRF2 occupancy at its target promoters, resulting in decreased cellular levels of ROS. Since ROS can lead to DNA damage (reviewed in [96]), positive regulation of NRF2 is another way in which PALB2 can promote the maintenance of a stable genome.
The LDEETGE motif is present at amino acids 88–94 in PALB2, so it is distinct from the coiled-coil domain at the N-terminus of this protein. Indeed, while mutation of the coiled-coil motif disrupts interactions with BRCA1, this does not disrupt the interaction of PALB2 with KEAP1 [73]. By independently interacting with BRCA1 and KEAP1, PALB2 may serve to coordinate DNA repair with detoxification of ROS. Further, while detoxification of ROS may contribute to the function of PALB2 as a tumor suppressor, it would appear that its role in DNA repair is more important in this regard. No cancer-associated mutants of the LDEETGE motif of PALB2 have been reported. Further, the majority of PALB2 mutations found in cancer disrupt interactions with proteins that are involved in HR [53, 58, 60, 69].
2.4. PALB2 binds DNA and may have a direct role in HR through its interaction with RAD51
Recent in vitro studies have yielded important insight into how PALB2 may function in DNA repair. Notably, PALB2 has a previously unrecognized DNA binding activity [92, 93]. Two DNA binding regions (amino acids 1–200 and 372–561), both within the N-terminal third of PALB2, have been identified. Along with the capacity of PALB2 to homo-oligomerize, these two binding sites could permit PALB2 to bind multiple DNA molecules simultaneously [93]. In this context, it should be noted that BRCA2 also binds DNA in vitro [97]. Interestingly, the PALB2-BRCA2 interaction can compensate for the loss of DNA binding by BRCA2 in mediating DSB-initiated HR in vivo [98].
Additionally, PALB2 can promote RAD51-dependent formation of a D loop [92, 93], in which invading single-strand DNA pairs with homologous DNA within an intact duplex (Fig. 4). In this way, PALB2 may have a direct role in HR. PALB2 could have two activities in promoting D loop formation. The first of these is a potential role in stabilizing the association of RAD51 with the invading single-strand DNA [93]. This is an activity that PALB2 shares with BRCA2 [99]. Additionally, PALB2 can inhibit the association of RPA with the invading DNA, thereby promoting binding of RAD51 to DNA [93].
Figure 4.
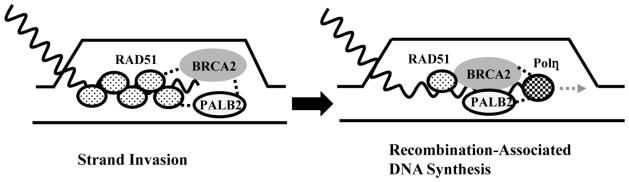
PALB2 and BRCA2 can directly bind RAD51 and pol η. The interaction of PALB2 and BRCA2 with RAD51 promotes formation of a D-loop during strand invasion (left), while interaction with pol η promotes recombination-associated DNA synthesis (right). PALB2 and BRCA2 may thereby coordinate these two steps during HR.
PALB2 has two sites which directly bind the RAD51 recombinase, one at the N-terminus, between amino acids 101–184, and another in the WD40 domain [92, 93]. It should be noted, however, that the WD40 domain of PALB2 appears to contain the primary binding site for RAD51 (Section 4.2). Whether these two RAD51-binding sites have distinct roles in mediating HR is unknown.
Interestingly, while BRCA2 and PALB2 are cancer susceptibility genes, RAD51 has not been identified as a tumor suppressor. Perhaps RAD51 has an essential function in HR and thus mutations which interfere with its function may not be viable. Support for this possibility comes from findings that RAD51 is overexpressed in BRCA1-deficient cells and is regulated by alternative mechanisms in BRCA2-deficient cells [100, 101]. Still, the RAD51 135G>C polymorphism has been shown to increase cancer risk in carriers of BRCA2 mutations [102], implicating some role for RAD51 in suppressing breast cancer.
While PALB2 alone can promote D-loop formation in vitro, it can synergize with RAD51AP1 [92]. In support of this finding, PALB2 and RAD51AP1 bind directly in vitro. Only a weak interaction has been found in cells, however, and it is not known what domain of PALB2 is involved. Still, these findings raise the possibility that a ternary complex of PALB2, RAD51 and RAD51AP1, or perhaps a higher-order complex that also contains BRCA2, may have an important function in HR.
3. The central portion of PALB2
The region at the center of PALB2, which we will define here as being from amino acids 395–790, has fewer defined interactions than the other two regions of PALB2. Still, the central third of PALB2 is of interest since it contains two regions that modulate the interaction of PALB2 with chromatin. This includes the chromatin-association motif (ChAM) at amino acids 395–446 (Fig. 1A) and a region (amino acids 611–764) that binds the MRG15 protein [71, 103] (Fig. 1C). A subsequent report determined that a smaller region of PALB2, from amino acids 562–629, binds to MRG15 [104]. Both the ChAM motif and the region that binds to MRG15 are distinct from the DNA binding domains that have been identified in the N-terminal third of PALB2 (Section 2.4).
3.1 The chromatin-association motif (ChAM)
As determined by comparing the proportion of the protein present in the chromatin fraction following a detergent-based extraction, the ChAM domain clearly promotes the association of PALB2 with chromatin [103]. This is the case either with or without exposure to DNA damage induced by IR or MMC. Deletion of the ChAM motif does not completely abrogate PALB2 association with chromatin, however. Further, defective chromatin association of the ΔChAM mutant of PALB2 can be partially rescued by overexpression of MRG15, suggesting that the ChAM motif of PALB2 and the interaction of PALB2 with MRG15 cooperate in localizing PALB2 to chromatin. Since PALB2 regulates the association of BRCA2 with chromatin [61, 65, 103], it is perhaps not surprising that the ChAM domain of PALB2 also promotes the association of BRCA2 with chromatin [103]. The ChAM domain has a more minor effect on the association of BRCA1 with chromatin, which is consistent with the finding that PALB2 is not required for the assembly of BRCA1 foci ([61, 63, 91] and Section 2.1).
Interestingly, while the ChAM motif promotes the association of PALB2 with chromatin, it is not involved in binding to DNA [92, 93, 103]. The association of PALB2 with chromatin may instead be promoted by the apparent capacity of the ChAM motif to interact with nucleosomal histones [103].
The ChAM motif clearly has a role in the function of PALB2 in the cellular response to DNA damage [103]. For example, this motif promotes the recruitment of PALB2, and of RAD51 downstream, to sites of DNA damage. Further, a mutant of PALB2 in which the ChAM motif was deleted conferred no resistance to MMC when expressed in PALB2-deficient cells. Interestingly, cells with a genetic deficiency for PALB2 are sensitive to inhibition of poly-ADP ribose polymerase (PARP) [57, 58, 93, 103], but deletion of the ChAM motif yielded only an intermediate sensitivity to the PARP inhibitor, olaparib, as compared to cells lacking reconstitution [103]. It is currently unknown why the ChAM motif is fully required for resistance to MMC but not olaparib. One possibility is that these agents induce DNA double-strand breaks (DSBs) after collapse of the replication fork, but by different mechanisms [103].
3.2 PALB2 interacts with MRG15, a component of various histone-modifying complexes
The interaction of PALB2 with MRG15 has been identified both in protein complexes isolated from cells, using mass spectrometry [65, 71], and through yeast-two hybrid screens [72]. This interaction is of particular interest given the potential challenges of recognizing and repairing DNA damage in the context of highly packaged chromatin (reviewed in [105, 106]). Indeed, the interaction of PALB2 and MRG15 provides a potential interface between chromatin regulation and DNA damage repair by HR.
MRG15/MORF4L1 and its yeast homolog, EAF3, are components of the Tip60/NuA4 histone acetyltransferase complex that is conserved from yeast to humans (reviewed in [107, 108]). The Tip60 complex is an important chromatin regulator that has been implicated in transcriptional regulation, as well as in DNA repair [108]. In particular, Tip60-dependent acetylation of histones H3 and H4 can mediate ubiquitination of H2A and recruitment of the RAD51 recombinase [109, 110].
Interestingly, while MRG15 readily associates with PALB2, other componenents of the Tip60 complex are in low abundance, or are absent, in PALB2-containing complexes [65, 71]. Thus, MRG15 may have PALB2-related functions that are independent of the Tip60 complex. In this context, it should be noted that while MRG15 is also a component of histone deacetylase (HDAC) 1 and 2 complexes [111], and the KDM5A histone demethylase complex [112], components of these other MRG15-related complexes were also not specifically detected in the PALB2 complex.
Multiple reports suggest that MRG15 has some role in PALB2-dependent repair of DNA by HR, but differ in the purported role of MRG15 in this process [65, 71, 72]. Sy et al. (2009) [71] suggest that knockdown of MRG15, or deletion of a broad region of PALB2 (amino acids 611–764) that contains the MRG15-interacting domain, leads to hyper-recombination. They therefore propose that MRG15 is a suppressor of HR. Consistent with this possibility, deletion of the region of PALB2 which interacts with MRG15 resulted in increased sister chromatid exchange (SCE). Depletion of MRG15, or reconstitution of PALB2-deficient cells with the MRG15-interaction mutant, did not alter sensitivity to MMC in this study [71].
In contrast, Hayakawa et al. (2010) [65] suggest that RNAi-mediated depletion of MRG15 decreases DNA DSB-initiated HR, but not to the same degree as depletion of PALB2 or BRCA2. They also reported that deletion of MRG15 sensitizes cells to MMC. Opposite from Sy et al., and consistent with a positive role in HR, these authors instead found that depletion of MRG15 decreases SCE [65].
Thus, together, these two reports support a role for the interaction of MRG15 with PALB2 in regulating HR, but disagree about whether MRG15 is a positive or negative regulator. While resolution of this question is important for understanding the mechanistic contribution of MRG15 to HR, the basis for these reported differences is currently unclear.
Consistent with a role for MRG15 in promoting HR, Hayakawa et al. (2010) also found that depletion of MRG15 leads to decreased assembly of PALB2 and RAD51 foci, both in untreated populations and following exposure to IR. This is of interest, since the presence of a chromodomain in MRG15, and its association with various histone modifying complexes, suggests an intimate relationship of MRG15 with chromatin. It should be noted, however, that while the chromodomain of MRG15 can bind histone H3 methylated at Lysine 36 [113], the role of this chromodomain in the regulation of PALB2 behavior during HR is currently unknown.
An additional report also demonstrated an association between MRG15 and PALB2 [72]. Consistent with Hayakawa et al. (2010), murine embryonic fibroblasts, in which Mrg15 is disrupted, display decreased Rad51 foci and a moderate sensitivity to IR [72]. Given the apparent physical and functional relationship of MRG15 to PALB2, which is the product of a breast cancer gene, MRG15 was considered to be a strong candidate cancer gene. Martrat et al. (2011) found no alterations in the MRG15 gene in familial breast cancer [72]. Meanwhile, another study identified variants in patients with inherited breast cancer but these were determined to be unlikely to be disease-causing [114]. Further, while PALB2 is also a FA gene, MRG15 was not identified as a FA gene in the screen by Martrat et al.
4. The C-terminus of PALB2
The carboxy-terminal one-third of PALB2 (approximately amino acids 791–1186) is dominated by a seven-bladed WD40-type β-propeller from amino acids 853–1186 (Fig. 1A). WD40 domains are well known to mediate protein interactions (reviewed in [115]). Indeed, it was recently discovered that the WD40 domain of PALB2 mediates direct interactions with many of the key proteins involved in HR, including BRCA2 and RAD51, as well as the RAD51 paralogs RAD51C and XRCC3 [58]. Additionally, binding of the WD40 domain of PALB2 to pol η appears to mediate recombination-associated DNA synthesis [116]. Thus, the C-terminal WD40 domain may coordinate the activities of these key effectors of HR (Fig. 4). Since truncation of even the last four amino acids of PALB2 destabilizes the entire WD40 domain [68], and given the important role of HR in tumor suppression, the WD40 domain appears to be critical to the role of PALB2 in preventing tumor formation.
4.1 BRCA2 functions are mediated by direct binding to the PALB2 WD40 domain
Reflective of the important functional relationship between PALB2 and BRCA2, PALB2 was named “partner and localizer of BRCA2” [61]. Few BRCA2-interacting proteins had been identified, so the discovery of PALB2 yielded important new insight into the function and regulation of BRCA2.
It has been found that PALB2 is required for the recruitment of BRCA2 to nuclear foci that assemble either spontaneously or in response to DNA damage [61, 63]. Further, PALB2 stabilizes the chromatin-associated pool of BRCA2 [61, 65]. BRCA2 has a well-characterized role in the assembly of the RAD51 recombinase into nuclear foci [80]. Thus, the fact that PALB2 also regulates RAD51 foci [61, 63], strengthens the conclusion that it is a functional partner of BRCA2. Additional support for this possibility comes from the finding that PALB2 has a role in DSB-initiated HR and in resistance to MMC which is shared with BRCA2 [61]. Additionally, PALB2 appears to mediate the BRCA2-dependent intra-S phase checkpoint in response to IR [61], and PALB2 and BRCA2 cooperate in regulating the G2 checkpoint in response to IR [117].
Importantly, mutants of PALB2 found in breast cancer or Fanconi anemia patients, which are defective for interaction with BRCA2, are deficient for DSB-initiated HR and for resistance to MMC [24, 53, 60]. Cells from FA patients with biallelic mutation of BRCA2 have similar defects in the DNA damage response [26, 81]. The close functional relationship of PALB2 and BRCA2 is also reflected by the fact that they are associated with similar spectra of cancers when mutated. Both are breast [8, 52, 53, 60, 118] and pancreatic [119, 120] cancer genes. Additionally, BRCA2 is a well-known ovarian cancer gene [8], and predicted deleterious mutations or epigenetic silencing of PALB2 are similarly observed in ovarian cancer [121, 122]. Further, biallelic mutation of PALB2 and BRCA2 in Fanconi anemia patients is associated with an especially early onset of acute myeloid leukemia, and with a high risk of medulloblastoma and Wilms tumor that is rarely seen in other FA subtypes [25, 123, 124].
Truncation mutants of PALB2 typically remove a portion, or all, of its WD40 domain. Defects of such mutants for DNA repair by HR were initially attributed to perturbation of BRCA2 function [24, 53, 60]. This was reasonable, since mutations of BRCA2 and PALB2 result in highly similar phenotypes in FA patients [25]. However, since the PALB2 WD40 domain binds a number of HR proteins, and inactivation of these proteins leads to similar defects in the DNA damage response, it is now unsure how much of the phenotypes associated with truncation of the PALB2 WD40 domain are directly due to perturbation of the interaction with BRCA2.
4.2 PALB2 interacts with RAD51 through a major binding site in its WD40 domain
Available evidence would suggest that the C-terminal WD40 domain of PALB2, rather than the N-terminal region (Section 2.4), is the primary binding site for RAD51. For example, truncation or elimination of the WD40 domain leads to a dramatic reduction in the binding of PALB2 to RAD51 [58, 63]. Additionally, deletion of exon 4 leads to reversion of a PALB2 mutation in EUFA1341 cells from a Fanconi anemia patient and results in deletion of amino acids 71–561 [24]. While this mutant lacks the N-terminal RAD51-binding domain, it is largely resistant to MMC. In contrast, the original Y551X mutant, present in parental EUFA1341 cells, which eliminates the WD40 domain but retains the N-terminal binding site for RAD51, confers no resistance to MMC.
Disruption of the WD40 domain abrogates the assembly of RAD51 foci [24, 63]. The function of PALB2 upstream of RAD51 is also supported by the finding that key regulators of PALB2, including BRCA1 and MRG15, are required for optimal assembly of RAD51 foci [63, 65].
Although both RAD51 and BRCA2 can directly bind to the WD40 domain of PALB2 [58, 68, 93], whether they bind simultaneously in a ternary complex has not been determined. This possibility is consistent, however, with the finding that RAD51 and BRCA2 have different patterns of interactions with missense mutants of the PALB2 WD40 domain [58]. Thus, each protein may have different points of contact with PALB2. Further, it has been demonstrated that WD40 domains on other proteins can bind to multiple substrates at the same time [125]. Consistent with the formation of such a ternary complex, PALB2 and a BRCA2 chimer promote RAD51-dependent strand invasion in vitro [93].
4.3 PALB2 interacts with the RAD51 paralog, RAD51C, through its WD40 domain
Five somatic paralogs of RAD51, including RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3, have been identified in vertebrate cells (reviewed in [17]). These paralogs have a similar size as RAD51, and also possess Walker A and B nucleotide binding motifs, but have an overall homology with RAD51 of 20–30% [17, 126]. Like RAD51, the paralogs have a role in HR and the maintenance of genome stability. For example, deletion of any of the paralogs in chicken DT40 cells compromises HR and sensitizes cells to MMC [55].
The role of the RAD51 paralogs is not well understood. Disruption of any of the genes in DT40 cells severely attenuates IR-induced assembly of RAD51 foci [55], suggesting a role early in HR prior to strand invasion. But some of the paralogs have also been reported to have functions late in HR [127–129]. This includes the finding that RAD51C foci persist longer at DSBs than do RAD51 foci [127], and the finding that RAD51C and XRCC3 can mediate resolution of Holliday junctions in vitro [129].
RAD51B-RAD51C-RAD51D-XRCC2 and RAD51C-XRCC3 complexes have been identified in cells [130–132], and direct interactions between various RAD51 paralogs have been identified using yeast 2- and 3-hybrid studies [133]. However, the recent finding that RAD51C, as well as XRCC3, can bind to the PALB2 WD40 domain is a strong indication that these RAD51 paralogs may function in HR outside of paralog-only complexes [58].
In a recent study, PALB2 and BRCA2 were the only proteins besides RAD51 and RAD51 paralogs that were identified in RAD51C protein complexes by mass spectrometry [58]. This was interesting considering that, like PALB2 and BRCA2, RAD51C had been identified as a breast/ovarian cancer gene [6, 7].
Further, RAD51C was identified as a putative FA gene [30], similar to PALB2 and BRCA2. Several affected children within a single family had a biallelic missense mutation in RAD51C (R258H). The only surviving child displayed cellular phenotypes, including DNA interstrand crosslink (ICL)-induced G2-M accumulation and chromosome instability, and developmental abnormalities, which are characteristic of FA. The surviving patient did not have cancer or display hematological abnormalities through age ten, which is not unusual in FA [30]. The finding that RAD51C directly binds to PALB2 further suggests a function for this RAD51 paralog in the FA-BRCA pathway of DNA repair. Consistent with this possibility, the RAD51C missense mutant from a FA patient, R258H, displays defective co-immunoprecipitation of PALB2 and BRCA2, as well as RAD51 and XRCC3 [58, 134]. Missense mutants of RAD51C from breast cancer patients, including L138F and D159N, display similar defects.
RAD51C directly binds to the WD40 domain of PALB2 [58]. Consistent with this observation, truncation of the WD40 domain abrogates binding to BRCA2, RAD51C and XRCC3, and largely diminishes binding to RAD51 [58]. Thus, the PALB2 WD40 domain could coordinate, or scaffold, a complex of key components of the machinery for HR. These proteins might cooperate to mediate DNA repair by HR and thereby promote genome stability.
Alternatively, it is possible that PALB2 binds to specific HR proteins at different steps in HR. In this way, PALB2 might coordinate various steps involved in this process, including strand invasion, recombination-dependent DNA synthesis, and resolution of the Holliday junction. As an example, the L1143P missense mutant of the PALB2 WD40 domain, which has a clearly diminished interaction with RAD51C, both in vitro and in vivo, leads to a modest but significant increase in the assembly of BRCA2 foci in response to IR [58]. Thus, it is possible that RAD51C acts subsequent to BRCA2 and displaces it from its interaction with PALB2. Further work will be needed to resolve these questions.
Since truncation of the PALB2 WD40 domain abrogates interactions with numerous HR proteins, to better analyze how each protein binds PALB2, missense mutants of the WD40 domain that were identified in breast cancer patients [135] were tested instead [58]. Interestingly, RAD51C has a different pattern of interaction with the L939W, T1030I and L1143P mutants of the PALB2 WD40 domain in vitro, than do BRCA2 or XRCC3, or RAD51. Thus, multiple HR proteins could possibly bind to the WD40 domain of PALB2 simultaneously.
4.4 PALB2 interacts with the RAD51 paralog, XRCC3, through its WD40 domain
XRCC3 also directly binds to the WD40 domain of PALB2 [58]. The pattern of interaction of XRCC3 with PALB2 WD40 missense mutants resembles that of BRCA2, rather than that of RAD51C. This is of potential importance since RAD51C and XRCC3 have been reported to form a complex [130–132]. This suggests that each protein may independently bind to PALB2 rather than one of the proteins in the RAD51C-XRCC3 complex simply having an interaction with PALB2. One possibility is that RAD51C and XRCC3 bind to the PALB2 WD40 domain at different steps during HR-dependent repair of DNA.
Interestingly, PALB2-deficient cells show diminished levels of the RAD51C-XRCC3 complex, as compared to complemented cells [58]. Thus, binding of PALB2 to one or both components of the RAD51C-XRCC3 complex may compete with a RAD51C-XRCC3 complex that is devoid of PALB2. While the RAD51C-XRCC3 complex has Holliday junction resolvase activity in vitro [136], whether the PALB2-free complex formed by these proteins has any function in cells is presently unknown.
The observation that RAD51C is a FA gene and breast/ovarian cancer susceptibility gene, like PALB2, yields some support for the possibility that the RAD51C protein has critical functions in complex with PALB2. But XRCC3 has not been identified as a FA gene or as a cancer gene, and the role of the complex it forms with PALB2 is currently unknown.
4.5 PALB2 binds pol η and promotes recombination-dependent DNA synthesis
An additional interaction of the PALB2 WD40 domain, with pol η, has recently been reported [116]. Pol η directly binds to PALB2 and also to the BRC repeats of BRCA2 [116]. It was already known that PALB2 promotes strand invasion during HR [92, 93], thereby forming a D-loop intermediate. The 3′ tail end ssDNA is then utilized as a primer for recombination-associated DNA synthesis. Thus, the ability of PALB2 to interact with both RAD51 and with pol η may coordinate strand invasion and subsequent synthesis (Fig. 4). Indeed, PALB2 and BRCA2 promote DNA synthesis by pol η on D loop substrates in vitro [116]. Further, PALB2, as well as BRCA2, are required for the assembly of pol η into foci at blocked replication forks and may recruit it to the 3′ end ssDNA at the D loop. Consistent with a key role in recombination-associated DNA synthesis, deletion of pol η in DT40 cells impairs HR [137].
Pol η is a translesion polymerase which can bypass thymidine dimers that are induced by ultraviolet radiation. A deficiency for this activity, resulting from biallelic mutation of the RAD30 gene, greatly increases the risk of sunlight-induced skin cancer in Xeroderma pigmentosum variant (XP-V) patients [138, 139]. While this demonstrates the role of pol η as a tumor suppressor, biallelic mutation of PALB2 or BRCA2 in Fanconi anemia does not confer a particular predisposition to skin cancer. Thus, the contribution of defects in pol η-dependent recombination-associated DNA synthesis to the development of cancer is less clear than the role of defects in pol η-dependent repair of thymidine dimers. The latter activity is related to nucleotide excision repair rather than HR.
5. Implications of the PALB2 network for cancer biology and cancer management
The finding that PALB2 is involved in a network of tumor suppressors that regulate or mediate HR has already yielded important insight into how this repair pathway maintains genome stability and thereby prevents cancer. The fact that BRCA1, PALB2, BRCA2 and RAD51C are all breast/ovarian cancer genes underlines the importance of the interaction of the corresponding proteins in a common pathway of DNA repair. The observation that mutation of many of these HR genes leads to a similar spectrum of tumors suggests the importance of particular DNA lesions, including DSBs and stalled/collapsed replication forks, to the genesis of breast/ovarian cancer. Importantly, further investigation of protein interactions within the PALB2 network should continue to improve the understanding of mechanistic steps that are involved in HR. This may ultimately lead to the identification of novel biomarkers for diagnosing and treating cancer, and could identify novel therapeutic targets.
5.1 How missense mutations in PALB2 affect DNA repair
With the exception of the G1145R missense mutant of PALB2, which was found in a breast cancer patient and does not diminish HR or cellular resistance to MMC [53], until recently no functional studies had been conducted on PALB2 missense mutants. A recent study, however, found that the L939W and L1143P missense variants/mutants of the PALB2 WD40 domain are stably expressed in PALB2-deficient fibroblasts and have modest but significant decreases in HR and in cellular resistance to IR [58]. This is consistent with the fact that altered, but not abrogated interactions with BRCA2, RAD51 and RAD51C, are observed [58]. In contrast, truncation mutants of PALB2, found in FA or breast cancer, which remove the WD40 domain, have no HR activity and confer no resistance to MMC [24, 53, 60]. Thus, missense mutants of the PALB2 WD40 domain may, in general, be associated with less genome instability than the truncation mutants. Although less than that conferred by truncation mutants, these missense mutants may still confer an increased cancer risk. Importantly, the recent study by Park et al. has established an assay system for further functional evaluation of PALB2 missense variants [58].
Interestingly, a missense mutant, T1030I, which was examined by Park et al. could not be stably expressed in PALB2-deficient fibroblasts and appears to be degraded in a proteasome-dependent manner [58]. Thus, there are at least two mechanisms by which missense mutants of the PALB2 WD40 domain may act. The first mechanism involves instability of PALB2, and thus lack of a functional complex with BRCA2, RAD51 and RAD51C, as seen for PALB2-T1030I. The second mechanism involves altered but not abrogated interactions with these HR proteins, as seen for PALB2-L939W and PALB2-L1143P. Unstable missense mutants are expected to have a larger defect in DNA repair and, thus a greater risk of developing cancer.
Similarly, truncation mutations of PALB2 also appear to act by at least two mechanisms. Truncations which remove the WD40 domain lead to stable mutants [24, 53, 60], whereas the Y1183X mutant of PALB2 interferes with closure of the WD40 structure and is unstable [25]. Either type of truncation mutant is expected to fully abrogate PALB2 function, however.
5.2 Role of the PALB2 network in preventing genome instability and replication defects
Genome instability enables the development of cancer by leading to a series of genetic changes, including amplifications, deletions, rearrangements and substitutions, which result in genetic diversity (reviewed in [140]). This diversity can lead to clonal selection and the development of more aggressive cancers, and even to resistance to therapy [141]. The PALB2 network is thought to suppress tumor formation by promoting DNA repair, thereby preventing genome instability and the initiation of cancer. Consistent with this thinking, BRCA1, PALB2, BRCA2, and RAD51C all prevent chromosome instability characterized by chromosome breaks or rearrangements that occur spontaneously, or in response to DNA interstrand crosslinking agents or PARP inhibitors [21, 26, 30, 81, 142].
There is also evidence that PALB2 and related proteins may maintain genome stability by promoting fidelity in DNA replication. Heterozygous mutation of PALB2 is associated with a six-fold increase in the risk of developing malignant disease [143]. Interestingly, cells that are heterozygous for PALB2 mutations display increased chromosome instability. Further, this increased chromosome instability is associated with perturbation of DNA replication, including increased origin firing and decreased distances between active origins [143]. Loss of heterozygosity would be expected to yield even greater chromosome instability and might similarly be related to defects at replication forks.
As a further indication of the potential role of the PALB2 network in mediating chromosome stability through a role in DNA replication, BRCA1 and BRCA2 prevent the degradation of newly replicated DNA at stalled forks [144, 145]. Evidence would suggest that their action in maintaining the stability of DNA at the replication fork is separable from their role in DSB repair [144]. The function of BRCA1 and BRCA2 in the response to replication stress is relevant both to understanding the genesis of cancer and the response to treatment. For example, BRCA1 is required for the stability of common fragile sites, which are loci at which replication stalling occurs more frequently [146]. Also, chemotherapeutic agents that result in ICLs induce replication stress and/or fork stalling [49, 145, 147]; BRCA1- [21] and BRCA2-deficient cells [26], as well as PALB2- [24, 53, 60, 61, 63] and RAD51C-deficient cells [6, 30, 55], are all hypersensitive to ICLs. While the role of PALB2 was not specifically tested, one might anticipate that it is also involved in this process.
5.3 Implications of the PALB2 network for the treatment of cancer
The identification of a network of tumor suppressors that includes PALB2 may have important consequences for treating cancer. For example, defects in DSB repair are typically associated with increased sensitivity to radiation. Additionally, because the DSB-initiated HR pathway can rescue defects in base excision repair, mutation of genes in this network are typically also associated with hypersensitivity to PARP inhibitors [57, 58, 93, 103, 148–150]. Further, DNA interstrand crosslinking agents, including platinum compounds and nitrogen mustards are a major class of chemotherapeutic agents utilized to treat cancer [49]. BRCA1, PALB2, BRCA2 and RAD51C, which are key components of the HR machinery, are required for cellular resistance to ICLs [6, 21, 26, 61]. Thus, there are numerous therapeutic options which may be utilized to treat tumors with deficiencies in the PALB2-based network of HR.
At present, there is abundant interest in inhibitors of histone methyltransferases and histone demethylases for treating cancer [151, 152]. Thus, the fact that MRG15 possesses a chromodomain and influences HR could potentially be utilized to rationally design combination treatments for cancer. For example, inhibitors which modulate histone methylation might be utilized to suppress recruitment of MRG15, and MRG15-dependent recruitment of PALB2 and RAD51, and thereby inhibit HR. This would be expected to sensitize cells to DNA damage induced by radiation or DNA interstrand crosslinking agents.
5.4 Assays for defective assembly of RAD51 foci to predict deficiencies in PALB2 network proteins
The majority of the tumor suppressors in the PALB2 network, including BRCA1, PALB2, BRCA2, and RAD51C, are involved in the assembly of RAD51 foci in response to DNA damage [30, 55, 61, 63, 80, 153]. Thus, it may be possible to utilize assembly of RAD51 foci as a functional assay to identify a defect in the central PALB2 network, without having information on the presence or clinical significance of mutations in any of the associated genes. Since this network of HR proteins mediates resistance to radiation, as well as to ICLs, a defect in RAD51 foci may be predictive of a better clinical response to such treatments [154]. For example, there have been reports that mutations in BRCA1 or BRCA2 lead to increased response of ovarian cancers to platinum compounds and/or increased survival [155–158]. An approach for ex vivo irradiation of living tumor tissue to assess levels of RAD51 foci in breast cancer tissue has recently been described [159].
6. Conclusions
Eight proteins, BRCA1, KEAP1, MRG15, BRCA2, RAD51, RAD51C, XRCC3, and pol η, have been identified which directly bind to PALB2 [58, 63–66, 68, 71, 73, 92, 93, 116]. The majority of these proteins, except for MRG15, RAD51 and XRCC3, have been demonstrated to have tumor suppressor activities [6, 8, 9, 75, 138]. Additionally, RAP80, Abraxas, and RAD51AP1 have functional interactions with PALB2 [62, 92]. Among these, RAP80 and Abraxas are also tumor suppressors [4, 160]. Thus, PALB2 appears to function in a network of tumor suppressors. PALB2 may act as the hub of this network based upon its numerous interactions and its apparent function near the center of the network. PALB2 recruitment to sites of DNA damage is downstream of ubiquitin-dependent signaling and of BRCA1. PALB2 recruitment also involves MRG15, which is a component of histone remodeling complexes [62, 65, 71]. Once recruited, though, PALB2 then has a role in regulating the response to oxidative stress through its interaction with KEAP1 [73], and appears to coordinate DNA repair by HR through its interactions with BRCA2, RAD51, RAD51C, XRCC3, and pol η[58, 92, 93, 116].
Loss of function of tumor suppressors in the PALB2 network is associated with a shared pre-disposition to breast and/or ovarian cancer via mutation of RAP80, Abraxas, BRCA1, PALB2, BRCA2 or RAD51C [4, 6, 8, 9, 52, 53, 160]. Understanding why loss of function of proteins related to DSB-initiated HR specifically predisposes individuals to breast and/or ovarian cancer may provide insight into risk factors for these cancers and could ultimately lead to improved cancer prevention. Also, further analysis of protein complexes which contain proteins in the PALB2 network may likely identify additional tumor suppressor genes.
Finally, as discussed in sections 5.3 and 5.4, the identification of the PALB2 network of DNA damage response proteins may have important implications for treating cancer, including the potential for identification of biomarkers to guide the selection of therapeutic options. Definition of this network has also provided a framework for a better understanding of DNA repair by HR and this insight could, ultimately, lead to novel therapeutic targets.
Acknowledgments
This work was supported by NIH R01 HL085587 (PRA).
Abbreviations
- BRCA1
Breast cancer susceptibility 1
- BRCA2
Breast cancer susceptibility 2
- DNA
Deoxyribonucleic acid
- DSB
DNA double-strand break
- FA
Fanconi anemia
- HR
Homologous recombination
- ICL
DNA interstrand crosslink
- IR
Ionizing radiation
- KEAP1
Kelch-like ECH-associated protein 1
- MMC
Mitomycin C
- MMR
Mismatch repair
- PALB2
Partner and Localizer of BRCA2
- PARP
poly-ADP ribose polymerase
- RAD51AP1
RAD51-associated protein 1
- RAP80
Receptor associated protein 80
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature reviews Genetics. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 2.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature reviews Molecular cell biology. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer cell. 2007;11:103–105. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Solyom S, Aressy B, Pylkas K, Patterson-Fortin J, Hartikainen JM, Kallioniemi A, Kauppila S, Nikkila J, Kosma VM, Mannermaa A, Greenberg RA, Winqvist R. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Science translational medicine. 2012;4:122ra123. doi: 10.1126/scitranslmed.3003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park DJ, Lesueur F, Nguyen-Dumont T, Pertesi M, Odefrey F, Hammet F, Neuhausen SL, John EM, Andrulis IL, Terry MB, Daly M, Buys S, Le Calvez-Kelm F, Lonie A, Pope BJ, Tsimiklis H, Voegele C, Hilbers FM, Hoogerbrugge N, Barroso A, Osorio A, Giles GG, Devilee P, Benitez J, Hopper JL, Tavtigian SV, Goldgar DE, Southey MC. Rare mutations in XRCC2 increase the risk of breast cancer. American journal of human genetics. 2012;90:734–739. doi: 10.1016/j.ajhg.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K, Deissler H, Engel C, Muller-Myhsok B, Neveling K, Kiechle M, Mathew CG, Schindler D, Schmutzler RK, Hanenberg H. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nature genetics. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 7.Vuorela M, Pylkas K, Hartikainen JM, Sundfeldt K, Lindblom A, von Wachenfeldt Wappling A, Haanpaa M, Puistola U, Rosengren A, Anttila M, Kosma VM, Mannermaa A, Winqvist R. Further evidence for the contribution of the RAD51C gene in hereditary breast and ovarian cancer susceptibility. Breast cancer research and treatment. 2011;130:1003–1010. doi: 10.1007/s10549-011-1677-x. [DOI] [PubMed] [Google Scholar]
- 8.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 9.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25:1329–1333. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104:2807–2816. doi: 10.1002/cncr.21536. [DOI] [PubMed] [Google Scholar]
- 12.Moran A, O’Hara C, Khan S, Shack L, Woodward E, Maher ER, Lalloo F, Evans DG. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Familial cancer. 2012;11:235–242. doi: 10.1007/s10689-011-9506-2. [DOI] [PubMed] [Google Scholar]
- 13.Lal G, Liu G, Schmocker B, Kaurah P, Ozcelik H, Narod SA, Redston M, Gallinger S. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer research. 2000;60:409–416. [PubMed] [Google Scholar]
- 14.Scully R, Xie A. Double strand break repair functions of histone H2AX. Mutation research. 2013;750:5–14. doi: 10.1016/j.mrfmmm.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Molecular and cellular biology. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mimitou EP, Symington LS. DNA end resection: Many nucleases make light work. DNA Repair. 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Seminars in cell & developmental biology. 2011;22:898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. The Journal of biological chemistry. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 19.Coleman KA, Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. The Journal of biological chemistry. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes & development. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer research. 2001;61:4842–4850. [PubMed] [Google Scholar]
- 22.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature reviews Cancer. 2012;12:587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 23.Barker CA, Powell SN. Enhancing radiotherapy through a greater understanding of homologous recombination. Seminars in radiation oncology. 2010;20:267–273. e263. doi: 10.1016/j.semradonc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nature genetics. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 25.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature genetics. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 26.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 27.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nature genetics. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 28.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature genetics. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 29.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, Fraternali F, Freund M, Hartmann L, Grimwade D, Roberts RG, Schaal H, Mohammed S, Rahman N, Schindler D, Mathew CG. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nature genetics. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 31.Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, Jaspers NG, Bettecken T, Joenje H, Schindler D, Rouse J, de Winter JP. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nature genetics. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- 32.Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nature genetics. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillon J, Ramirez MJ, Pujol R, Casado JA, Banos R, Rio P, Knies K, Zuniga S, Benitez J, Bueren JA, Jaspers NG, Scharer OD, de Winter JP, Schindler D, Surralles J. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. American journal of human genetics. 2013;92:800–806. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Winter JP, Leveille F, van Berkel CG, Rooimans MA, van Der Weel L, Steltenpool J, Demuth I, Morgan NV, Alon N, Bosnoyan-Collins L, Lightfoot J, Leegwater PA, Waisfisz Q, Komatsu K, Arwert F, Pronk JC, Mathew CG, Digweed M, Buchwald M, Joenje H. Isolation of a cDNA representing the Fanconi anemia complementation group E gene. American journal of human genetics. 2000;67:1306–1308. doi: 10.1016/s0002-9297(07)62959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Winter JP, Rooimans MA, van Der Weel L, van Berkel CG, Alon N, Bosnoyan-Collins L, de Groot J, Zhi Y, Waisfisz Q, Pronk JC, Arwert F, Mathew CG, Scheper RJ, Hoatlin ME, Buchwald M, Joenje H. The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nature genetics. 2000;24:15–16. doi: 10.1038/71626. [DOI] [PubMed] [Google Scholar]
- 36.de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CG, Bosnoyan-Collins L, Alon N, Carreau M, Bender O, Demuth I, Schindler D, Pronk JC, Arwert F, Hoehn H, Digweed M, Buchwald M, Joenje H. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nature genetics. 1998;20:281–283. doi: 10.1038/3093. [DOI] [PubMed] [Google Scholar]
- 37.Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, Schindler D, Arwert F, Pals G, Mathew CG, Waisfisz Q, de Winter JP, Joenje H. Identification of the Fanconi anemia complementation group I gene, FANCI. Cellular oncology: the official journal of the International Society for Cellular Oncology. 2007;29:211–218. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fanconi anaemia/Breast cancer, Positional cloning of the Fanconi anaemia group A gene. Nature genetics. 1996;14:324–328. doi: 10.1038/ng1196-324. [DOI] [PubMed] [Google Scholar]
- 39.Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen DF, Savoia A, Cheng NC, van Berkel CG, Strunk MH, Gille JJ, Pals G, Kruyt FA, Pronk JC, Arwert F, Buchwald M, Joenje H. Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nature genetics. 1996;14:320–323. doi: 10.1038/ng1196-320. [DOI] [PubMed] [Google Scholar]
- 40.Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nature structural & molecular biology. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 41.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, Elledge SJ. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature. 1992;356:763–767. doi: 10.1038/356763a0. [DOI] [PubMed] [Google Scholar]
- 43.Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D’Andrea AD, Moses R, Grompe M. Positional cloning of a novel Fanconi anemia gene, FANCD2. Molecular cell. 2001;7:241–248. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- 44.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W. A novel ubiquitin ligase is deficient in Fanconi anemia. Nature genetics. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 45.Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W, Joenje H. X-linked inheritance of Fanconi anemia complementation group B. Nature genetics. 2004;36:1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 46.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature genetics. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. The Journal of clinical investigation. 2012;122:3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andreassen PR, Ren K. Fanconi anemia proteins, DNA interstrand crosslink repair pathways, and cancer therapy. Current cancer drug targets. 2009;9:101–117. doi: 10.2174/156800909787314011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nature structural & molecular biology. 2011;18:500–503. doi: 10.1038/nsmb.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 52.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nature genetics. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, Kallioniemi A, Pylkas K, Karppinen SM, Rapakko K, Miron A, Sheng Q, Li G, Mattila H, Bell DW, Haber DA, Grip M, Reiman M, Jukkola-Vuorinen A, Mustonen A, Kere J, Aaltonen LA, Kosma VM, Kataja V, Soini Y, Drapkin RI, Livingston DM, Winqvist R. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 54.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N C. Breast Cancer Susceptibility. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nature genetics. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 55.Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Molecular and cellular biology. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abbott DW, Freeman ML, Holt JT. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. Journal of the National Cancer Institute. 1998;90:978–985. doi: 10.1093/jnci/90.13.978. [DOI] [PubMed] [Google Scholar]
- 57.Ghosh S, Sur S, Yerram SR, Rago C, Bhunia AK, Hossain MZ, Paun BC, Ren YR, Iacobuzio-Donahue CA, Azad NA, Kern SE. Hypersensitivities for acetaldehyde and other agents among cancer cells null for clinically relevant Fanconi anemia genes. The American journal of pathology. 2014;184:260–270. doi: 10.1016/j.ajpath.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park JY, Singh TR, Nassar N, Zhang F, Freund M, Hanenberg H, Meetei AR, Andreassen PR. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene. 2013 doi: 10.1038/onc.2013.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peng M, Litman R, Jin Z, Fong G, Cantor SB. BACH1 is a DNA repair protein supporting BRCA1 damage response. Oncogene. 2006;25:2245–2253. doi: 10.1038/sj.onc.1209257. [DOI] [PubMed] [Google Scholar]
- 60.Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, van Beers EH, Li L, Khalil T, Quenneville LA, Omeroglu A, Poll A, Lepage P, Wong N, Nederlof PM, Ashworth A, Tonin PN, Narod SA, Livingston DM, Foulkes WD. Analysis of PALB2/FANCN-associated breast cancer families. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6788–6793. doi: 10.1073/pnas.0701724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 62.Zhang F, Bick G, Park JY, Andreassen PR. MDC1 and RNF8 function in a pathway that directs BRCA1-dependent localization of PALB2 required for homologous recombination. Journal of cell science. 2012;125:6049–6057. doi: 10.1242/jcs.111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Molecular cancer research: MCR. 2009;7:1110–1118. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Current biology: CB. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayakawa T, Zhang F, Hayakawa N, Ohtani Y, Shinmyozu K, Nakayama J, Andreassen PR. MRG15 binds directly to PALB2 and stimulates homology-directed repair of chromosomal breaks. Journal of cell science. 2010;123:1124–1130. doi: 10.1242/jcs.060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sy SM, Huen MS, Zhu Y, Chen J. PALB2 regulates recombinational repair through chromatin association and oligomerization. The Journal of biological chemistry. 2009;284:18302–18310. doi: 10.1074/jbc.M109.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO reports. 2009;10:990–996. doi: 10.1038/embor.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer research. 2010;70:7353–7359. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nikkila J, Coleman KA, Morrissey D, Pylkas K, Erkko H, Messick TE, Karppinen SM, Amelina A, Winqvist R, Greenberg RA. Familial breast cancer screening reveals an alteration in the RAP80 UIM domain that impairs DNA damage response function. Oncogene. 2009;28:1843–1852. doi: 10.1038/onc.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sy SM, Huen MS, Chen J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. The Journal of biological chemistry. 2009;284:21127–21131. doi: 10.1074/jbc.C109.023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martrat G, Maxwell CM, Tominaga E, Porta-de-la-Riva M, Bonifaci N, Gomez-Baldo L, Bogliolo M, Lazaro C, Blanco I, Brunet J, Aguilar H, Fernandez-Rodriguez J, Seal S, Renwick A, Rahman N, Kuhl J, Neveling K, Schindler D, Ramirez MJ, Castella M, Hernandez G, Easton DF, Peock S, Cook M, Oliver CT, Frost D, Platte R, Evans DG, Lalloo F, Eeles R, Izatt L, Chu C, Davidson R, Ong KR, Cook J, Douglas F, Hodgson S, Brewer C, Morrison PJ, Porteous M, Peterlongo P, Manoukian S, Peissel B, Zaffaroni D, Roversi G, Barile M, Viel A, Pasini B, Ottini L, Putignano AL, Savarese A, Bernard L, Radice P, Healey S, Spurdle A, Chen X, Beesley J, Rookus MA, Verhoef S, Tilanus-Linthorst MA, Vreeswijk MP, Asperen CJ, Bodmer D, Ausems MG, van Os TA, Blok MJ, Meijers-Heijboer HE, Hogervorst FB, Goldgar DE, Buys S, John EM, Miron A, Southey M, Daly MB, Harbst K, Borg A, Rantala J, Barbany-Bustinza G, Ehrencrona H, Stenmark-Askmalm M, Kaufman B, Laitman Y, Milgrom R, Friedman E, Domchek SM, Nathanson KL, Rebbeck TR, Johannsson OT, Couch FJ, Wang X, Fredericksen Z, Cuadras D, Moreno V, Pientka FK, Depping R, Caldes T, Osorio A, Benitez J, Bueren J, Heikkinen T, Nevanlinna H, Hamann U, Torres D, Caligo MA, Godwin AK, Imyanitov EN, Janavicius R, Sinilnikova OM, Stoppa-Lyonnet D, Mazoyer S, Verny-Pierre C, Castera L, de Pauw A, Bignon YJ, Uhrhammer N, Peyrat JP, Vennin P, Ferrer SF, Collonge-Rame MA, Mortemousque I, McGuffog L, Chenevix-Trench G, Pereira-Smith OM, Antoniou AC, Ceron J, Tominaga K, Surralles J, Pujana MA. Exploring the link between MORF4L1 and risk of breast cancer. Breast cancer research: BCR. 2011;13:R40. doi: 10.1186/bcr2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KI, Ganesan S, Kong AN, Zhang DD, Xia B. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Molecular and cellular biology. 2012;32:1506–1517. doi: 10.1128/MCB.06271-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, Hayes DN, Major MB. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer research. 2013;73:2199–2210. doi: 10.1158/0008-5472.CAN-12-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends in biochemical sciences. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 76.Fruehauf JP, Meyskens FL., Jr Reactive oxygen species: a breath of life or death? Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 77.Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, MELL Oxidative stress and cancer: an overview. Ageing research reviews. 2013;12:376–390. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 78.Lupas A. Coiled coils: new structures and new functions. Trends in biochemical sciences. 1996;21:375–382. [PubMed] [Google Scholar]
- 79.Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 80.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer research. 1999;59:3547–3551. [PubMed] [Google Scholar]
- 81.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Molecular cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 82.Mermershtain I, Glover JN. Structural mechanisms underlying signaling in the cellular response to DNA double strand breaks. Mutation research. 2013;750:15–22. doi: 10.1016/j.mrfmmm.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 86.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 87.Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 90.Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nature structural & molecular biology. 2007;14:710–715. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- 91.Buisson R, Masson JY. PALB2 self-interaction controls homologous recombination. Nucleic acids research. 2012;40:10312–10323. doi: 10.1093/nar/gks807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, Yu X, Galkin VE, Liu D, Tsai MS, Sy SM, Schild D, Egelman E, Chen J, Sung P. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nature structural & molecular biology. 2010;17:1255–1259. doi: 10.1038/nsmb.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nature structural & molecular biology. 2010;17:1247–1254. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Molecular and cellular biology. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes & development. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicologic pathology. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 97.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 98.Siaud N, Barbera MA, Egashira A, Lam I, Christ N, Schlacher K, Xia B, Jasin M. Plasticity of BRCA2 function in homologous recombination: genetic interactions of the PALB2 and DNA binding domains. PLoS genetics. 2011;7:e1002409. doi: 10.1371/journal.pgen.1002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nature structural & molecular biology. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- 100.Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer research. 2007;67:9658–9665. doi: 10.1158/0008-5472.CAN-07-0290. [DOI] [PubMed] [Google Scholar]
- 101.Yata K, Lloyd J, Maslen S, Bleuyard JY, Skehel M, Smerdon SJ, Esashi F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Molecular cell. 2012;45:371–383. doi: 10.1016/j.molcel.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Antoniou AC, Sinilnikova OM, Simard J, Leone M, Dumont M, Neuhausen SL, Struewing JP, Stoppa-Lyonnet D, Barjhoux L, Hughes DJ, Coupier I, Belotti M, Lasset C, Bonadona V, Bignon YJ, Rebbeck TR, Wagner T, Lynch HT, Domchek SM, Nathanson KL, Garber JE, Weitzel J, Narod SA, Tomlinson G, Olopade OI, Godwin A, Isaacs C, Jakubowska A, Lubinski J, Gronwald J, Gorski B, Byrski T, Huzarski T, Peock S, Cook M, Baynes C, Murray A, Rogers M, Daly PA, Dorkins H, Carriers BM, Schmutzler RK, Versmold B, Engel C, Meindl A, Arnold N, Niederacher D, Deissler H, Ovarian C, Spurdle AB, Chen X, Waddell N, Cloonan N, Kirchhoff T, Offit K, Friedman E, Kaufmann B, Laitman Y, Galore G, Rennert G, Lejbkowicz F, Raskin L, Andrulis IL, Ilyushik E, Ozcelik H, Devilee P, Vreeswijk MP, Greene MH, Prindiville SA, Osorio A, Benitez J, Zikan M, Szabo CI, Kilpivaara O, Nevanlinna H, Hamann U, Durocher F, Arason A, Couch FJ, Easton DF, Chenevix-Trench G B.M.C.S. Genetic Modifiers of Cancer Risk in, B. Epidemiological Study of, B. German Consortium for Hereditary, C. Kathleen Cuningham Consortium for Research into Familial Breast, B. Consortium of Investigators of Modifiers of. RAD51 135G-->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. American journal of human genetics. 2007;81:1186–1200. doi: 10.1086/522611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bleuyard JY, Buisson R, Masson JY, Esashi F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO reports. 2012;13:135–141. doi: 10.1038/embor.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie T, Graveline R, Kumar GS, Zhang Y, Krishnan A, David G, Radhakrishnan I. Structural basis for molecular interactions involving MRG domains: implications in chromatin biology. Structure. 2012;20:151–160. doi: 10.1016/j.str.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smeenk G, van Attikum H. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annual review of biochemistry. 2013;82:55–80. doi: 10.1146/annurev-biochem-061809-174504. [DOI] [PubMed] [Google Scholar]
- 106.Gospodinov A, Herceg Z. Shaping chromatin for repair. Mutation research. 2013;752:45–60. doi: 10.1016/j.mrrev.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 107.Chen M, Tominaga K, Pereira-Smith OM. Emerging role of the MORF/MRG gene family in various biological processes, including aging. Annals of the New York Academy of Sciences. 2010;1197:134–141. doi: 10.1111/j.1749-6632.2010.05197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sapountzi V, Logan IR, Robson CN. Cellular functions of TIP60. The international journal of biochemistry & cell biology. 2006;38:1496–1509. doi: 10.1016/j.biocel.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 109.Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K, Kimura H, Ikura M, Nishikubo S, Ito T, Muto A, Miyagawa K, Takeda S, Fishel R, Igarashi K, Kamiya K. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Molecular and cellular biology. 2007;27:7028–7040. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature cell biology. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 111.Yochum GS, Ayer DE. Role for the mortality factors MORF4, MRGX, and MRG15 in transcriptional repression via associations with Pf1, mSin3A, and Transducin-Like Enhancer of Split. Molecular and cellular biology. 2002;22:7868–7876. doi: 10.1128/MCB.22.22.7868-7876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hayakawa T, Ohtani Y, Hayakawa N, Shinmyozu K, Saito M, Ishikawa F, Nakayama J. RBP2 is an MRG15 complex component and down-regulates intragenic histone H3 lysine 4 methylation. Genes to cells: devoted to molecular & cellular mechanisms. 2007;12:811–826. doi: 10.1111/j.1365-2443.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- 113.Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, Huang Q, Liu Q, Hao Q, Ding J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic acids research. 2006;34:6621–6628. doi: 10.1093/nar/gkl989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rio Frio T, Haanpaa M, Pouchet C, Pylkas K, Vuorela M, Tischkowitz M, Winqvist R, Foulkes WD. Mutation analysis of the gene encoding the PALB2-binding protein MRG15 in BRCA1/2-negative breast cancer families. Journal of human genetics. 2010;55:842–843. doi: 10.1038/jhg.2010.112. [DOI] [PubMed] [Google Scholar]
- 115.Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends in biochemical sciences. 2010;35:565–574. doi: 10.1016/j.tibs.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 116.Buisson R, Niraj J, Pauty J, Maity R, Zhao W, Coulombe Y, Sung P, Masson JY. Breast Cancer Proteins PALB2 and BRCA2 Stimulate Polymerase eta in Recombination-Associated DNA Synthesis at Blocked Replication Forks. Cell reports. 2014;6:553–564. doi: 10.1016/j.celrep.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Menzel T, Nahse-Kumpf V, Kousholt AN, Klein DK, Lund-Andersen C, Lees M, Johansen JV, Syljuasen RG, Sorensen CS. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO reports. 2011;12:705–712. doi: 10.1038/embor.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Phelan CM, Lancaster JM, Tonin P, Gumbs C, Cochran C, Carter R, Ghadirian P, Perret C, Moslehi R, Dion F, Faucher MC, Dole K, Karimi S, Foulkes W, Lounis H, Warner E, Goss P, Anderson D, Larsson C, Narod SA, Futreal PA. Mutation analysis of the BRCA2 gene in 49 site-specific breast cancer families. Nature genetics. 1996;13:120–122. doi: 10.1038/ng0596-120. [DOI] [PubMed] [Google Scholar]
- 119.Teng DH, Bogden R, Mitchell J, Baumgard M, Bell R, Berry S, Davis T, Ha PC, Kehrer R, Jammulapati S, Chen Q, Offit K, Skolnick MH, Tavtigian SV, Jhanwar S, Swedlund B, Wong AK, Kamb A. Low incidence of BRCA2 mutations in breast carcinoma and other cancers. Nature genetics. 1996;13:241–244. doi: 10.1038/ng0696-241. [DOI] [PubMed] [Google Scholar]
- 120.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P. Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer research. 2008;68:998–1002. doi: 10.1158/0008-5472.CAN-07-2418. [DOI] [PubMed] [Google Scholar]
- 122.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, King MC, Swisher EM. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hirsch B, Shimamura A, Moreau L, Baldinger S, Hag-alshiekh M, Bostrom B, Sencer S, D’Andrea AD. Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood. 2004;103:2554–2559. doi: 10.1182/blood-2003-06-1970. [DOI] [PubMed] [Google Scholar]
- 124.Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, Ben-Porat L, Mah K, Batish SD, Kutler DI, MacMillan ML, Hanenberg H, Auerbach AD. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. 2004;103:3226–3229. doi: 10.1182/blood-2003-09-3138. [DOI] [PubMed] [Google Scholar]
- 125.Makde RD, England JR, Yennawar HP, Tan S. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 2010;467:562–566. doi: 10.1038/nature09321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miller KA, Sawicka D, Barsky D, Albala JS. Domain mapping of the Rad51 paralog protein complexes. Nucleic acids research. 2004;32:169–178. doi: 10.1093/nar/gkg925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, Suram A, Jaco I, Benitez J, Herbig U, Blasco MA, Jonkers J, Tarsounas M. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nature structural & molecular biology. 2010;17:1461–1469. doi: 10.1038/nsmb.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chun J, Buechelmaier ES, Powell SN. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Molecular and cellular biology. 2013;33:387–395. doi: 10.1128/MCB.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu Y, Masson JY, Shah R, O’Regan P, West SC. RAD51C is required for Holliday junction processing in mammalian cells. Science. 2004;303:243–246. doi: 10.1126/science.1093037. [DOI] [PubMed] [Google Scholar]
- 130.Liu N, Schild D, Thelen MP, Thompson LH. Involvement of Rad51C in two distinct protein complexes of Rad51 paralogs in human cells. Nucleic acids research. 2002;30:1009–1015. doi: 10.1093/nar/30.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Masson JY, Tarsounas MC, Stasiak AZ, Stasiak A, Shah R, McIlwraith MJ, Benson FE, West SC. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes & development. 2001;15:3296–3307. doi: 10.1101/gad.947001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wiese C, Collins DW, Albala JS, Thompson LH, Kronenberg A, Schild D. Interactions involving the Rad51 paralogs Rad51C and XRCC3 in human cells. Nucleic acids research. 2002;30:1001–1008. doi: 10.1093/nar/30.4.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schild D, Lio YC, Collins DW, Tsomondo T, Chen DJ. Evidence for simultaneous protein interactions between human Rad51 paralogs. The Journal of biological chemistry. 2000;275:16443–16449. doi: 10.1074/jbc.M001473200. [DOI] [PubMed] [Google Scholar]
- 134.Somyajit K, Subramanya S, Nagaraju G. Distinct roles of FANCO/RAD51C protein in DNA damage signaling and repair: implications for Fanconi anemia and breast cancer susceptibility. The Journal of biological chemistry. 2012;287:3366–3380. doi: 10.1074/jbc.M111.311241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hellebrand H, Sutter C, Honisch E, Gross E, Wappenschmidt B, Schem C, Deissler H, Ditsch N, Gress V, Kiechle M, Bartram CR, Schmutzler RK, Niederacher D, Arnold N, Meindl A. Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Human mutation. 2011;32:E2176–2188. doi: 10.1002/humu.21478. [DOI] [PubMed] [Google Scholar]
- 136.Liu Y, Tarsounas M, O’Regan P, West SC. Role of RAD51C and XRCC3 in genetic recombination and DNA repair. The Journal of biological chemistry. 2007;282:1973–1979. doi: 10.1074/jbc.M609066200. [DOI] [PubMed] [Google Scholar]
- 137.Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N, Takeda S. Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Molecular cell. 2005;20:793–799. doi: 10.1016/j.molcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 138.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 139.Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. The EMBO journal. 1999;18:3491–3501. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 141.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bowman-Colin C, Xia B, Bunting S, Klijn C, Drost R, Bouwman P, Fineman L, Chen X, Culhane AC, Cai H, Rodig SJ, Bronson RT, Jonkers J, Nussenzweig A, Kanellopoulou C, Livingston DM. Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8632–8637. doi: 10.1073/pnas.1305362110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nikkila J, Parplys AC, Pylkas K, Bose M, Huo Y, Borgmann K, Rapakko K, Nieminen P, Xia B, Pospiech H, Winqvist R. Heterozygous mutations in PALB2 cause DNA replication and damage response defects. Nature communications. 2013;4:2578. doi: 10.1038/ncomms3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Arlt MF, Xu B, Durkin SG, Casper AM, Kastan MB, Glover TW. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Molecular and cellular biology. 2004;24:6701–6709. doi: 10.1128/MCB.24.15.6701-6709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.De Haro LP, Wray J, Williamson EA, Durant ST, Corwin L, Gentry AC, Osheroff N, Lee SH, Hromas R, Nickoloff JA. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic acids research. 2010;38:5681–5691. doi: 10.1093/nar/gkq339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 149.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 150.Min A, Im SA, Yoon YK, Song SH, Nam HJ, Hur HS, Kim HP, Lee KH, Han SW, Oh DY, Kim TY, O’Connor MJ, Kim WH, Bang YJ. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Molecular cancer therapeutics. 2013;12:865–877. doi: 10.1158/1535-7163.MCT-12-0950. [DOI] [PubMed] [Google Scholar]
- 151.Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nature reviews Drug discovery. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 152.Zagni C, Chiacchio U, Rescifina A. Histone methyltransferase inhibitors: novel epigenetic agents for cancer treatment. Current medicinal chemistry. 2013;20:167–185. doi: 10.2174/092986713804806667. [DOI] [PubMed] [Google Scholar]
- 153.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. The Journal of biological chemistry. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 154.Swisher EM, Taniguchi T, Karlan BY. Molecular scores to predict ovarian cancer outcomes: a worthy goal, but not ready for prime time. Journal of the National Cancer Institute. 2012;104:642–645. doi: 10.1093/jnci/djs203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, Levine DA, Cannistra SA. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:3555–3561. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, Agnew KJ, Pritchard CC, Scroggins S, Garcia RL, King MC, Swisher EM. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tu be, and peritoneal carcinomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:764–775. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, Friedlander M, Fox S, Bowtell D, Mitchell G. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:2654–2663. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang D, Khan S, Sun Y, Hess K, Shmulevich I, Sood AK, Zhang W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA: the journal of the American Medical Association. 2011;306:1557–1565. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Willers H, Taghian AG, Luo CM, Treszezamsky A, Sgroi DC, Powell SN. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Molecular cancer research: MCR. 2009;7:1304–1309. doi: 10.1158/1541-7786.MCR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yin Z, Menendez D, Resnick MA, French JE, Janardhan KS, Jetten AM. RAP80 is critical in maintaining genomic stability and suppressing tumor development. Cancer research. 2012;72:5080–5090. doi: 10.1158/0008-5472.CAN-12-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]