

Abstract
Reactive oxygen species and the NADPH oxidase (NOX) enzyme are both up-regulated after spinal cord injury (SCI) and play significant roles in promoting post-injury inflammation. However, the cellular and temporal expression profile of NOX isotypes, including NOX2, 3 and 4, after SCI is currently unclear. The purpose of this study was to resolve this expression profile and examine the effect of inhibition of NOX on inflammation after SCI. Briefly, adult male rats were subjected to moderate contusion SCI. Double immunofluorescence for NOX isotypes and CNS cellular types was performed at 24 hours, 7 days and 28 days post-injury. NOX isotypes were found to be expressed in neurons, astrocytes and microglia, and this expression was dependent on injury status. NOX2 and 4 were found in all cell types assessed, while NOX3 was positively identified in neurons only. NOX2 was the most responsive to injury, increasing in both microglia and astrocytes. The biggest increases in expression were observed at 7 days post-injury and increased expression was maintained through 28 days. NOX2 inhibition by systemic administration of gp91ds-tat at 15 minutes, 6 hours or 7 days after injury reduced both pro-inflammatory cytokine expression and evidence of oxidative stress in the injured spinal cord. This study therefore illustrates the regional and temporal influence on NOX isotype expression and the importance of NOX activation in SCI. This information will be useful in future studies of understanding reactive oxygen species production after injury and therapeutic potentials.
Keywords: Animal studies, cytokines, gp91ds-tat, NOX2, oxidative stress
Introduction
In the United States alone, there are over 12,000 new cases of spinal cord injury (SCI) reported each year, contributing to over 273,000 people currently living with disabilities caused by these types of injuries[1, 2]. In SCI, secondary tissue damage and cell death follows the initial mechanical insult, expanding from the primary injury site and contributing to permanent motor, autonomic, and sensory function loss. Inflammation, including activation of microglia and invasion of peripheral macrophages, and oxidative stress play central roles in this secondary injury.
Reactive oxygen species (ROS) are important signaling messengers as well as normal by products of cellular metabolism. However, an overabundance of ROS can be cytotoxic and leads to modification of lipids, proteins and DNA (for review, [3]). The NADPH oxidase (NOX) enzyme is a major source of ROS in injury and disease conditions [4, 5]. The NOX family is comprised of seven known isotypes: NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1 and DUOX2. The NOX enzymes are transmembrane electron carriers that facilitate the transfer of electrons from NADPH to molecular oxygen to yield superoxide (O2• -).
Cellular expression of NOX in the central nervous system (CNS) appears to be isotype dependent[6]. In particular, the NOX2, 3 and 4 isotypes have been identified in CNS cells, particularly after stimulation or activation. For example, Savchenko et al. demonstrated that expression of NOX1 and NOX2 are altered in microglia and neurons after stimulation in vitro [7]. Studies have also shown that NOX2 is constitutively expressed in microglia and up-regulated upon activation in cases of multiple sclerosis [8], ischemia [9] and traumatic brain injury (TBI)[6,10]. We have demonstrated that NOX2 components are up-regulated [11-13] and oxidative stress has been noted to be elevated by 3 hours and persists for weeks after SCI [14]. Additionally, NOX2 derived ROS has been implicated in neuropathic pain following peripheral nerve injury[15]. In addition, we have identified NOX3 and 4 in neurons, astrocytes and microglia in brain before and after traumatic injury [6].
Activation of NOX2 is contingent upon the association of its subunits, and several studies have found that blocking assembly NOX2 can reduce inflammation and improve recovery after injury. For example, apocynin prevents the migration of p47PHOX to the membrane and has been shown to reduce superoxide anion generation[16], increase neuronal survival [17], reduce inflammation [18] and improve sensorimotor recovery after a brain injury [19]. Further, inhibition of NOX activity with dephenyleneiodonium (DPI), a flavoprotein inhibitor that prevents electron flow, reduces lesion volume after SCI in adult rats [12]. However, while both of these inhibitors are effective in inhibiting ROS production by NOX, neither is specific for the NOX2 isotype and have been shown to have activities on other, non-NOX enzymes. Gp91ds-tat, on the other hand, specifically manipulates NOX2 activation. This 9 amino acid chimeric peptide attaches to the p47 PHOX binding site and inhibits its association with gp91PHOX[20]. Previous studies have shown that gp91ds-tat reduces neuronal damage and edema following TBI[18]
To date, the temporal and cellular expression of NOX isotypes in the spinal cord after injury has not been clarified. Therefore, the aim of this study was to identify the temporal profile and cellular localization of the NOX2, 3 and 4 in the normal and injured spinal cord. Additionally, this study aimed to examine the functional impact of NOX2 utilizing the NOX2 specific inhibitor gp91ds-tat.
Methods
Animal Handling and Surgical Methods
Adult male Sprague Dawley rats (275 – 325g) were used for all experiments. Rats were dual housed and received food and water ad libitum with a 12:12 hour light cycle. All experiments complied fully with the principles set forth in the “Guide for the Care and Use of Laboratory Animals” prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council (DHEW pub. No. (NIH) 85-23, 2985) and were approved by the Uniformed Services University IACUC.
Moderate contusion SCI was performed in rats that were anesthetized with ketamine/xylazine (0.1ml/100g, I.P.; Characterization study) or isoflurane (4% induction, 2% maintenance; NOX inhibition study). A moderate injury was induced using an Infinite Horizons Impactor (160.7+/-10.4kdynes; Precision Systems and Instrumentation, Fairfax Station, VA) positioned over the exposed spinal cord at vertebral level T-9. Sham injured rats underwent the same experimental procedures, but received a laminectomy only. Animals were allowed to recover on heating pads and received acetaminophen (200mg/kg) in drinking water for 72 hours post-injury. Manual bladder expression was performed twice per day until normal bladder expression returned.
Characterization of NOX
Immunohistochemistry
At 24 hours (n = 4 injured, 2 sham), 7 days (n = 4 injured, 2 sham), and 28 days (n = 5 injured, 1 sham, 1 naïve), rats were anesthetized (Euthasol, 0.22ml/kg, I.P.) and intracardially perfused with 100 ml of 0.9% saline followed by 300 ml of 10% buffered formalin. A 10mm section of spinal cord centered at the lesion epicenter, T-9, was then dissected, post-fixed in 10% buffered formalin overnight and cryoprotected in 30% sucrose for at least 48 hours.
Standard single or double fluorescent or DAB-based immunohistochemistry on serial 20 μm thick coronal sections was performed as described previously [11]. Antibodies included NOX2/gp91PHOX (1μg/ml, Abcam, Cambridge, MA), NOX3 (5μl/ml, Santa Cruz, Santa Cruz, CA), NOX4 (1μg/ml, Abcam, Cambridge, MA), Iba-1 (Ionized calcium-binding adapter molecule 1, micrgolia/macrophage specific marker; 1μg/ml, Wako, Richmond, VA), NeuN (Neuronal nuclei, neuronal specific marker; 1:100, Millipore, Billerica, MA), GFAP (Glial fibrillary acidic protein, astrocyte specific marker; 1:1000, Abcam, Cambridge, MA), and CD11b (cluster of differentiation molecule 11B, microglia/macrophage specific marker; 2μg/ml, Serotec, Oxford, UK). It should be noted that a number of NOX specific antibodies from a number of sources were tested. NOX antibodies have a history of lack of specificity and fidelity. Each antibody was tested in conjunction with cell specific antibodies and with western blotting to confirm that each resulted in appropriate, expected staining and expected band sizes in western blots (data not shown), as described in our previous work[6].
Appropriate secondary antibodies linked to AlexaFluor dyes (Invitrogen, Carlsbad, CA) were incubated with tissue sections for 1 hour at room temperature. Slides were coverslipped using mounting media containing DAPI to counterstain for nuclei (Vector Labs, Burlingame, CA). For NOX3 immunohistochemistry, the Vector ABC kit with DAB and/or NovaRed reaction products were utilized due to poor resolution with fluorescence secondaries (Vector Labs, Burlingame, CA).
To ensure accurate and specific staining, negative controls were used in which the primary antibody was not applied to sections from injured tissue, and only staining of cells that were double-labeled with expected cell markers or had expected labeling patterns (i.e., classic microglia morphology) was confirmed as positive labeling. Immunofluorescence was imaged using an Olympus DP72 microscope with Olympus cell Sens microscopy software (Olympus, Center Valley, PA) and the Hamamatsu NanoZoomer-RS Digital Slide Scanner (Hamamatsu Photonics, Bridgewater, NJ).
NOX Inhibition
Treatment
Rats received either gp91ds-tat or scrambled peptide (solubilized in distilled, pathogen free water; Anaspec, Fremont, CA) at 1mg/kgvia I.P. injection at 15 minutes, 6 hours or 7 days post-injury (n = 4 per treatment per time point). Naïve rats (n = 2) were used to assess non-injured responses in outcome measures and underwent no surgery or treatment.
At 24 hours post treatment, animals were anesthetized (Euthasol, 0.22ml/kg, I.P.) and intracardially perfused with 100mL of 0.9% saline. A 10mm section of spinal cord centered at the lesion epicenter, T-9, was dissected, weighed and homogenized in 20 vol T-PER (Thermo Scientific, Waltham, MA) containing 0.1% Halt Protease Inhibitor Cocktail (Thermo Scientific, Waltham, MA). The tissue was then centrifuged at 15,000 rpm for 10 minutes, after which the pellet was discarded. The protein content of the supernatant was determined using Coomassie Plus Protein Assay Reagent (Thermo Scientific, Waltham, MA).
Cytokine Expression and Oxidative Stress Detection
A Rat Proteome Profiler Cytokine Antibody Array kit (R&D Systems, Minneapolis, MN) was used to determine the relative expression of 29 cytokines and chemokines. 250-500μL of protein lysates were analyzed per sample, and expression values were normalized to controls in the array as per the manufacturer’s instructions.
Carbonylation of proteins, as a measure of oxidative stress, was determined using the Oxyblot kit (Millipore, Billerica, MA). 15μg of protein isolated at 24 hours post-treatment as described above was used for the Oxyblot kit using the provided reagents as per the manufacturer’s instructions. The samples were loaded into precast polyacrylamide gels, subjected to gel electrophoresis and transferred to nitrocellulose membranes. Carbonylation, which occurs when carbonyl groups are introduced into proteins during periods of oxidative stress, was visualized with a Fujifilm LAS 3000 (Fujifilm, Stamford, CT), and subsequently analyzed using Image J (NIH, Bethesda, MD). Band densities were normalized to β-actin (Abcam, Cambridge, MA).
Western blot
Twenty-five μg of protein from samples obtained 24 hours after a 6 hour post-injury treatment were run in an SDS polyacrylamide gel electrophoresis and blotted onto a nitrocellulose membrane. The blot was then probed with antibodies against CD11b (2:1000, single band at 50kD), NeuN (2:1000, double band at 47 and 66kD) and GFAP (2:10,000, single band at 70kD). Immune complexes were detected with appropriate secondary antibodies and chemiluminescence reagents (Pierce, Rockford, IL). COX-IV (Mitochondria complex IV, 16kD, 1:1000, Abcam) was used as a control for gel loading and protein transfer. NIH Image J was used to assess pixel density of resultant blots for quantitation.
Statistics
Quantitative data are presented as mean +/- standard error of the mean. All assays were performed by an investigator blinded to treatment group. Proteome Profiler data are presented as pg/ml and were analyzed using repeated t-tests within each group. Oxyblot data were analyzed using one-way ANOVA with Tukey post-test. All statistical tests were performed using the GraphPad Prism Program, Version 5.0 for Windows (GraphPad Software, San Diego, CA). A p value < 0.05 was considered statistically significant.
Results
NOX isotypes were evaluated in tissue from sham or naïve rats or from rats that had received a SCI 24 hours, 7 days or 28 days previously. H&E tissue staining showed an evolution of the lesion over time (Fig. 1). Tissue showed an initial swelling, followed by slight cavitation and tissue disorganization, then larger cavitation at 28 days post-injury. All measurement of NOX isotypes were made in the dorsal column white matter or dorsal horn gray matter of the lesion epicenter and perilesional region (+/- 1 – 3 mm rostral/caudal from epicenter) (Fig. 1). Because no marked differences were observed in sham tissue over time or in comparison to naïve tissue, all sham and naïve tissue has been combined into one group to reduce animal number requirements, and is referred to as ‘naïve’ in figures.
Figure 1.
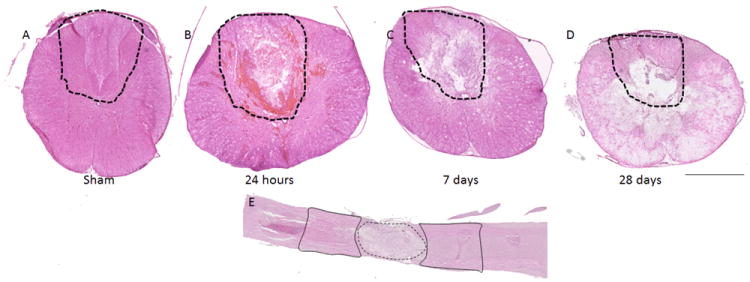
Evolution of the spinal cord injury. H&E stained spinal cord tissue prior to injury (A) and at 24 hours (B), 7 days (C) and 28 days (D) after a moderate contusion injury in the adult male Sprague Dawley rat. Areas assessed are outlined in the dashed line. Panel E demonstrates a longitudinal section of injured spinal cord to indicate ‘lesion’ (dotted line) and ‘perilesional’ (solid line) regions. Bar = 1mm (A – D); 2mm (E).
Microglia/Macrophages show NOX2 and NOX4 expression
Iba1+ cells, representing both microglia and macrophages, were positive for NOX2 only in injured spinal cord (Figure 2), not in naïve tissue. Colabeling of NOX2 and Iba1 increased at 7 days post-injury, but was reduced by 28 days post-injury. Because available NOX4 antibodies were produced in the same species as Iba1, an alternative microglia/macrophage marker, CD11b, was utilized for double-staining with NOX4. CD11b+/NOX4+ cells were faintly seen in uninjured spinal cord and increased by 24 hours, but were no longer observed by 7 or 28 days post injury (Figure 3). NOX3+ was not found to be co-expressed on Iba1+ cells (data not shown).
Figure 2.
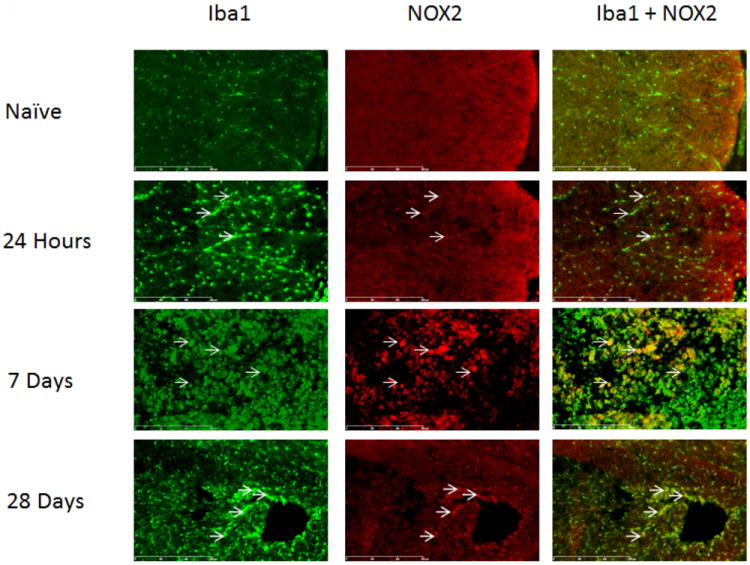
Microglia/macrophages expressed NOX2 in the spinal cord at the lesion epicenter, within regions formerly occupied by white matter. Cells labeled with the microglial marker Iba1 (green) were positive for NOX2 (red, arrows) in injured spinal cord tissue. NOX2 staining increased by 7 days-post injury, but decreased by 28 days. Bar=300μm.
Figure 3.
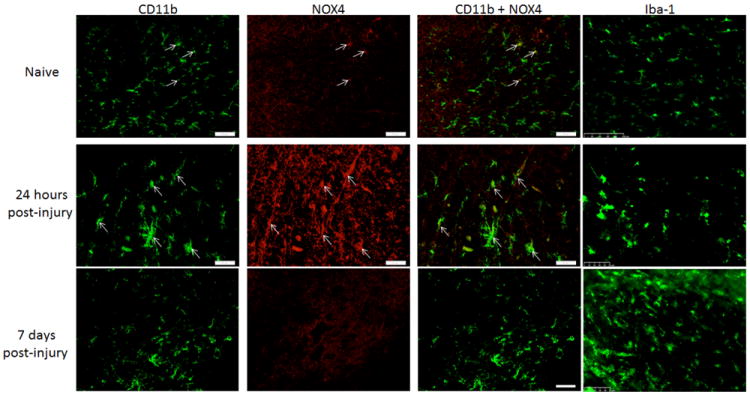
Microglia/macrophages expressed NOX4 in the spinal cord at the lesionepicenter, within regions formerly occupied by white matter. Both naïve and injured animals had CD11b positive (green) and NOX4 positive (red) cells (arrows). When merged, colabeled cells were yellow. To demonstrate consistent microglia/macrophage labeling with CD11b, adjacent sections were immunolabeled with Iba-1 and imaged at the same magnification, demonstrating similar labeling patterns. Bar= 50μm.
Neurons show NOX2 and NOX3 expression
NeuN+ cells were also NOX2+ and NOX3+in both injured and uninjured spinal cord sections (Figure 4), but not NOX4+ (data not shown). Injury led to a loss of NeuN+ cells at the lesion epicenter24 hours and 7 days post-injury, so that double-labeling for NOX2 was not observed. However, in regions caudal to the lesion epicenter, NeuN+/NOX3+ cells were observed at all time points post-injury (Figure 4).
Figure 4.
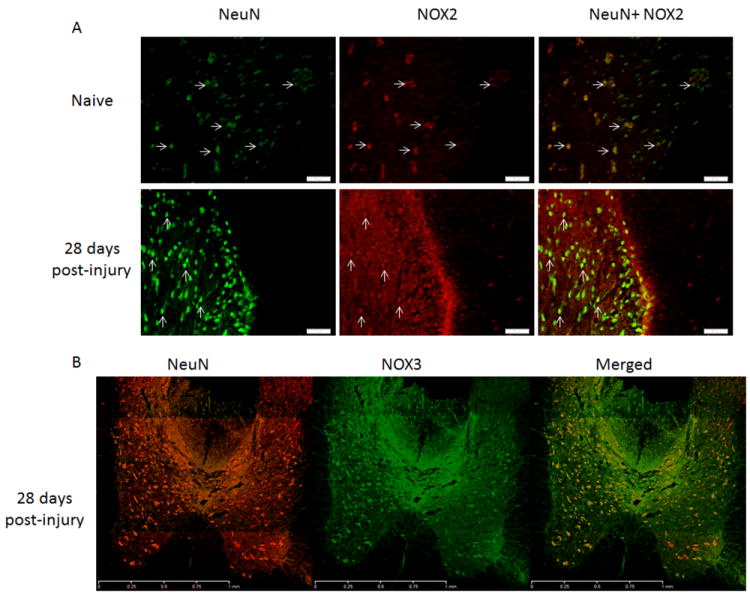
Neurons in the spinal cord expressed NOX2 and NOX3 in the dorsal horns 3mm caudalto the injury site. NOX2 (red) positive cells were also NeuN+ (green, arrows) in naïve and 28 day injured spinal cord tissue (A; bar = 50μm). NeuN positive cells (red) were found to express NOX3 (green) at all time points post-injury, with no obvious change in expression profile. Image B shows the gray matter 3mm caudal to the injury site at 28 days post-injury; bar = 1mm).
Astrocytes show NOX2 and NOX4 expression
GFAP positive cells in spinal cord sections were NOX3- (data not shown), but NOX2+ and NOX4+. The relative density of GFAP+/NOX2+ cells was elevated at 24 hours and 7 days post-injury in comparison to uninjured tissue, and decreased by 28 days (Figure 5). GFAP+/NOX4+ remained consistent over time (Figure 6).
Figure 5.
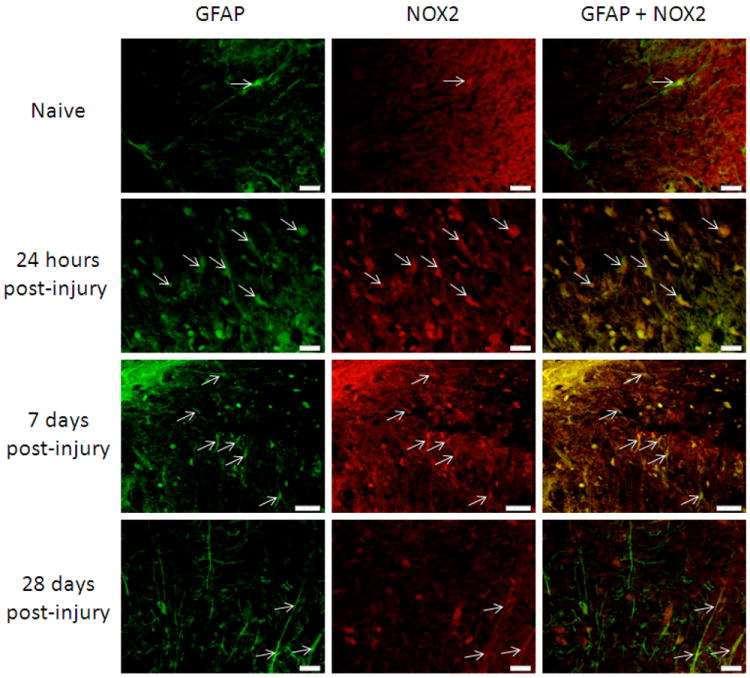
Astrocytes in the spinal cord were NOX2 positive in the spinal cord 3mm rostral to the lesion site, in white matter regions. GFAP (green) positive cells were also stained with an antibody against NOX2 (red; arrows) in naïve tissue and at 24 hours, 7 days and 28 days post-injury. Qualitatively, there appeared to be an increase in GFAP+/NOX2+ cells by 24 hours and 7 days post-injury that decreased by 28 days post-injury. Bar=20μm.
Figure 6.
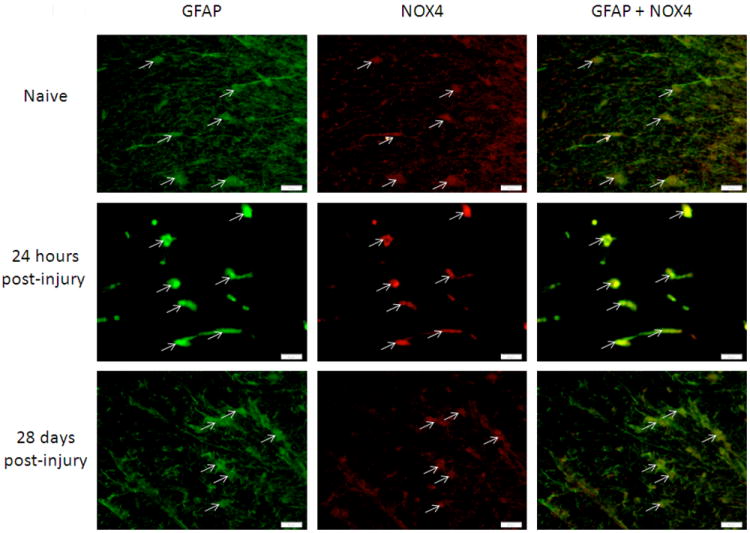
Astrocytes in the spinal cord were NOX4 positive in the spinal cord 3mm rostral to the lesion site, in white matter regions. GFAP+ (green) and NOX4+ (red; arrows) cell presence was consistent in injured and uninjured spinal cord tissue. Bar=20μm.
Inhibition of NOX2 after Spinal Cord Injury Reduces Inflammation and Oxidative Stress
NOX2 inhibition after SCI was found to have no significant effect on the neuronal or astrocytic population in the injured tissue at 24 hours after injury (Fig. 7). However, treatment with gp91ds-tat at 6 hours resulted in a significant reduction in the microglia/macrophage marker CD11b at 24 hours post-injury (Fig. 7), suggesting an early anti-inflammatory effect of the treatment.
Figure 7.
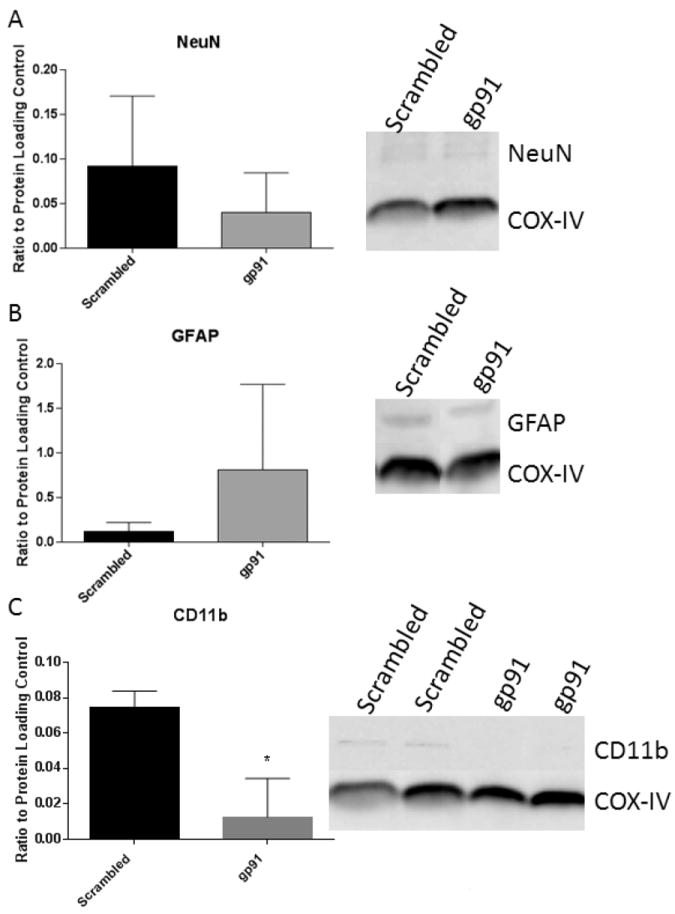
NOX2 inhibition alters acute microglial response without significantly altering other cellular presence. Western blot analysis of neuronal (A), astrocytic (B) and microglial (C) markers at 24 hours after gp91ds-tat treatment at 6 hours post-injury shows no significant effect of treatment on neuronal or astrocyte presence, but a significant inhibition of microglial markers. Bars represent mean +/- SEM. Blots are representative of an n of 4/group.
In support of this data, inhibition of NOX2 using gp91ds-tat resulted in significant suppression of several pro-inflammatory cytokines. Analysis of the Proteome Profiler indicated reduced cytokine expression when gp91ds-tat was administered at 15 minutes post-injury and assessed at 24 hours post-injury, with significant reduction in 13 of the 29 cytokines and chemokines represented (Figure 8). Gp91ds-tat administration at 6 hours and 7 days post-injury did not result in significant differences in cytokine expression (data not shown).
Figure 8.
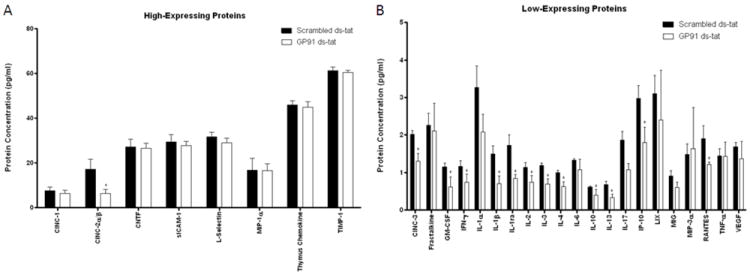
Quantitative analysis of relative cytokine expression in rats treated with 1 mg/kg gp91ds-tat or scrambled peptide 15 minutes following moderate contusion SCI. Two groups of cytokines were found: (A) high-expressing proteins, with more than 15pg/ml concentrations, and (B) low-expressing proteins, with expression of less than 5pg/ml. Significant reductions in several cytokines were observed, particularly in the low expression group (B); overall 13 cytokines were found reduced with gp91ds-tat treatment. N = 4/group, *p < 0.05.
Assessment of oxidative stress using the Oxyblot protein assay demonstrated that SCI induced a marked increase in carbonylation of protein in comparison to naïve tissue. Gp91ds-tat treatment at 15 minutes and 7 days post-injury induced a trend toward reduction this response, measured 24 hours after treatment. However, treatment at 6 hours resulted in a significant reduction in carbonylation of proteins at 24 hours after treatment (Figure 9).
Figure 9.
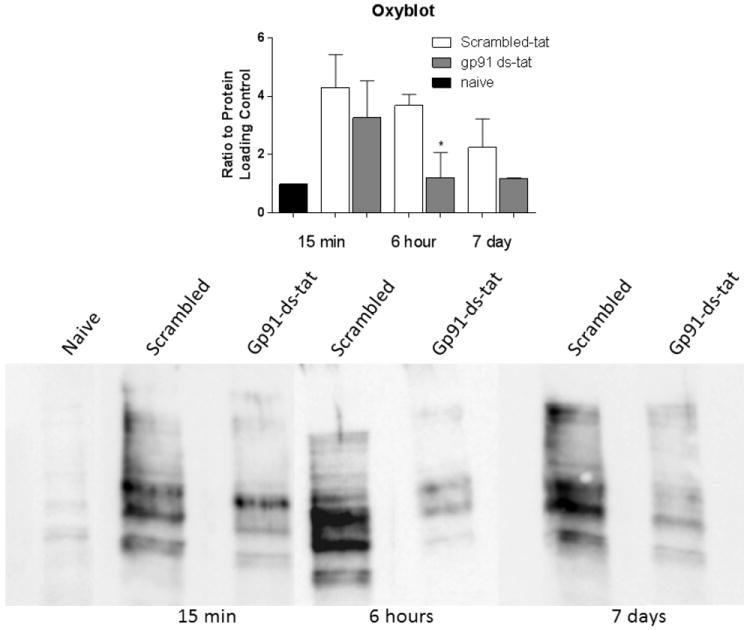
Quantitative analysis and representative blot of carbonylation modification via oxyblot of rats treated with gp91ds-tat or scrambled peptide at 15 minutes, 6 hours, and 7 days following a moderate contusion SCI. Injury demonstrated a marked increase in oxidative damage in comparison to naïve tissue. Gp91ds-tat induced a trend for decreased oxidative modification in all three groups. A significant (*) reduction can be observed in the 6 hours group. N = 4/treatment/group. *p<0.05.
Discussion
We now demonstrate that NOX isotypes are expressed in a variety of cell types in the spinal cord and this expression is dependent on injury status. Of the NOX isotypes studied, all were identified in spinal cord before and after injury. While NOX2 was found in all cell types investigated, NOX4 was expressed in both astrocytes and microglia, but not in neurons, while NOX3 was positively identified only in neurons. NOX3 may also have been present in microglia, but staining confirmation was complicated. NOX2 was the most responsive to injury, increasing post-injury in microglia and astrocytes.
Previous work has demonstrated an increase in NOX expression after spinal cord injury [11, 13, 21], with an increase through 6 months after injury in rodents [12]. ROS production has been reported from 6 hours to several months post-injury in humans [21, 22]. In the current study we observed the biggest increases in expression of NOX2 at 7 days post-injury, and up-regulation was maintained through 28 days post-injury.
In this study, the qualitative degree of NOX2+/Iba1+ cells was increased in the spinal cord after injury, with a peak in expression at 7 days post-injury and continued expression at 28 days post-injury, which is in agreement with previous studies [10-12]. Interestingly, NOX2 was not observed in resting microglia in the spinal cord, unlike in brain, where NOX2 was observed in naïve and injured tissue[6]. This may reflect a regional difference in NOX expression in microglial cells. It is also important to note that Iba1does not distinguish between microglia and macrophages, so observed changes in NOX expression may be a result of either endogenous microglia or invading macrophages.
Like microglia, neurons also express NOX2. Because of the loss or damage of neurons in the lesion epicenter at 24 hours and 7 days after injury, double labeling of NeuN and NOX2 was not observed, but NeuN+/NOX2+ cells were observed at 28 days post-injury. NOX3, on the other hand, was found to be expressed in neurons constitutively, regardless of injury state. In spinal cord tissue, NOX2 has been previously reported in neurons, and this expression has been noted to increase with LPS stimulation [23]. Functionally, neuronal NOX2 has been shown to contribute to both intra- and extracellular release of ROS following NMDA stimulation [7, 24], while NOX3 plays a role in induction of apoptosis of neurons in the inner ear in response to stress[25].
Astrocytic NOX expression was restricted to the NOX2 and 4 isotypes. Functionally, NOX2 has been shown to play a role in potentiating inflammation and enhancing the release of matrix metalloproteins[26], while the function of NOX4 in astrocytes is unclear[27]. After spinal cord injury, NOX4 was observed at all time points and in naïve tissue.
Inhibition of NOX activity, through non-specific approaches using apocynin or specific inhibitors of NOX2, has been shown to reduce neuronal death after SCI [23, 28], TBI [17, 18], and cortical ischemia [29, 30]. Impellizzeri et al. [28] also demonstrated a marked reduction in pro-inflammatory cytokines and neutrophil invasion and a significant improvement in motor function after SCI in mice following acute administration of the non-specific NOX inhibitor apocynin. Inhibition of NOX activation in microglia, via administration of apocynin or knockout of the p47PHOX subunit, not only reduces inflammatory responses, but polarizes microglia into the M2 or alternative activation phenotype [31], which may have marked therapeutic implications. In addition, specifically targeting NOX2 via knockout also reduces neuronal death in a rodent TBI model [10]. Further, 10 mg/kg pretreatment with the NOX2 specific inhibitory peptide gp91ds-tat significantly diminished neuronal damage and edema following TBI[18]. In agreement with these studies, we now show that a 1 mg/kg dose of gp91ds-tat can greatly reduce pro-inflammatory cytokine expression as well as oxidative modification following SCI. It is currently unclear if the other NOX isoforms play a significant role in post-injury inflammation and cellular damage, but our results and other suggest that targeting NOX2 specifically can reduce these effects. Future work will explore the contribution of the other isoforms in SCI secondary damage.
NOX activation in microglia/macrophages can exacerbate the post-injury inflammatory response via activity on transcription factors that ultimately increase pro-inflammatory cytokine expression [32, 33]. These pro-inflammatory cytokines are short lived: up-regulated within the first few hours post injury and returning to baseline within 1 day[34, 35]. As such, while our study only shows a significant decrease in pro-inflammatory cytokines with treatment 15 minutes post-injury, it is possible that cytokine levels have already dropped considerably when assessed after a 7 day treatment. Though the expression tapers off very quickly, the acute increase in pro-inflammatory cytokines is responsible for a spectrum of cytotoxic activities as well as the recruitment, proliferation and activation of microglia to the injury site[36]. This is supported by our data demonstrating that 6 hour post-injury NOX2 inhibition results in marked reduction in microglial/macrophage marker expression in the injury site at 24 hours (Fig. 7). Further, one study found that following spinal nerve transection in mice, CD11b mRNA expression was up-regulated 9-fold in the wildtype, versus a mere 2-fold in NOX2-deficient mice[15]. This influx of microglia and their activation at the injury site can then lead to further ROS damage and inflammation. It is important to note that gp91ds-tat treatment at 1mg/kg 15 minutes post-injury significantly reduced both M1 and M2 related cytokines, including IL1β, IL4 and IL10. This was a somewhat unexpected finding, and suggests a global suppression in microglial number rather than a specific inhibition of one particular phenotype of cell. Administration of gp91ds-tat in TBI studies has been shown to reduce immunolabeling for microglial cells, but phenotypic orientation or priming was not evaluated [18]. However, future research is necessary to investigate this effect in more detail.
In addition to its effects on cytokine expression, the gp91ds-tat treatment also resulted in decreased oxidative stress at all three time points, as measured by carbonylation of proteins. The data show a trend for reduction at 15 minutes and 7 days, and a significant difference within the 6 hour group. Previous literature has found an increase in oxidative activity 12-48 hours and 1 week post SCI, with a peak at 24 hours[21]. Based on our immunostaining work, it is clear that the largest number of activated microglia, as well as the highest expression of NOX2, occurs at 7 days post injury. Though treatment at 7 days post-injury decreased ROS production and protein carbonylation slightly, this time point may have been too late to prevent much of the oxidative modification. Moreover, due to the high level of NOX2 activation, the dosage may not have been adequate to sufficiently suppress ROS production. Future work will further pursue this avenue.
It is important to note that NOX2 is not the only source of ROS in the spinal cord, and our work now shows that NOX3 and 4 may play a role in oxidative stress following injury. However, inhibition of just NOX2 at 6 hours post injury did significantly reduce ROS production, suggesting that NOX2 activity is accountable for a notable portion of the oxidative damage.
Conclusion
In summary, we now show that NOX isotypes have a temporal and cellular dependence that has not been observed previously. These differences in expression suggest different mechanisms of activation of the isotypes in each cell. In addition, we report that inhibition of NOX2 through gp91ds-tat treatment following SCI reduces free radical production as well as pro-inflammatory cytokine expression, which may have important therapeutic implications.
Acknowledgments
The authors would like to thank Ms. GuzalKhayrullin, Ms. Sara Bermudez and Ms. Ramona von Leden for technical and editorial assistance.
This work was funded by the Uniformed Services University Intramural Program and the NINDS/NIH (Grant number 1R01NS073667-01A1).
Footnotes
Declaration of Interest Statement
No competing financial interests exist.
References
- 1.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention; 2010. [Google Scholar]
- 2.Center TNSS. Spinal cord injury: Facts and Figures. 2009.
- 3.Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40:959–75. doi: 10.1016/s0028-3908(01)00019-3. [DOI] [PubMed] [Google Scholar]
- 4.Quinn MT, Ammons MC, Deleo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci (Lond) 2006;111:1–20. doi: 10.1042/CS20060059. [DOI] [PubMed] [Google Scholar]
- 5.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 6.Cooney SJ, Bermudez-Sabogal SL, Byrnes KR. Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J Neuroinflammation. 2013;10:155. doi: 10.1186/1742-2094-10-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savchenko VL. Regulation of NADPH oxidase gene expression with PKA and cytokine IL-4 in neurons and microglia. Neurotox Res. 2013;23:201–13. doi: 10.1007/s12640-012-9327-6. [DOI] [PubMed] [Google Scholar]
- 8.Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012;135:886–99. doi: 10.1093/brain/aws012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCann SK, Dusting GJ, Roulston CL. Early increase of Nox4 NADPH oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J Neurosci Res. 2008;86:2524–34. doi: 10.1002/jnr.21700. [DOI] [PubMed] [Google Scholar]
- 10.Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation. 2010;7:41. doi: 10.1186/1742-2094-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrnes KR, Garay J, Di Giovanni S, De Biase A, Knoblach SM, Hoffman EP, Movsesyan V, Faden AI. Expression of two temporally distinct microglia-related gene clusters after spinal cord injury. Glia. 2006;53:420–33. doi: 10.1002/glia.20295. [DOI] [PubMed] [Google Scholar]
- 12.Byrnes KR, Washington PM, Knoblach SM, Hoffman E, Faden AI. Delayed inflammatory mRNA and protein expression after spinal cord injury. J Neuroinflammation. 2011;8:130. doi: 10.1186/1742-2094-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pajoohesh-Ganji A, Knoblach SM, Faden AI, Byrnes KR. Characterization of inflammatory gene expression and galectin-3 function after spinal cord injury in mice. Brain Res. 2012;1475:96–105. doi: 10.1016/j.brainres.2012.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrico KM, Vaishnav R, Hall ED. Temporal and spatial dynamics of peroxynitrite-induced oxidative damage after spinal cord contusion injury. J Neurotrauma. 2009;26:1369–78. doi: 10.1089/neu.2008-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci U S A. 2010;107:14851–6. doi: 10.1073/pnas.1009926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Touyz RM. Apocynin, NADPH oxidase, and vascular cells: a complex matter. Hypertension. 2008;51:172–4. doi: 10.1161/HYPERTENSIONAHA.107.103200. [DOI] [PubMed] [Google Scholar]
- 17.Choi BY, Jang BG, Kim JH, Lee BE, Sohn M, Song HK, Suh SW. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012;1481:49–58. doi: 10.1016/j.brainres.2012.08.032. [DOI] [PubMed] [Google Scholar]
- 18.Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 2012;7:e34504. doi: 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo W, Bravo T, Jadhav V, Titova E, Zhang JH, Tang J. NADPH oxidase inhibition improves neurological outcomes in surgically-induced brain injury. Neurosci Lett. 2007;414:228–32. doi: 10.1016/j.neulet.2006.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–14. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 21.Bao F, Bailey CS, Gurr KR, Bailey SI, Rosas-Arellano MP, Dekaban GA, Weaver LC. Increased oxidative activity in human blood neutrophils and monocytes after spinal cord injury. Exp Neurol. 2009;215:308–316. doi: 10.1016/j.expneurol.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 22.Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–69. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- 23.Li B, Guo YS, Sun MM, Dong H, Wu SY, Wu DX, Li CY. The NADPH oxidase is involved in lipopolysaccharide-mediated motor neuron injury. Brain Res. 2008;1226:199–208. doi: 10.1016/j.brainres.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 24.Reyes RC, Brennan AM, Shen Y, Baldwin Y, Swanson RA. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J Neurosci. 2012;32:12973–8. doi: 10.1523/JNEUROSCI.1597-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukherjea D, Jajoo S, Kaur T, Sheehan KE, Ramkumar V, Rybak LP. Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid Redox Signal. 2010;13:589–98. doi: 10.1089/ars.2010.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin CC, Hsieh HL, Shih RH, Chi PL, Cheng SE, Chen JC, Yang CM. NADPH oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Commun Signal. 2012;10:35. doi: 10.1186/1478-811X-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khanna A, Guo M, Mehra M, Royal W., 3rd Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J Neuroimmunol. 2013;254:69–75. doi: 10.1016/j.jneuroim.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Impellizzeri D, Mazzon E, Esposito E, Paterniti I, Bramanti P, Cuzzocrea S. Effect of Apocynin, an inhibitor of NADPH oxidase, in the inflammatory process induced by an experimental model of spinal cord injury. Free Radic Res. 2010 doi: 10.3109/10715762.2010.526604. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–9. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 30.Tang XN, Cairns B, Cairns N, Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience. 2008;154:556–562. doi: 10.1016/j.neuroscience.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju KD, Lim JW, Kim KH, Kim H. Potential role of NADPH oxidase-mediated activation of Jak2/Stat3 and mitogen-activated protein kinases and expression of TGF-beta1 in the pathophysiology of acute pancreatitis. Inflamm Res. 2011;60:791–800. doi: 10.1007/s00011-011-0335-4. [DOI] [PubMed] [Google Scholar]
- 33.Simonyi A, He Y, Sheng W, Sun AY, Wood WG, Weisman GA, Sun GY. Targeting NADPH oxidase and phospholipases A2 in Alzheimer’s disease. Mol Neurobiol. 2010;41:73–86. doi: 10.1007/s12035-010-8107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Jones NR, Blumbergs PC, Van Den Heuvel C, Moore EJ, Manavis J, Sarvestani GT, Ghabriel MN. Severity-dependent expression of pro-inflammatory cytokines in traumatic spinal cord injury in the rat. J Clin Neurosci. 2005;12:276–84. doi: 10.1016/j.jocn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 35.Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur J Neurosci. 1997;9:1422–38. doi: 10.1111/j.1460-9568.1997.tb01497.x. [DOI] [PubMed] [Google Scholar]
- 36.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–8. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 37.Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, Loison F, Sarraj B, Kasorn A, Jo H, Blanchard C, Zirkle D, McDonald D, Pai SY, Serhan CN, Luo HR. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci U S A. 2010;107:3546–51. doi: 10.1073/pnas.0914351107. [DOI] [PMC free article] [PubMed] [Google Scholar]