

Abstract
The effect of force on the thermodynamics and kinetics of reactions is described. The key parameters are the difference in end-to-end distance between reactant and product for thermodynamics, and the distance to the transition state for kinetics. I focus the review on experimental results on force unfolding of RNA. Methods to measure Gibbs free energies and kinetics for reversible and irreversible reactions are described. The use of the worm-like-chain model to calculate the effects of force on thermodynamics and kinetics is illustrated with simple models. The main purpose of the review is to describe the simple experiments that have been done so far, and to encourage more people to enter a field that is new and full of opportunities.
Keywords: single molecules, RNA thermodynamics, RNA kinetics, laser tweezers, biological macromolecules
INTRODUCTION
Chemical and biochemical reactions are often studied by varying temperature, solvent, or pressure. Photochemical reactions depend on light, and some more specialized reactions are affected by electric or magnetic fields. These variables can change the equilibria among reactants and products, and they can change the rates of the reactions. Mechanical force can also affect the thermodynamics and kinetics of reactions, but force was not a practical experimental variable until it became possible to manipulate and detect single molecules. Force could now be applied to one or a few reactants, leaving the rest of the system unperturbed. Thus force is not only a novel way to affect reactions, it is also a type of variable different from the usual temperature, pressure, and solvent variables. Force directly perturbs only the reactant, or the part of the reactant chosen; it is a local perturbation, unlike the global perturbations traditionally used.
Mechanical force has been applied to individual molecules with atomic force microscopes, laser tweezers, and magnetic tweezers (9, 25, 49). The reactions studied have been noncovalent interactions, including breaking biotin-avidin (18) and antigen-antibody (23) bonds, initiating chair-boat transitions in polysaccharides (32), unfolding proteins (11, 27) and RNA (30, 35), unzipping DNA (4), analyzing DNA-polymerase interactions (19) and DNA-topoisomerase interactions (48), and many others. Force affects both the thermodynamics and kinetics of reactions. In this review I concentrate on the magnitudes of the forces required to unfold an RNA and on the rates of unfolding at these forces. The key parameters that characterize the effects are (a) the change in length (extension) between reactants and products for thermodynamics; and (b) the change in extension between reactants and transition state for kinetics. Clearly, the same methods apply to the unfolding of proteins and of any other polymers.
It is important to understand how macromolecules unfold and refold in biological cells. In a cell, RNA folds rapidly as it is synthesized, but it unfolds (and refolds) repeatedly during its life cycle. A random sequence RNA is 50% base paired; thus, every messenger RNA must be unfolded to single strand during its translation to protein. The ribosome uses chemical energy to pull the messenger RNA into its decoding site to read each codon. Viral RNAs that are copied by RNA-dependent RNA polymerase or reverse transcriptase are unfolded as nucleotides are added to the newly synthesized complementary strand. RNA helicases are involved in unfolding RNA during assembly of ribosomes and spliceosomes. The forces found experimentally to unfold RNA are in the range of 10 to 25 pN (30, 35), well within the range exerted by known enzymes that operate on nucleic acids (19, 45, 57).
I first review the theory of force as a thermodynamic variable, and then as a specific example, I compare the unfolding of an RNA molecule by force and by temperature. Methods for measuring the Gibbs free energy of mechanical unfolding for reversible and irreversible reactions are discussed. For a reversible reaction the Gibbs free energy is equal to the mechanical work (force times distance). For irreversible reactions the free energy can still be obtained from an appropriate average of many iterations of the irreversible work values. Next I compare the kinetics of unfolding/refolding at zero force and as a function of force. Various methods for measuring and analyzing the kinetics are described. My goal is to describe simple systems that illustrate the differences between single-molecule and ensemble methods, and to highlight the unique information obtainable from force. (A related review, “Mechanical Processes in Biochemistry,” by C. Bustamante et al. is scheduled to appear in Volume 73 of the Annual Review of Biochemistry.)
THERMODYNAMICS AND FORCE
I consider a system consisting of a single molecule surrounded by solvent, with a force applied to the single molecule, for example, by manipulation of beads attached to the ends of the molecule. The change in energy, dU, of the system depends on the sum of heat and work inputs. For a reversible process the equation is:
| 1 |
Force is a vector, so the differential work, dw = F · dX, depends on the component of the force, F, in the direction of the change of extension, dX. The positive sign of FdX in contrast with the negative sign of −PdV means that force is positive when it is pulling on the system, and negative when it is pushing. To add mechanical work, FdX, to the general thermodynamic equations for changes in energy, one must use only reversible paths linking equilibrium states of the system. Note that from Equation 1 the reversible mechanical work is equal to the change of energy at constant entropy and volume.
Gibbs free energy was defined by a Legendre transform as G ≡ U − PV − TS to produce a variable of state dependent on intrinsic variables T and P instead of extensive variables S and V.
| 2 |
Now, the reversible mechanical work is equal to the change of Gibbs free energy at constant temperature and pressure, a much more practical result. However, by this definition free energy still depends on an extensive variable, X. That is why [X] is explicitly added in Equation 2 to emphasize that G is a function of X. By a further Legendre transform
| 3 |
one obtains
| 4 |
where G[F] depends on intensive variables T, P, and F. Both G[F] ≡ U − PV − TS − FX and G[X] ≡ U − PV − TS are useful definitions of Gibbs free energy for a molecule under force.
Force is applied to a single molecule by attaching the molecule to a surface and to the tip of an atomic force microscope, or by attaching two beads to the molecule. In laser tweezer experiments, one bead is held in a laser trap (a laser beam focused to a spot), the other bead is held on a micropipet. The trap or the micropipet is moved so as to stretch and unfold the molecule. The force is monitored by the deviation of a bead from the center of the laser spot. Experiments can be done at constant force (by moving the micropipet to keep the bead a fixed distance from the center of the trap) or at constant extension (by moving the micropipet to keep the beads a fixed distance apart). Clearly, which definition of free energy (Equation 2 or Equation 4) is most convenient to use depends on the experiment. Either value of free energy is easily obtained from the other using Equation 3, and the two definitions obviously are equal at zero force.
At constant T and P the change in Gibbs free energy, ΔG = G2 − G1, can be obtained from the reversible work:
| 5 |
It may be more convenient to calculate the change in the force representation of Gibbs free energy, ΔG[F], by integrating −XdF instead:
| 6 |
The two values of free energies are related by
| 7 |
with Δ(FX) = F2X2 − F1X1.
Addition of force and extension as thermodynamic variables affect the criteria for equilibrium.
| 8 |
| 9 |
Thus, in the presence of a force, a criterion for equilibrium is that G[X] is a minimum and ΔG[X] is zero at constant T, P, and X. A more useful criterion is that G[F] is a minimum and ΔG[F] is zero at constant T, P and F.
RNA THERMODYNAMICS
Thermal Unfolding
It is convenient to consider the folded structure of an RNA molecule to be made up of secondary structure and tertiary structure. Secondary structure consists of double-stranded helices, hairpin loops, internal loops, and bulges; tertiary structure involves interactions among the secondary structure elements (8). The free energies of RNA secondary structure (relative to the single strand) can be reasonably approximated as a sum of negative contributions from helices plus positive contributions from the loss of entropy when loops and bulges form. The free energies of the helices are calculated as a sum of nearest-neighbor contributions from the base pairs (53). The thermodynamics of RNA helices, loops, and bulges has come mainly from absorbance melting curves of oligonucleotides, with some data from calorimetry. A convenient web site, the Mfold server (http://www.bioinfo.rpi.edu/~zukerm/), provides the calculated free energy of RNA secondary structure in a solvent of 1 M NaCl from an input sequence. Predicted secondary structures can be obtained from the calculated free-energy minimum and free energies near the minimum. Alternatively, base pairing can be specified to obtain the thermodynamics for a chosen secondary structure.
An RNA structure that thermally unfolds to single strand in a two-state process can be characterized by a melting temperature, the temperature where the free energies of the folded and unfolded (single-strand) species are equal (ΔG° = 0). The melting temperature, Tm, is obtained from the ratio of enthalpy to entropy, analogous to a phase transition.
| 10 |
Reversible Force Unfolding
When force is used to unfold an RNA, the free-energy change is measured from the mechanical work required. For a reversible process the reversible mechanical work is equal to the Gibbs free energy (Equation 2) at constant temperature and pressure. Figure 1a shows a set of four unfolding/refolding force versus extension curves for P5ab, a 22-bp hairpin, in 250 mM NaCl, 10 mM Mg2+ at 25°C. The RNA hairpin is held between DNA · RNA handles attached to polystyrene beads (30), with one bead in a laser light trap and the other on a micropipet. As the beads are moved apart, the force increases continuously as the handles are extended. At approximately 14.5 pN the RNA unfolds from hairpin to single strand, causing an abrupt “rip” in the smooth curve. After the abrupt transition the force increases further as the handles plus the single-stranded RNA extend further. When the extension is decreased, the refolding curve follows the same path as the unfolding. Thus, the unfolding of the RNA is a two-state process and there is no hysteresis. Experimentally, the process is reversible and all-or-none. The slope of the rip at the melting force is due to the properties of the laser light trap. When the RNA unfolds, the force drops rapidly until the distance between the beads adjusts to the new unfolded state. In a laser trap, force is measured from the distance moved by the bead from the center of the trap. Force and extension are thus coupled by the spring constant of the trap. In Figure 1 the spring constant was 0.1 pN/nm; the slope of the rip is equal to this value. (The slope of the noise seen throughout the force-extension curve has this same value.) The reversible unfolding/refolding process is characterized as occurring under a constant effective force, Fm, at the rip.
Figure 1.
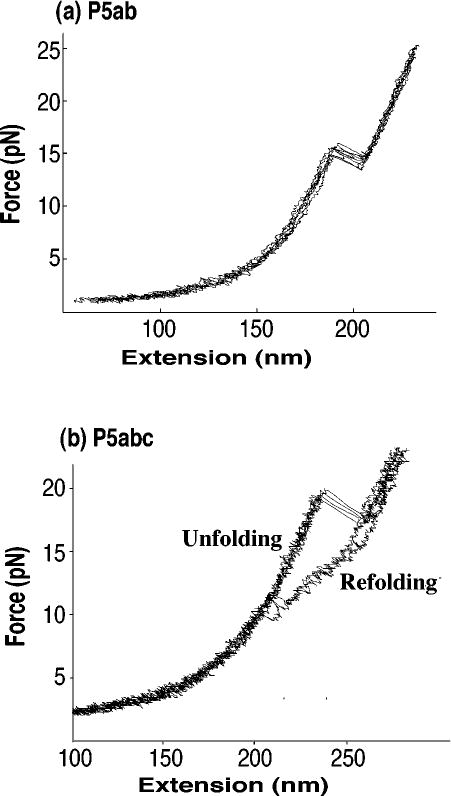
(a) Force-extension curves for the unfolding and refolding of an RNA hairpin, P5ab (sequence is in Figure 2), at 25°C, 250 mM NaCl, 10 mM Mg2+. Four curves are shown. The pulling rate is 2–3 pN s−1; at this rate the unfolding of the RNA is reversible. No hysteresis is seen. (b) Force-extension curves for the unfolding and refolding of a three-helix junction with an A-rich bulge, P5abc (sequence is P5ab with the addition of a stem loop and a bulge). Conditions are the same as in (a), but now the unfolding/refolding is irreversible. There is significant hysteresis. The curves are from Reference 30.
The free-energy change is directly measured as the reversible work at constant T,P,F.
| 11 |
Here ΔX is the difference in end-to-end distance—the difference in extension—between the unfolded state and the folded state. In the force representation, the free-energy change is zero for a process at equilibrium at constant T,P,F.
| 12 |
To compare the free energies measured at two different forces, a thermodynamic cycle joining the reactions at the two forces is used.
| 13 |
| 14 |
The equations can be used to relate the free energy at the melting force, the force, Fm, where the free energies of the folded and unfolded species are equal, to the standard free energy of the reaction at zero force, ΔG°. I write the equations specifically for the unfolding of a hairpin to a single strand, thus from Equation 13:
| 15 |
The standard free energy of unfolding at zero force is equal to the free energy of unfolding at the melting force minus the work required to stretch the single strand to its extension at Fm. I assume that the extension of the hairpin—the distance between the 5′ end and 3′ end—is essentially constant in comparison to the change in extension of the single strand. Equation 15 shows that the free energy of unfolding RNA is always less at zero force than at any other force, because of the extra work that must be done in stretching the single strand. The corresponding expression from Equation 14 is:
| 16 |
Equations 15 and 16 are equal from the identity ∫ d(Fx) = ∫ Fdx + ∫ xdF.
To apply Equations 15 and 16, one needs an expression for the extension of a single strand of RNA as a function of force. I have used a simple approximation for the worm-like-chain model (10), but other models can be used (7). The wormlike-chain model is characterized by two parameters: the persistence length, P, and the contour length, L.
| 17 |
In this approximation the force is inversely proportional to the persistence length and depends on the ratio of end-to-end distance, x, to contour length. Integration of Equation 17 gives
| 18 |
with R equal to the gas constant. The analogous equation for ∫ xdF is obtained by solving Equation 17 for x (a cubic equation), and integrating with respect to F.
Not many results on force unfolding of RNA have been published. They include a 49-nucleotide hairpin (P5ab) of 22 base pairs (30), and the approximately 400-nucleotide ribozyme (L-21) from Tetrahymena thermophila (35). The unfolding of the latter RNA shows one reversible transition (out of 8 total transitions) assigned to a 36-nucleotide hairpin (P9.2) with 14 base pairs. The standard free energies of both hairpins (ΔG° values at 25°C in 1 M NaCl) are easily calculated from the Mfold server. To illustrate the differences in free energies of unfolding under force and at zero force, I used the value of ΔG° from Mfold and Equation 15 to estimate the melting force. A constant value of extension, XHP = 2 nm, was used for the hairpin, and the extension of the single strand, XSS, was varied to provide a value of melting force, Fm, and to satisfy Equations 15 and 17. The parameters for the worm-like-chain, Equation 17, are P = 1 nm, L = 0.59 nm/nucleotide. Table 1 gives the calculated values: the melting force, Fm; the free energy of unfolding, FmΔX; the stretching free energy, ∫Fdx; and the change in extension, ΔX. There are many approximation and assumptions in these calculations; the results are presented only as examples of the magnitudes of the contributions from stretching a single strand of RNA and breaking the base pairs in a hairpin. For these examples, roughly one third of the free energy of unfolding an RNA at 25°C by force comes from stretching the single strand. Table 1 compares the calculated forces and extensions with experiment; the agreement is reasonable.
TABLE 1.
Experimental and calculated thermodynamic parameters for unfolding RNA hairpins
| Name | Tm(°C)a | ΔG° unfold (kcal/mol)a |
ΔG[X] at Fm |
ΔG stretch |
Fm(expt)b (pN) |
Fm(calc)b (pN) |
ΔX(expt)c (nm) |
ΔX(calc)c (nm) |
|---|---|---|---|---|---|---|---|---|
| P5abd 49 nt | 97 | 33.8 | 47.4 | 13.6 | 14.5 ± 1 | 17.0 | 18 ± 2 | 19.3 |
| P9.2e 36 nt | 90 | 17.3 | 25.8 | 8.5 | 12±2 | 13.8 | 15 ±2 | 13.0 |
The melting temperature, Tm, is the temperature where the hairpin and single strand have equal concentrations; the Tm and standard Gibbs free energy at 25°C and zero force, ΔG°, in 1 M NaCl were obtained from Mfold 2.3 at http://www.bioinfo.rpi.edu/~zukerm/. The free energy (ΔG[X]) of unfolding at the melting force (Fm) and the free energy of stretching (ΔGstretch) the single strand to this force were fit to ΔG°.
Fm is the melting force at 25°C.
The change in end-to-end distance on unfolding the RNA is ΔX.
See Reference 30.
See Reference 35.
Table 1 also shows that forces in the range from 10 pN to 20 pN suffice to unfold RNA hairpins, which require temperatures above 80°C to unfold thermally. These forces are well within the range of forces produced by DNA polymerases (31), DNA-dependent RNA polymerases (19, 57), and other DNA-dependent molecular motors (45). Mechanical unfolding of RNA in a biological cell can thus be accomplished by natural processes.
The Free-Energy Landscape
The force unfolding of the RNA hairpins listed in Table 1 was found experimentally to be all-or-none. This result can be understood by calculating the free energy of each partially unfolded species at Fm to find the relative amount of each species at equilibrium. The results are easiest to visualize in the force representation of free energy, where the free energies are equal at Fm for the intact hairpin and the single strand. Each partially unfolded species consists of a hairpin with one or more base pairs broken from the end. For illustrative purposes only, I treat the single strands at the ends as a worm-like chain even for only one nucleotide at each end (one base pair broken). The free energy of the hairpin (assumed to have a constant end-to-end distance) is −FXHP. The free energy (relative to the intact hairpin) of each species as a function of force is thus:
| 19 |
ΔG°(F = 0) is the (positive) standard free energy of breaking one or more base pairs from the end of the intact hairpin. The integral is the (positive) stretching free energy of the single strands in each species. The last term is the (negative) free energy of the single-strand portion of the partially unfolded species. The magnitude of XSS obviously increases with each base pair broken; its value is obtained from the contour length of the single strand portion and the force. Because breaking the final base pair leaves only a single strand with no hairpin contribution to its length, for this species the last term in Equation 19 is replaced by —F(XSS-XHP).
The calculated free-energy values for the partially melted species of P5ab at zero force and at Fm are shown in Figure 2. At zero force the free energies, ΔG°(F = 0), are calculated from the nearest-neighbor parameters for base pair breaking (53). At Fm Equation 19 is used. As the force increases, the species with the larger lengths of single-strand portions are increasingly stabilized until at Fm the free energies of the intact hairpin and the pure single strand are equal. The unfolding process appears all-or-none because the only appreciable species are the intact hairpin, the partially unfolded species with three base pairs broken (releasing the bulged U), and the single strand. The intact hairpin and the species with three base pairs broken are similar enough in end-to-end distance to appear as a single species.
Figure 2.

Calculated free-energy landscape at 25°C for P5ab at zero force and at Fm. At zero force the free energies are calculated from nearest-neighbor values; at the Fm the worm-like-chain model is used to obtain the effect of force on each partially unfolded species. At zero force the hairpin is the only species present. At Fm the species present in significant amounts are now the hairpin, the single strand, and the hairpin with three base pairs broken caused by the destabilizing effect of the bulged U.
In contrast to force unfolding with successive base pairs breaking from the end of the stem, thermal melting occurs both from the stem end and from the loop. All the base pairs become less stable as the temperature is raised; thus, the number of partially folded species is larger. However, the main difference between thermal melting and force unfolding is the nature of the single strand. In force unfolding the single strand is stretched out; it has reduced conformational variability with an end-to-end distance approaching the contour length of the polymer. In thermal unfolding the single strand is more like a random coil; it has a high entropy and is essentially a species with properties different from the single strand with ends held fixed. Unfolding an RNA by applying a local perturbation at the ends by force, instead of melting the RNA with a global thermal perturbation, creates a different process. It simplifies the analysis of unfolding, and it is more like the naturally occurring unfolding inside cells.
Irreversible Force Unfolding
When the kinetics of a reaction are slower than the loading rate—the rate at which the force changes—force versus extension curves show hysteresis (see Figure 1b) between unfolding and refolding curves. The amount of hysteresis and the forces at which the unfolding/refolding occur depend on the loading rate. Current laser tweezer experiments are usually done with loading rates in the range of 1–10 pN s−1; slower rates lead to excessive drift and unreliable measurements. This means that molecular relaxation times must be shorter than a few seconds to allow a reversible reaction to occur. For longer relaxation times the forces at which unfolding and refolding take place are kinetically controlled. The molecular processes are stochastic, so there is a range of forces for the transitions, and the loading rate has significant effects. The kinetics of the reaction determine the force-extension curves. Tertiary interactions in RNA slow the kinetics of unfolding. Magnesium ions directly bound to phosphate oxygens and base nitrogens often stabilize tertiary interactions in RNA. The rearrangements of the RNA that occur on ion binding are slower than simple base pair opening. However, even without tertiary interactions, the kinetics of RNA unfolding are sometimes too slow for measurement of a reversible process. Several G · C base pairs in a row stabilize a helix and slow the kinetics of unfolding significantly.
The Gibbs free energies for irreversible processes cannot be obtained from the mean work. However, by exponentially averaging the different irreversible work trajectories for many repetitions of the experiments, one can obtain the Gibbs free energy. Jarzynski (26) showed that the free energy could be obtained from irreversible processes by appropriate averaging (see also Reference 24).
| 20 |
In principle the exponential average, ⟨⟩, in Equation 20 must be done for an infinite number of repetitions of the experiment. This is equivalent to the requirement that a reversible process must proceed infinitely slowly—quasistatically. In practice the number of work trajectories that must be measured depends on the average work dissipated, ⟨wdiss⟩, a measure of the irreversibility of the process. The number of experiments required (40) to obtain a value of free energy within a chosen uncertainty is proportional to . As the average dissipated work becomes greater than 10 kT, the number of experiments required for a ±(1/2) kT error becomes impractical. The Jarzynski equation has been tested for RNA unfolding (29) and for the motion of small particles in a solvent (56). In both tests the process could be done reversibly to measure ΔG directly, and then irreversibly to test Equation 20. The RNA force unfolding was first done at a force loading rate of 2–5 pN s−1, in which the unfolding showed no hysteresis. The mean reversible work—equal to the change in Gibbs free energy for unfolding—was 60.2 ± 1.6 kT. The loading rate was then increased to 34 pN s−1, and then to 52 pN s−1. The unfolding was clearly irreversible and the mean work increased with loading rate, as expected from the second law of thermodynamics. The spread of the measured work values also increased with increasing loading rate. Application of Equation 20 with 100 to 150 repetitions of the experiment gave 59.6 ± 0.2 kT for the free energy from the irreversible reactions. The agreement is excellent.
An alternative method to obtain free energies from irreversible processes (C. Jarzynski, V. Pande & F. Ritort, personal communication) is to find the intersection of the distribution of work trajectories for the forward reaction (unfolding) and backward reaction (refolding). For Equation 20 to be valid, some of the unfolding work trajectories must have values less than the reversible work that must be done to unfold the RNA. Similarly, some of the folding work trajectories must have values greater than the reversible work obtained when the RNA refolds. The second law of thermodynamics states that the average work done on a system must be greater than the free-energy increase and that the average work obtained is less than the free-energy decrease. However, a particular folding or unfolding work trajectory can be greater than, less than, or equal to the free-energy change. The work trajectories corresponding to a reversible process (reversible work = free-energy change) are rare for an irreversible stochastic process. However, at the intersection of the distributions of folding and unfolding trajectories, the measured work values are the same. This is the criterion for a reversible process; thus, the work at the intersection is equal to the Gibbs free energy.
RNA KINETICS
Kinetics at Zero Force
The kinetics of unfolding and refolding RNA were measured in the 1970s by Pörschke (36–38) and others (15, 39, 55) using temperature-jump methods. The increase in absorbance (when stacking decreased as base pairs were broken in double strands) was used to follow the reactions. The data and references have been compiled by Turner et al. (54). More recently both absorbance and fluorescence (with fluorescent 2-amino purine replacing adenine in the RNA) have been used to study RNA hairpin unfolding (1, 2, 6, 33). Kinetic studies of more complex RNAs have concentrated on the interactions of secondary structures to form tertiary structure. The process is one of folding, but instead of single-strand to double-strand transitions, helices and loops interact to form higher-order structures. Folding reactions are initiated by adding Mg2+, and unfolding reactions are initiated by adding divalent ion scavengers, such as EDTA. The processes have been followed by using a wide variety of methods including chemical reactivity of accessible nucleotides (43, 44, 50), small-angle X-ray scattering (41), and single-molecule fluorescence (5, 42, 60). Recent reviews summarize much of the work (46, 52).
Kinetics in the Presence of a Force
It seems reasonable that force favors the equilibrium species with the longest extension in the direction of force. As the rate of a reaction between two species depends on the free-energy change between reactant and the transition state, it is reasonable that increasing force increases the kinetics. The increase in rate depends on the distance from the reactant to the transition state. The effect is exponential.
| 21 |
Here k(F) is the rate constant at force, F; k(0) is the rate constant at zero force; and X‡ is the distance to the transition state. As the distance to the transition state depends on force, and the mechanism of the reaction may well be different at zero force, Equation 21 in general is not a practical way to obtain values of the rate constant at zero force. However, the distance to the transition state can be obtained from the slope of a plot of ln k versus force, and extrapolations can provide the rate constants at nearby forces. An example of a ln k versus force plot for the unfolding of an RNA is shown in Figure 3.
Figure 3.
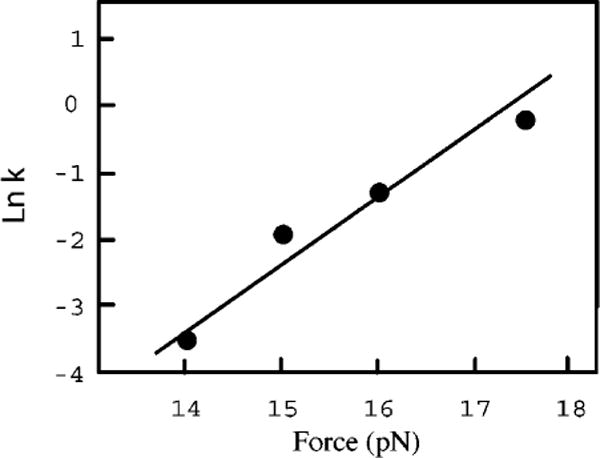
A plot of the logarithm of the rate constant versus force for unfolding a three-helix RNA junction that is the binding site for S15 ribosomal protein from Bacillus stearothermophilus (3). The slope of the line, which is equal to X‡/kT, gives a value of X‡ = 3.5 nm. The solvent is 4 mM Mg2+, pH 8 HEPES, 50 mM KCl; the temperature is 25°C. The rate constants were measured by force-jump experiments; the data are from unpublished work of Dr. D. Collin.
Three methods have been used to measure rate constants for folding/unfolding as a function of force: force-jump measurements, in which the force is rapidly increased or decreased and then held constant (analogous to temperature-jump kinetics) (34); force-ramp measurements, in which the force is increased linearly with time (17, 34); and hopping experiments, in which the reaction is near equilibrium and the molecule transits between two states (30).
The kinetics for a single molecule occurs stochastically. For a given set of conditions (force, temperature, and solvent), there is a range of lifetimes for a given species. The change in probability, P, that the reaction has not occurred is
| 22 |
Thus the probability that a species has not reacted decreases exponentially from 1 to 0 as time increases.
| 23a |
with k(F) as the time-independent rate constant. The logarithm of the probability is linear in time.
| 23b |
For force-jump experiments the force is raised or lowered rapidly compared with the molecular process, and then held constant. The lifetime of the species is measured (the time when the reaction occurs), and the experiment is repeated. The value of k can be obtained from the slope of a plot of the logarithm of P versus t. The fraction of trials with lifetimes greater than t is equal to the probability, P, that the reaction has not occurred before time t. Figure 4 shows data from the unfolding of the RNA described in Figure 3. Alternatively, the value of k can be obtained simply as the reciprocal of the average lifetime of the species.
Figure 4.
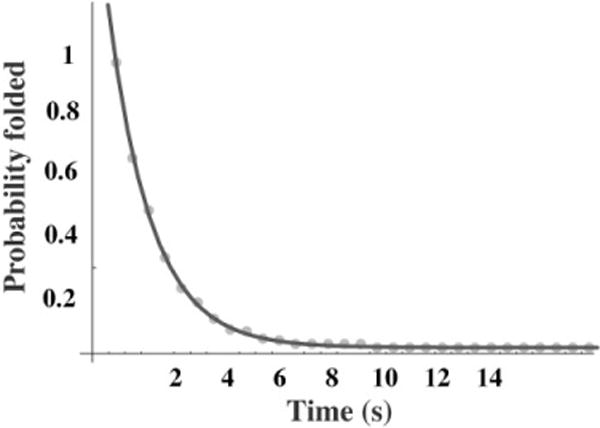
A plot of the fraction of experiments with lifetimes greater than time t versus t. The force was jumped to 17.5 pN and held constant until the RNA unfolded. The lifetime was measured and the experiment was repeated 238 times. The data fit a single exponential with a relaxation time of 1.3 s corresponding to a rate constant of 0.77 s−1. The data are from unpublished work of Dr. D. Collin.
In the force-ramp method the force is increased linearly with time (F = rt) during the experiment. There is a corresponding range of forces where the transition to a different species occurs. The distribution of transition forces depends on the loading rate, r (the increase in force with time), and the distance to the transition state, X‡. The equation is easily derived by explicitly adding the time dependence for k(F) to Equation 22.
| 24 |
Here b = X‡/kT. Integration of Equation 24 gives the desired result.
| 25 |
It is convenient to obtain X‡ and k(0) from the slope and intercept of a double logarithmic plot using the approximation that ebF ≫ 1.
| 26 |
Force-extension curves (see Figure 1) are obtained at a constant loading rate, r, and the transition force is measured for successive trials. The fraction of experiments in which no reaction has occurred at forces less than F provides the probability used in Equation 26. Data from the RNA studied in Figures 3 and 4 are shown in Figure 5. The slope (equal to X‡/kT) provides the distance to the transition state, X‡; the intercept (equal to k(0)kT/X‡) provides the rate constant at zero force. As discussed above, its value is not a good measure of the kinetics at zero force. Instead it should be considered a parameter for calculating rate constants at forces near the measured data. That is, the force-dependent kinetics apply to the range of forces that the molecular motors in biological cells are capable of exerting.
Figure 5.
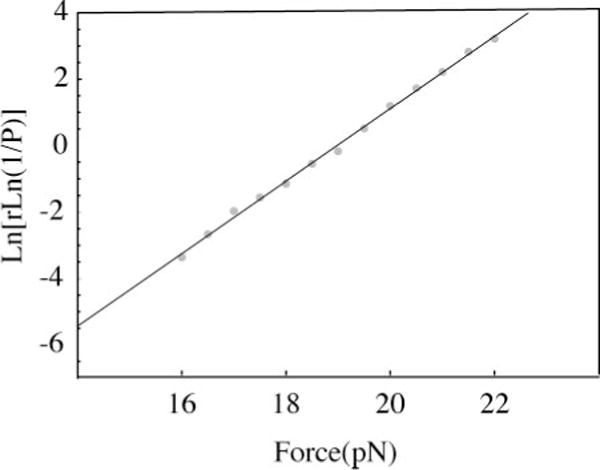
A plot that provides the rate constants as a function of force and the distance to the transition state. In this force-ramp method many force-extension curves are measured at a constant loading rate, and the transition force is noted for each trial. The fraction of trials with transition forces less than F is used in Equation 26. The loading rate here was 7.1 pN s−1; the rate constants and distance to the transition state are in reasonable agreement with the data shown in Figures 3 and 4. The RNA is described in Figure 3, and the data are from unpublished work of Dr. D. Collin.
The hopping method of measuring rates is conceptually simple (30). The transitions between two states at constant force are followed as a function of time for as long as the molecule and instrumentation allow (20 to 100 transitions). The distributions of lifetimes for the forward and reverse reactions are used as in Figure 4 to get the corresponding rate constants. The hopping method is limited to forces near the melting force. At other forces either the forward or backward rate constants become too fast (or too slow) to see hopping.
Complex RNAs
In complex RNAs with many domains, slow unfolding kinetics leads to detectable intermediates in force-extension curves. The ribozyme from T. thermophila containing about 400 nucleotides was found to have 8 intermediates (35). The number of intermediates seen, the transition force between species, and the unfolding paths differed among repetitions of the experiment. The assignment of the intermediates, and the identification of the kinetic barriers that produced the intermediates, was done by studying mutants and subdomains of the RNA, and by adding oligonucleotides that competed with secondary and tertiary structure. It is encouraging that assignment of the transitions in RNAs containing hundreds of nucleotides can be made. The kinetic barriers were mainly caused by tertiary interactions stabilized by Mg2+. In the absence of Mg2+ the kinetic barriers disappeared, and no detectable intermediates were seen. Clearly, understanding the role of Mg2+ in formation and stabilization of folded domains is important. Further quantitative measurements of how the kinetics and thermodynamics change with Mg2+ or other relevant ligands may provide insight into the role of small molecules in the functions of RNA.
Simulation of Force Unfolding and Refolding
In addition to general reviews on the effect of force on reactions (16, 51), there have been several theoretical simulations and analyses of RNA unfolding in the presence of force (13, 14, 20, 21, 59). To illustrate the differences between force unfolding and thermal unfolding, I simulate the kinetics of unfolding a simple hairpin.
I consider a 5-bp hairpin with a loop of four nucleotides so that all the steps can be shown; such a small hairpin would be difficult to study experimentally with present equipment. When a thermally stable hairpin is unfolded by applying force to its ends, the only species that need be considered are those in which successive base pairs are broken from the end (see Figure 6). Thus, for a 5-bp hairpin only six species are present: the intact hairpin, four intermediates, and the single strand. In thermal unfolding, base pairs can break from either end of the stem; this adds 10 intermediates to the unfolding of a 5-bp hairpin. For longer stem regions where melting can also occur in the middle of the stem, and for structures with internal loops, bulges, or tertiary interactions, the number of intermediates is much greater in thermal unfolding than in force unfolding. Clearly, modeling and understanding will be simpler for force unfolding than for thermal unfolding.
Figure 6.
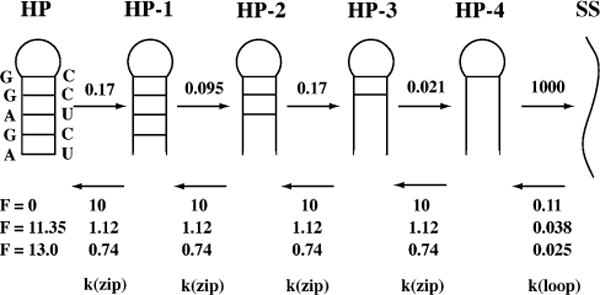
A mechanism for force unfolding of an RNA hairpin with four nucleotides in the loop. The force is applied to the ends of the RNA, so the base pairs break sequentially from the end of the hairpin. The base pair breaking rate constants, k(open), are given as independent of force; the base pair forming constants, k(close), are shown at three different forces (0 pN, 11.35 pN, and 13.0 pN) at 25°C. All rate constants have units of 106 s−1. I assume that the transition states for breaking base pairs are close to the intact base pair ; therefore, there is a negligible effect of force on the rate constants for breaking base pairs. The rate constants for forming base pairs decrease with increasing force.
There are four basic kinetic processes in the unfolding of a hairpin: breaking the last base pair in the hairpin, forming the first base pair in the hairpin, and breaking and forming successive base pairs. Forming the first base pair is special because it involves the loss of entropy for loop formation; it is expected to have the slowest elementary rate constant for forming a base pair. The other base pairs stack on the preceding pairs; these steps have faster rate constants. Because of the successful nearest-neighbor method of calculating thermodynamic stabilities of RNA secondary structures (53), there are equilibrium constant available for each of the elementary steps in the unfolding process. There are 10 equilibrium constants for the 10 nearest-neighbor Watson-Crick base pair sequences, and there are also equilibrium constants for loop formation that depend only on the number of nucleotides in the loop. Knowledge of the equilibrium constants provides the ratio of forward and backward rate constants, and thus the rate constants for breaking base pairs are defined once the rates for forming base pairs are chosen.
The four types of rate constants are k(loop close), k(loop open), k(zip close), k(zip open), as shown in Figure 6. The loop constants depend on the number of nucleotides in the loop, not on the sequence. The standard free energies at 25°C for loop formation are +6 ± 0.5 kcal/mol for loops containing 4 to 8 nucleotides (53). Thus the equilibrium constants (and ratio of rate constants) are in the range of 10−4 to 10−5.
| 27 |
There are 10 standard free energies at 25°C for adding a base pair that range from −1.0 to −3.5 kcal/mol depending on the nearest-neighbor sequences (53). The equilibrium constants (and ratio of rate constants) are thus in the range of 5 to 350.
| 28 |
The analysis can be simplified further by assuming that k(zip close) is independent of sequence. Breaking a base pair depends on the base pair and its neighbor but forming it does not. These assumptions mean that to model the kinetics of a hairpin, there are only two independent parameters; I chose k(loop open) and k(zip close). The other parameters are determined by the nearest-neighbor thermodynamic data. All these parameters of course apply at zero force; the effect of force depends on the distance to the transition state for each elementary step.
The model used for the force dependence is that of Cocco et al. (12). They assume that the distance to the transition state for breaking a base pair is 0.1 nm or less . The distance to the transition state for closing a base pair is
| 29 |
Here ΔX is the difference in extension for the species with the base pair open and with the base pair closed. As ΔX is at least a factor of 10 greater than Xopen‡, they use , and Xclose‡ = ΔX. This means k(loop open) and k(zip open) are independent of force. To calculate the effect of force on k(loop close) and k(zip close), I use the worm-like-chain model with the parameters that fit the equilibrium properties of singe-stranded RNA (30): persistence length = 1 nm, contour length per single-strand nucleotide = 0.59 nm.
Modelling the Kinetics of a Hairpin Unfolding/Refolding
I assume the mechanism of hairpin unfolding/refolding involves base pairs breaking and forming only from the ends of a double helix. For force unfolding, base pair breaking occurs sequentially from the stem end, and base pair forming occurs sequentially from the loop end. For thermal unfolding at zero force, base pair breaking can occur from either end. For thermal refolding at zero force the first base pair can form anywhere in the stem, followed by addition of successive base pairs. This is a simple model that contains the essence of the kinetics. For N base pairs in the stem, there are [N + 1] species for force unfolding, and [(N(N+1)/2)+1] species for unfolding at zero force.
The kinetics are governed by a set of coupled linear differential equations:
| 30 |
The vector A represents the fraction of each species present at any time; K is the matrix of rate constants. Methods for solving these equations are given in kinetics textbooks (47). The K matrix is diagonalized to obtain the diagonal matrix Λ with negative eigenvalues λ, and matrix P of eigenvectors. The time dependence of each species is:
| 31 |
The vector A0 is the distribution of species at zero time.
The kinetics of unfolding/refolding the hairpin (sequence shown in Figure 6) are modeled at three different forces: zero force where the hairpin is stable (Keq = [SS]/[HP] = 5.3 × 10−5), 11.35 pN where hairpin and single strand are equally stable (Keq = 0.98), and 13.0 pN where the single strand is the principal species (Keq = 7.8). At zero force two elementary rate constants were chosen: k(zip close) = 107 s−1 (rate constant for adding a base pair); and k(loop open) = 109 s−1 (rate constant for breaking the last base pair). The choice of values for these two rate constants is somewhat arbitrary, but the choice does not change any of the general conclusions. My goal is to show how force affects the kinetics and equilibrium of hairpin unfolding/refolding. At zero force the other rate constants are obtained from the equilibrium free-energy values using Equations 27 and 28. The force dependence of the rate constants are obtained using the worm-like chain for the single strands. All the values are shown in Figure 6. The worm-like chain is a poor approximation for such short chains, but the results illustrate the effect of force on the rates. Force slows the rate constants for forming base pairs and thus favors the single strand with increasing force.
Equation 31 was solved at the three forces for the sequential mechanism seen in Figure 6. Six relaxation times corresponding to the six differential equations were obtained. In addition, at zero force I applied the mechanism that allows base pair breaking from either end of the helix and allows the first base pair to form anywhere. This produces 16 species and 16 relaxation times. For all four models only one relaxation time, τ, dominates the kinetics. All the other relaxation times are at least 1000 times shorter. The values of the rate-determining relaxation times—the longest relaxation time for each model—are given in Table 2. The approach to equlibrium is fastest at zero force not only because force has not slowed the base forming steps, but also because of the multiple paths for folding/unfolding. The multiple paths increase the rate by a factor of 9.
TABLE 2.
The relaxation times τ, fraction of each species (HP, HP-1, SS are defined in Figure 6), equilibrium constants, and forward, kf, and backward, kb, rate constants for the RNA hairpin in Figure 6 at three different forces
| Force (pN) | τ (ms) | HP | HP-1 | SS | Keq | kf (s−1) | kb (s−1) |
|---|---|---|---|---|---|---|---|
| 0 (6 species) | 0.92 | 0.98 | 0.02 | 0.00 | 5.3 × 10−5 | 0.06 | 1.1 × 103 |
| 0 (16 species) | 0.12 | 0.98 | 0.02 | 0.00 | 5.3 × 10−5 | 0.45 | 8.5 × 103 |
| 11.35 | 13.2 | 0.47 | 0.07 | 0.46 | 0.98 | 38.2 | 39.2 |
| 13.0 | 7.9 | 0.11 | 0.03 | 0.86 | 7.8 | 112.1 | 14.7 |
The reaction can be treated approximately as a two-state process. Then the relaxation time is the reciprocal of the sum of effective forward and backward rate constants, and the equilibrium constant is their ratio.
| 32 |
The kf and kb parameters are linear combinations of the elementary rate constants in Figure 6; their values for the four models are given in Table 2. As expected, as the force increases, the forward rate constant (hairpin to single strand) increases and the backward rate constant decreases. The relaxation time is a maximum at the melting force, in which the forward and backward rate constants are equal.
FUTURE DEVELOPMENTS
The application of force to single molecules is a recent endeavor. Both the experimental methods and the theoretical analysis of the data are in their infancy. I hope that this review encourages people to improve the methods and to apply them to many other systems. In Berkeley, some of the obvious improvements are being implemented. Temperature variation (58) will allow entropy and enthalpy changes to be obtained from the temperature dependence of the free energy. I discussed the difference between thermal melting and force unfolding, but they become more similar as the temperature is raised. Addition of single-molecule fluorescence detection as force is applied (28) will allow FRET measurements to directly identify which parts of a molecule are unfolding. DNA · DNA handles can be applied to proteins to unfold single protein molecules in a manner similar to that done for RNA (C. Bustamante & S. Marqusee personal communication). The biggest challenge and the greatest reward is to apply single-molecule methods within biological cells. Laser tweezers can be used to manipulate macromolecules and to even perform surgery on them inside cells (22). I am sure that many more ideas are being considered and many more applications are underway. I await them eagerly.
Acknowledgments
Dr. Nancy Forde, Ms. Lisa Green, Dr. Raven Hanna, Dr. Pan Li, and Dr. Bibiana Onoa read the manuscript and made helpful suggestions. Dr. Delphine Collin kindly furnished her unpublished data. I am greatful for their help.
Footnotes
The Annual Review of Biophysics and Biomolecular Structure is online at http://biophys.annualreviews.org
LITERATURE CITED
- 1.Ansari A, Kuznetsov SV, Shen Y. Configurational diffusion down a folding funnel describes the dynamics of DNA hairpins. Proc Natl Acad Sci USA. 2001;98:7771–76. doi: 10.1073/pnas.131477798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansari A, Shen Y, Kuznetsov SV. Misfolded loops decrease the effective rate of DNA hairpin formation. Phys Rev Lett. 2002;88:069801. doi: 10.1103/PhysRevLett.88.069801. [DOI] [PubMed] [Google Scholar]
- 3.Batey RT, Williamson JR. Interaction of the Bacillus stearothermophilus ribosomal protein S15 with 16 S rRNA. I. Defining the minimal RNA site. J Mol Biol. 1996;261:536–49. doi: 10.1006/jmbi.1996.0481. [DOI] [PubMed] [Google Scholar]
- 4.Bockelmann U, Thomen P, Essevaz-Roulet B, Viasnoff V, Heslot F. Unzipping DNA with optical tweezers: high sequence sensitivity and force flips. Biophys J. 2002;82:1537–53. doi: 10.1016/S0006-3495(02)75506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bokinsky G, Rueda D, Misra VK, Rhodes MM, Gordus A, et al. Single-molecule transition-state analysis of RNA folding. Proc Natl Acad Sci USA. 2003;100:9302–7. doi: 10.1073/pnas.1133280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnet G, Krichevsky O, Libchaber A. Kinetics of conformational fluctuations in DNA hairpin-loops. Proc Natl Acad Sci USA. 1998;95:8602–6. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchiat C, Wang MD, Allemand J, Strick T, Block SM, Croquette V. Estimating the persistence length of a worm-like chain molecule from force-extension measurements. Biophys J. 1999;76:409–13. doi: 10.1016/s0006-3495(99)77207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burkard ME, Turner DH, Tinoco I., Jr . The interactions that shape RNA structure. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. Second. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1999. pp. 233–64. [Google Scholar]
- 9.Bustamante C, Bryant Z, Smith SB. Ten years of tension: single-molecule DNA mechanics. Nature. 2003;421:423–27. doi: 10.1038/nature01405. [DOI] [PubMed] [Google Scholar]
- 10.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 11.Carrion-Vazquez M, Oberhauser AF, Fisher TE, Marszalek PE, Li H, Fernandez JM. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog Biophys Mol Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 12.Cocco S, Marko JF, Monasson R. Slow nucleic acid unzipping kinetics from sequence-defined barriers. Eur Phys J. 2003;10:153–56. doi: 10.1140/epje/e2003-00019-8. [DOI] [PubMed] [Google Scholar]
- 13.Cocco S, Monasson R, Marko JF. Force and kinetic barriers to unzipping of the DNA double helix. Proc Natl Acad Sci USA. 2001;98:8608–13. doi: 10.1073/pnas.151257598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cocco S, Monasson R, Marko JF. Force and kinetic barriers to initiation of DNA unzipping. Phys Rev E Stat Nonlin Soft Matter Phys. 2002;65:041907. doi: 10.1103/PhysRevE.65.041907. [DOI] [PubMed] [Google Scholar]
- 15.Craig ME, Crothers DM, Doty P. Relaxation kinetics of dimer formation by self complementary oligonucleotides. J Mol Biol. 1971;62:383–401. doi: 10.1016/0022-2836(71)90434-7. [DOI] [PubMed] [Google Scholar]
- 16.Evans E. Probing the relation between force–lifetime–and chemistry in single molecular bonds. Annu Rev Biophys Biomol Struct. 2001;30:105–28. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 17.Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997;72:1541–55. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Florin EL, Moy VT, Gaub HE. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264:415–17. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 19.Forde NR, Izhaky D, Woodcock GR, Wuite GJ, Bustamante C. Using mechanical force to probe the mechanism of pausing and arrest during continuous elongation by Escherichia coli RNA polymerase. Proc Natl Acad Sci USA. 2002;99:11682–87. doi: 10.1073/pnas.142417799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerland U, Bundschuh R, Hwa T. Force-induced denaturation of RNA. Biophys J. 2001;81:1324–32. doi: 10.1016/S0006-3495(01)75789-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerland U, Bundschuh R, Hwa T. Mechanically probing the folding pathway of single RNA molecules. Biophys J. 2003;84:2831–40. doi: 10.1016/S0006-3495(03)70012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grier DG. A revolution in optical manipulation. Nature. 2003;424:810–16. doi: 10.1038/nature01935. [DOI] [PubMed] [Google Scholar]
- 23.Hinterdorfer P, Baumgartner W, Gruber HJ, Schilcher K, Schindler H. Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc Natl Acad Sci USA. 1996;93:3477–81. doi: 10.1073/pnas.93.8.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hummer G, Szabo A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc Natl Acad Sci USA. 2001;98:3658–61. doi: 10.1073/pnas.071034098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janshoff A, Neitzert M, Oberdorfer Y, Fuchs H. Force spectroscopy of molecular systems—single molecule spectroscopy of polymers and biomolecules. Angew Chem Int Ed. 2000;39:3213–37. doi: 10.1002/1521-3773(20000915)39:18<3212::aid-anie3212>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 26.Jarzynski C. Nonequilibrium equality for free energy differences. Phys Rev Lett. 1997;78:2690–93. [Google Scholar]
- 27.Kellermeyer MK, Smith SB, Bustamante C, Granzier HL. Mechanical fatigue in repetitively stretched single molecules of titin. Biophys J. 2001;80:852–63. doi: 10.1016/S0006-3495(01)76064-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang MJ, Fordyce PM, Block SM. Combined optical trapping and single-molecule fluorescence. J Biol. 2003;2:6. doi: 10.1186/1475-4924-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liphardt J, Dumont S, Smith SB, Tinoco I, Jr, Bustamante C. Equilibrium information from nonequilibrium measurements in an experimental test of Jarzynski’s equality. Science. 2002;296:1832–35. doi: 10.1126/science.1071152. [DOI] [PubMed] [Google Scholar]
- 30.Liphardt J, Onoa B, Smith SB, Tinoco I, Jr, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–37. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 31.Maier B, Bensimon D, Croquette V. Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc Natl Acad Sci USA. 2000;97:12002–7. doi: 10.1073/pnas.97.22.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marszalek PE, Li H, Oberhauser AF, Fernandez JM. Chair-boat transitions in single polysaccharide molecules observed with force-ramp AFM. Proc Natl Acad Sci USA. 2002;99:4278–83. doi: 10.1073/pnas.072435699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menger M, Eckstein F, Pörschke D. Dynamics of the RNA hairpin GNRA tetraloop. Biochemistry. 2000;39:4500–7. doi: 10.1021/bi992297n. [DOI] [PubMed] [Google Scholar]
- 34.Oberhauser AF, Hansma PK, Carrion-Vazquez M, Fernandez JM. Stepwise unfolding of titin under force-clamp atomic force microscopy. Proc Natl Acad Sci USA. 2001;98:468–72. doi: 10.1073/pnas.021321798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onoa B, Dumont S, Liphardt J, Smith SB, Tinoco I, Jr, Bustamante C. Identifying kinetic barriers to mechanical unfolding of the T. thermophila ribozyme. Science. 2003;299:1892–95. doi: 10.1126/science.1081338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pörschke D. A direct measurement of the unzippering rate of a nucleic acid double helix. Biophys Chem. 1974;2:97–101. doi: 10.1016/0301-4622(74)80029-3. [DOI] [PubMed] [Google Scholar]
- 37.Pörschke D. Model calculations on the kinetics of oligonucleotide double helix coil transitions. Evidence for a fast chain sliding reaction. Biophys Chem. 1974;2:83–96. doi: 10.1016/0301-4622(74)80028-1. [DOI] [PubMed] [Google Scholar]
- 38.Pörschke D. Thermodynamic and kinetic parameters of an oligonucleotide hairpin helix. Biophys Chem. 1974;1:381–86. doi: 10.1016/0301-4622(74)85008-8. [DOI] [PubMed] [Google Scholar]
- 39.Ravetch J, Gralla J, Crothers DM. Thermodynamic and kinetic properties of short RNA helices: the oligomer sequence AnGCUn. Nucleic Acids Res. 1974;1:109–27. doi: 10.1093/nar/1.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritort F, Bustamante C, Tinoco I., Jr A two-state kinetic model for the unfolding of single molecules by mechanical force. Proc Natl Acad Sci USA. 2002;99:13544–48. doi: 10.1073/pnas.172525099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, et al. Rapid compaction during RNA folding. Proc Natl Acad Sci USA. 2002;99:4266–71. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell R, Zhuang X, Babcock HP, Millett IS, Doniach S, et al. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci USA. 2002;99:155–60. doi: 10.1073/pnas.221593598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sclavi B, Sullivan M, Chance MR, Brenowitz M, Woodson SA. RNA folding at millisecond intervals by synchrotron hydroxyl radical footprinting. Science. 1998;279:1940–43. doi: 10.1126/science.279.5358.1940. [DOI] [PubMed] [Google Scholar]
- 44.Sclavi B, Woodson S, Sullivan M, Chance M, Brenowitz M. Following the folding of RNA with time-resolved synchrotron x-ray footprinting. Methods Enzymol. 1998;295:379–402. doi: 10.1016/s0076-6879(98)95050-9. [DOI] [PubMed] [Google Scholar]
- 45.Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DE, Bustamante C. The bacteriophage ϕ29 portal motor can package DNA against a large internal force. Nature. 2001;413:748–52. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- 46.Sosnick TR, Pan T. RNA folding: models and perspectives. Curr Opin Struct Biol. 2003;13:309–16. doi: 10.1016/s0959-440x(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 47.Steinfeld JI, Francisco JS, Hase WL. Chemical Kinetics and Dynamics. Englewood Cliffs, NJ: Prentice Hall; 1989. [Google Scholar]
- 48.Stone MD, Bryant Z, Crisona NJ, Smith SB, Vologodskii A, et al. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc Natl Acad Sci USA. 2003;100:8654–59. doi: 10.1073/pnas.1133178100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strick T, Allemand J, Croquette V, Bensimon A. The manipulation of single biomolecules. Phys Today. 2001 Oct;:46–51. [Google Scholar]
- 50.Thirumalai D, Lee N, Woodson SA, Klimov D. Early events in RNA folding. Annu Rev Phys Chem. 2001;52:751–62. doi: 10.1146/annurev.physchem.52.1.751. [DOI] [PubMed] [Google Scholar]
- 51.Tinoco I, Jr, Bustamante C. The effect of force on thermodynamics and kinetics of single molecule reactions. Biophys Chem. 2002:101–102. 513–33. doi: 10.1016/s0301-4622(02)00177-1. [DOI] [PubMed] [Google Scholar]
- 52.Treiber DK, Williamson JR. Beyond kinetic traps in RN folding. Curr Opin Struct Biol. 2001;11:309–14. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 53.Turner DH. Conformational changes. In: Bloomfield VA, Crothers DC, Tinoco I Jr, editors. Nucleic Acids: Structures, Properties, and Functions. Mill Valley, CA: Univ. Sci. Books; 1999. pp. 259–334. [Google Scholar]
- 54.Turner DH, Sugimoto N, Freier SM. Thermodynamics and kinetics of base-pairing and of DNA and RNA self-assembly and helix coil transitions. In: Saenger W, editor. Nucleic Acids. VII/1c. Landolt-Bornstein, Berlin: Springer-Verlag; 1990. pp. 201–27. [Google Scholar]
- 55.Uhlenbeck OC, Martin FH, Doty P. Self-complementary oligoribonucleotides: effects of helix defects and guanylic acid-cytidylic acid base pairs. J Mol Biol. 1971;57:217–29. doi: 10.1016/0022-2836(71)90342-1. [DOI] [PubMed] [Google Scholar]
- 56.Wang GM, Sevick EM, Mittag E, Searles DJ, Evans DJ. Experimental demonstration of violations of the second law of thermodynamics for small systems and short time scales. Phys Rev Lett. 2002;89:050601. doi: 10.1103/PhysRevLett.89.050601. [DOI] [PubMed] [Google Scholar]
- 57.Wang MD, Schnitzer MJ, Yin H, Landick R, Gelles J, Block SM. Force and velocity measured for single molecules of RNA polymerase. Science. 1998;282:902–7. doi: 10.1126/science.282.5390.902. [DOI] [PubMed] [Google Scholar]
- 58.Williams MC, Rouzina I, Bloomfield VA. Thermodynamics of DNA interactions from single molecule stretching experiments. Acc Chem Res. 2002;35:159–66. doi: 10.1021/ar010045k. [DOI] [PubMed] [Google Scholar]
- 59.Zhang WB, Chen S-J. RNA hairpin-folding kinetics. Proc Natl Acad Sci USA. 2002;99:1931–36. doi: 10.1073/pnas.032443099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, et al. A single-molecule study of RNA catalysis and folding. Science. 2000;288:2048–51. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]