

SUMMARY
Alternative splicing of the PKM2 gene produces two isoforms, M1 and M2, which are preferentially expressed in adult and embryonic tissues, respectively. The M2 isoform is reexpressed in human cancer and has nonmetabolic functions in the nucleus as a protein kinase. Here, we report that PKM2 is acetylated by p300 acetyltransferase at K433, which is unique to PKM2 and directly contacts its allosteric activator, fructose 1,6-bisphosphate (FBP). Acetylation prevents PKM2 activation by interfering with FBP binding and promotes the nuclear accumulation and protein kinase activity of PKM2. Acetylationmimetic PKM2(K433) mutant promotes cell proliferation and tumorigenesis. K433 acetylation is decreased by serum starvation and cell-cell contact, increased by cell cycle stimulation, epidermal growth factor (EGF), and oncoprotein E7, and enriched in breast cancers. Hence, K433 acetylation links cell proliferation and transformation to the switch of PKM2 from a cytoplasmic metabolite kinase to a nuclear protein kinase.
INTRODUCTION
Pyruvate kinase (PK) catalyzes the last and a rate-limiting step in glycolysis by transferring a phosphate group from phosphoenolpyruvate (PEP) to ADP to produce pyruvate and ATP. The human genome encodes two distinct PK genes, PKLR and PKM2, which express four PK isoforms: L, R, M1, and M2 (Mazurek, 2011). The L and R isoforms are expressed specifically in liver and red blood cells, respectively, from the PKLR gene through the use of different promoters (Noguchi et al., 1987), whereas M1 and M2 are expressed in most adult tissues and during embryogenesis, respectively, from the PKM2 gene by alternative RNA splicing (Noguchi et al., 1986). PKM1 differs from the other three PK isoforms in that it possesses a high level of activity without the need of allosteric activation by fructose 1,6-bisphosphate (FBP) (Vander Heiden et al., 2010). Notably, PKM2 is highly expressed in tumors of many different types (Mazurek et al., 2005; Yamada and Noguchi, 1995). The mechanism underlying the switch of PKM1-PKM2 alternative splicing remained elusive for a long time but was recently found to be regulated, in part, by Myc. In this study by David et al. (2010), three heterogenous nuclear ribonucleoproteins (hnRNPs), hnRNPA1, hnRNPA2, and hnRNPI (also known as PTB), were found to bind repressively to sequences flanking exon 9 of the PKM2 gene, resulting in exon 10 inclusion and the production of PKM2 mRNA. The expressions of the genes encoding for these three hnRNP are upregulated by Myc, linking the function of the Myc oncogene to the altered activity of this major metabolic enzyme (David et al., 2010).
The significance of selective expression of the M2 isoform in developing embryos and reexpression in tumor cells is not clear at present. There are two different views on how high levels of PKM2 would benefit actively proliferating embryonic and tumor cells. One holds that the switching from constitutive, highly active PKM1 to the FBP-regulated PKM2 allows cells to regulate the FBP binding, through either binding with phosphotyrosine (Christofk et al., 2008a; 2008b) or a conformational change induced by Y105 phosphorylation (Hitosugi et al., 2009), thereby yielding a means of decreasing the activity of PKM2 and the rate of glycolysis and accumulating more glycolytic intermediates for biosynthetic reactions to support cell growth and division.
The other proposes a glycolysis-independent function based on the recent findings that PKM2, but not PKM1, can enter the nucleus, where it acts as a protein kinase and a transcriptional coactivator. Luo et al. reported that PKM2 gene transcription is activated by hypoxia-inducible factor (HIF-1), and PKM2 protein in turn physically interacts with HIF-1α in the nucleus to promote transactivation of HIF-1 target genes, thereby constituting a positive feedback loop that can reprogram glucose metabolism in cancer cells (Luo et al., 2011). Separately, Yang et al. reported that activation of epidermal growth factor receptor (EGFR) induces translocation of PKM2, but not PKM1, into the nucleus, where it binds with β-catenin and is recruited by β-catenin to stimulate cyclin D1 expression (Yang et al., 2011). The pyruvate kinase activity of PKM2 does not seem to be involved in the function of PKM2 in the nucleus as a transcription cofactor. Instead, a different function of PKM2—as a protein kinase—is emerging as important. PKM2 normally presents in the cytoplasm in a homotetramer and acts as a metabolite kinase. Gao et al. reported that PKM2, when existing in a homodimer form, can use PEP as a phosphate donor to phosphorylate tyrosine residue in signal transducer and activator of transcription (STAT3) (Gao et al., 2012). More recently, it was found that PKM2 can directly bind to and phosphorylate histone H3 at residue T11 upon EGFR activation, leading to the dissociation of histone deacetylase 3 (HDAC3) from cyclin D1, as well as Myc promoters, and subsequent acetylation and activation of both growth- and proliferation-promoting oncogenes (Yang et al., 2012).
The mechanisms controlling the switch of PKM2 from a cytoplasmic metabolite kinase to a nuclear protein kinase and how this switch is linked to both mitogenic and oncogenic signaling pathways are not known. The current study is directed to answer these two questions.
RESULTS
PKM2 Is Acetylated at K433 by p300 Acetyltransferase
We and others have recently reported acetylation proteomic analyses and found that most metabolic enzymes in mammalian cells are acetylated, notably including K433 of PKM2 (Choudhary et al., 2009; Kim et al., 2006; Zhao et al., 2010). K433 is encoded by the M2-specific exon 10 and is conserved among all vertebrate PKM2, but not present in PKM1 (Figure S1A available online). K433 binds to the 6′-phosphate oxygen of FBP and is the only residue in the FBP-binding pocket that is different between PKM1 and PKM2 (Dombrauckas et al., 2005; Jurica et al., 1998). Substitution of an analogous FBP-binding residue in yeast PK (T403) by a negatively charged glutamate increased the dissociation constant (KD) for FBP by 540-fold (Bond et al., 2000). Molecular modeling suggests that acetylation at K433, if it occurs, could significantly neutralize the charge on the lysine side chain, causing a steric hindrance of the FBP binding (Figure 1A).
Figure 1. PKM2 Is Acetylated at K433 by p300 Acetyltransferase.
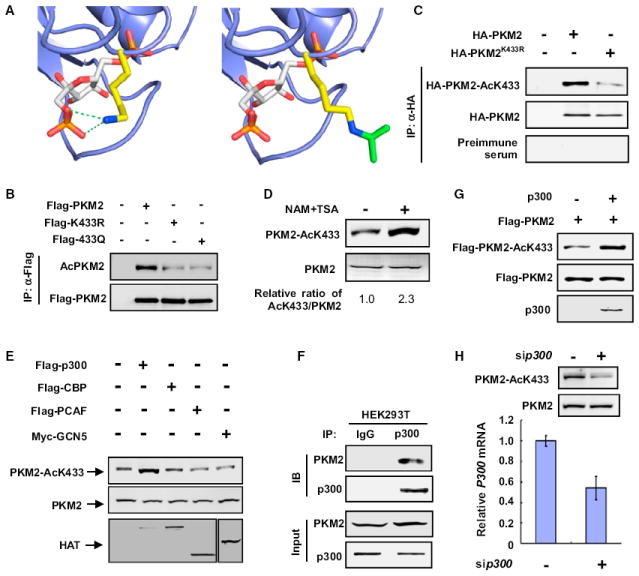
(A) Molecular modeling of acetylation of K433 in PKM2. The FBP binding site of a monomer of PKM2 (protein database [PDB] ID 1T5A) (Dombrauckas et al., 2005) is rendered in the slate blue cartoon. FBP is shown in sticks with carbon atoms colored white, oxygen atoms colored red, and phosphorus atoms colored orange. The side chain of K433 is rendered in stick mode with carbon atoms colored yellow and the nitrogen atom colored blue. The green dashed lines represent critical electrostatic interactions between the lysine side chain and the oxygens of the 6′-phosphate of FBP.
(B) Mutation of K433 decreases PKM2 acetylation. HEK293T cells were transfected with the indicated plasmids, and PKM2 protein was immunoprecipitated (IP), followed by western analysis using either an anti-pan-acetyl lysine or anti-Flag antibody to determine the levels of acetylated and total PKM2 proteins, respectively.
(C) Characterization of acetyl-PKM2 (K433) antibody. The indicated plasmids were transfected into HEK293T cells, and the acetylation level of immunoprecipitated HA-PKM2 was determined by immunoblot using the acetylation site (K433)-specific antibody (AcPKM2[K433]).
(D) Endogenous PKM2 is acetylated at K433. HEK293T cells were treated with TSA and NAM. Endogenous PKM2 protein and acetylation levels were determined by western blotting with the indicated antibodies. The relative ratio of K433-acetylated PKM2 versus total PKM2 was quantified, and the value derived from untreated cells was set to 1.
(E) p300 expression increases PKM2 acetylation at K433. HEK293T cells were transfected with the indicated plasmids, and PKM2 acetylation at K433 was determined by western blot.
(F) Endogenous p300 binds with PKM2. Endogenous coimmunoprecipitation (coIP) of p300 and PKM2 was performed as indicated in the lysate of HEK293T cells, and the association was examined by western analysis.
(G) p300 acetylates PKM2 at K433 in vitro. The recombinant PKM2 was incubated with purified p300 in vitro, the HAT assay kit was purchased from Millipore, and the PKM2 K433 acetylation level was analyzed by western blotting using the antibody recognizing K433-acetylated PKM2.
(H) Knocking down p300 decreases endogenous K433 acetylation of PKM2. HEK293T cells were transfected with siRNA oligonucleotide targeting p300, and the levels of PKM2 protein and K433 acetylation were determined by western blotting. p300 knockdown efficiency was determined by quantitative PCR (qPCR). See also Figure S1.
To confirm that K433 of PKM2 is acetylated, we first replaced K433 with either acetyl-mimetic glutamine (K433Q) or nonacetylatable arginine (K433R) and found that both mutations significantly reduced the overall acetylation of PKM2 when compared with the wild-type protein, suggesting that K433 is a major acetylation site for PKM2 (Figure 1B). To further investigate K433 acetylation, we generated an antibody specific to acetylated K433 and verified its specificity (Figures 1C, S1B, S1C, and S1D). K433-acetylated PKM2 can be readily detected (Figure 1C) and was increased by the treatment of cells with deacetylase inhibitors trichostatin A (TSA) and nicotinamide (NAM) (Figure 1D), demonstrating endogenous acetylation of PKM2 at K433 in cultured cells.
To identify the enzyme responsible for PKM2 K433 acetylation, we examined several acetyltransferases, including p300 (E1A binding protein, 300 kDa), CBP (CREB binding protein), PCAF (p300/CBP-associated factor), and GCN5 (KAT2A). We found that ectopic expression of p300, but not the other three acetyltransferases, increased PKM2 K433 acetylation (Figure 1E). Both ectopically expressed and endogenous p300 bound to PKM2 (Figures 1F and S1E). Pull down experiment further confirmed that p300-PKM2 binding was direct (Figure S1F). p300 directly acetylates PKM2 at K433 in vitro (Figure 1G). Moreover, knocking down p300 significantly reduced K433 acetylation of the endogenous PKM2 (Figure 1H). Together, these results demonstrate that p300 is the acetyltransferase of PKM2 at K433.
Acetylation at K433 Inhibits FBP Binding and Reduces PKM2 Activity
To determine how acetylation of K433 affects PKM2 activity, we immunopurified wild-type and K433 mutants of PKM2 from transfected human embryonic kidney 293T (HEK293T) cells and measured PKM2 activity. We found that FBP potently activated the wild-type PKM2 by 2-fold but had significantly less of an effect (55% increase) on the K433R mutant and virtually no effect on the K433Q mutant (Figure 2A). Both the wild-type and K433R mutant PKM2 bound to FBP (Figure 2B). Notably, substitution of K433 with an acetylation-mimetic glutamine (K433Q) completely disrupted FBP binding. To strengthen this finding, we purified PKM2 from cells cotransfected with p300 and found that coexpression of p300 caused a significant increase of PKM2 acetylation and nearly completely blocked FBP activation of hyperacetylated PKM2 (Figure 2C). Together, these results demonstrate a strong effect of K433 acetylation in blocking FBP binding to and activation of PKM2.
Figure 2. Acetylation at K433 Inhibits FBP Binding to Prevent Dimer-to-Tetramer Transition of PKM2.
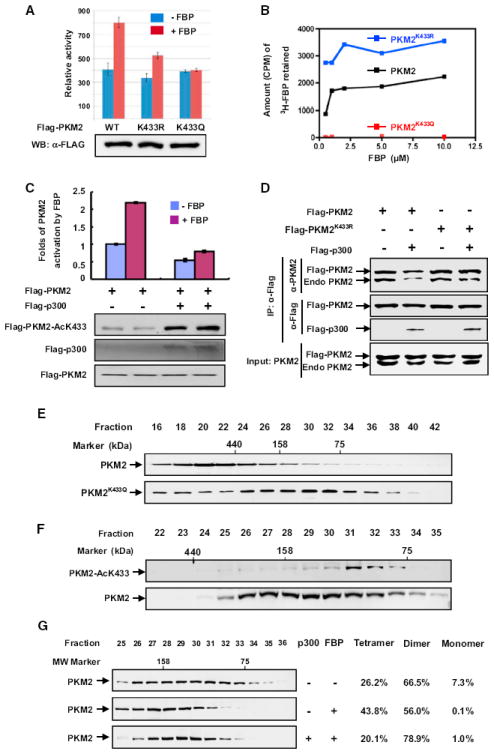
(A) K433 mutations block FBP-induced activation of pyruvate kinase activity of PKM2. Flag-tagged wild-type (WT) and mutant PKM2 proteins were expressed in HEK293T cells and purified by immunoprecipitation. The enzyme activity was determined in the presence (500 μM) or absence of FBP and normalized against protein level. Mean values of relative enzyme activity of triplicate experiments ± SD are presented.
(B) Acetylation-mimetic mutation at K433 disrupts PKM2-FBP binding. FBP-PKM2 binding assays were carried out using 14C-FBP and recombinant PKM2 purified from E.coli as described in the Experimental Procedures. Recombinant PKM2 was incubated with 14C-FBP, and the samples were dialyzed before scintillation counting.
(C) p300 inhibits FBP-induced activation of pyruvate kinase activity of PKM2. Flag-PKM2 was cotransfected with p300 expression plasmid into HEK293T cells. PKM2 was immunopurified, and FBP-induced PKM2 activity was assayed. The mean values of triplicates ± SD are presented.
(D) K433R mutation blocks p300-promoted PKM2 tetramer-dimer transition. Flag-tagged wild-type and K433R mutant PKM2 were transfected into cells, and their interaction with endogenous PKM2 was analyzed by IP-western.
(E) Acetylation-mimetic K433Q mutation is enriched in PKM2 dimer fractions. Recombinant wild-type and K433Q mutant PKM2 proteins were purified from E. coli and separated by gel filtration, followed by western analysis.
(F) The K433-acetylated PKM2 are exclusively present in the dimer fractions. Total cell lysate derived from HEK293T cells was separated by gel filtration, followed by western analysis for total and K433-acetylated PKM2 proteins.
(G) p300 acetyltransferase prevents tetramerization of PKM2. The p300 plasmid was transfected into HEK293T cells, and cell lysate was separated by gel filtration, followed by western analysis. PKM2 tetramer (fractions 25–27), dimer (fractions 28-33), and monomer (fractions 34–36) were quantified using the ImageQuant TL program.
Acetylation at K433 Prevents Dimer-to-Tetramer Transition of PKM2
Whereas PKM1, PKR, and PKL present predominantly in cells as homotetramer, PKM2 presents in both homotetrameric and homodimeric forms. The tetrameric form of PK has significantly higher affinity toward the substrate PEP and is highly active under physiological PEP concentration, while the dimeric form is almost inactive. FBP allosterically activates PKM2 by causing rapid (within minutes) and reversible dimer-to-tetramer conversion in response to glucose (Ashizawa et al., 1991; Kato et al., 1989). The ratio of dimer to tetramer is positively correlated with cell proliferation, cell growth, survival, and transformation (Mazurek, 2011). How cells regulate the dimer-tetramer ratio in sensing nutrient condition or growth factors is not known. To determine if acetylation at K433 regulates FBP-mediated dimer-tetramer conversion, we first determined whether expression of p300 affects PKM2 tetramerization by examining the interaction between ectopically expressed, Flag-tagged wild-type and K433R mutant PKM2 with endogenous PKM2. We found that while overexpression of p300 decreased the interaction between wild-type PKM2, it had no effect on the PKM2(K433R) mutant (Figure 2D). Next, we performed gel filtration analysis of wild-type and acetylation-mimetic K433Q mutant of PKM2 purified from E. coli. Wild-type PKM2 displayed tetramer as the major form, while K433Q mutant PKM2 existed mostly as dimer or monomer (Figure 2E). We then fractionated HEK293T cell lysates by gel filtration and determined the distribution of endogenous K433-acetylated PKM2 by western blotting. While the distribution of total PKM2 spread throughout multiple fractions (ranging from monomer and dimer to tetramer), K433-acetylated PKM2 was only detected in a low-molecular-weight fraction corresponding to dimer (Figure 2F).
These two results indicate a strong effect of acetylation at K433 in preventing the conversion of monomer or dimer to tetramer. Furthermore, while the addition of FBP (as expected) increased the tetrameric form of PKM2 from 26.2% to 43.8%, with a concomitant decrease of the dimeric form from 66.5% to 56.0% and an almost complete disappearance of the monomeric form, it had very little effect on hyperacetylated PKM2 purified from cells overexpressing p300, which reduced the tetrameric form to 20.1% and increased the dimeric form to 78.9% even in the presence of FBP (Figure 2G). Together, these results support the conclusion that acetylation at K433 of PKM2 inhibits FBP binding and conversion of inactive dimer to active tetramer.
PKM2 K433 Acetylation Is Associated with Cell Proliferation
To determine whether K433 acetylation is regulated by cell proliferation, we first examined the levels of PKM2 K433 acetylation in response to serum deprivation and stimulation. We found that serum deprivation of human H1299 non-small cell lung carcinoma cells dramatically reduced PKM2 K433 acetylation, causing a clear reduction within 24 hr and reducing K433 acetylation to a barely detectable low level after 4 days (Figure 3A). Conversely, readdition of serum to serum-starved H1299 cells increased K433 acetylation within 1 hr. Associated with the reduction of K433 acetylation, serum starvation enhanced the activation of wild-type, but not K433R mutant PKM2, by FBP (Figure 3B). Moreover, serum deprivation also increased PKM2 in the tetramer fractions (Figure 3C). To determine how a specific mitogenic signal affects K433 acetylation, we treated cells with EGF and found that EGF treatment significantly increased PKM2 K433 acetylation (Figure 3D). We then examined how K433 acetylation is affected by cell density. H1299 cells were seeded at different densities. and K433 acetylation of PKM2 was determined 2.5 days later. The level of PKM2 K433 acetylation was high in sparse cells, decreased when cells came into contact with each other, and was reduced to an almost undetectably low level after cells reached high density (Figures 3E and S2). Taken together, these results demonstrate that K433 acetylation of PKM2 is decreased in quiescent or contact-inhibited cells and is stimulated upon mitogenic stimulation.
Figure 3. PKM2 K433 Acetylation Is Associated with Cell Growth Stimulation.
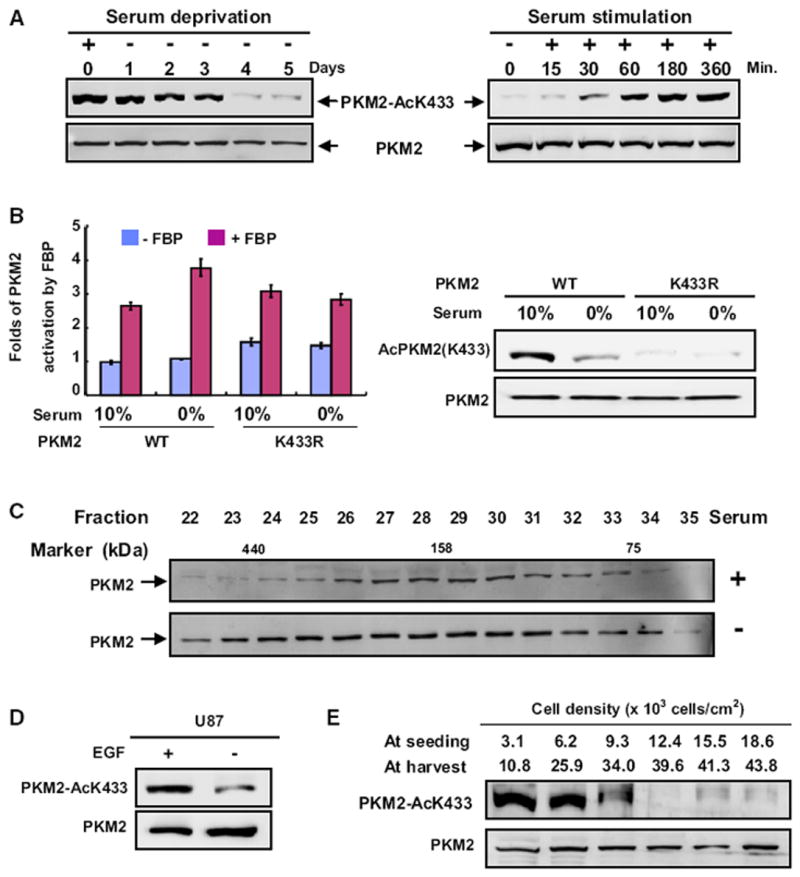
(A) Serum deprivation and stimulation decrease and increase PKM2 K433 acetylation, respectively. H1299 cells were serum starved (0% FBS) for different times or stimulated with the addition of 10% FBS after 2 days of serum starvation. Total cell lysates were analyzed by western analysis as indicated.
(B) Serum starvation enhances FBP activation of the pyruvate kinase activity of wild-type, but not the K433R mutant PKM2. WT and K433R mutant PKM2 proteins were immunopurified from transfected 293T cells cultured in the presence of 0% or 10% FBS, followed by enzyme activity in the presence or absence of FBP. The mean values of triplicates ± SD are presented.
(C) Serum deprivation prevents PKM2 dimerization. H1299 cells were cultured with or without serum for two days, and cell lysates were separated by gel filtration, followed by western analysis.
(D) EGF promotes K433 acetylation of PKM2. U87 cells were starved for 24 hr with 0.5% serum, followed by treatment with EGF (100 ng/ml) for 6 hr, and the PKM2 protein and K433 acetylation levels were determined by western analysis.
(E) PKM2 K433 acetylation inversely correlates with cell density. H1299 cells were seeded at different densities as indicated, cultured for 2.5 days, and the levels of both PKM2 and K433-acetylated PKM2 were analyzed by western blotting. See also Figure S2.
Oncoprotein HPV16-E7 Promotes K433 Acetylation and Inhibits PKM2 Tetramerization
E7 oncoprotein encoded by high-risk human papilloma virus (e.g., HPV16-E7) was previously found to bind with PKM2 (our confirmatory results in Figure S3B) and, through an unknown mechanism, to shift PKM2 to the inactive dimeric state and decrease glycolytic flux rate even in the presence of a high level of FBP (Mazurek et al., 2001; Zwerschke et al., 1999). We found that the expression of HPV16-E7 significantly increased (2.3-fold) the K433 acetylation of PKM2 in a dose-dependent manner (Figure 4A). Consistent with our finding that p300 binds to and acetylates PKM2 (Figure 1) and an early report that E7 binds to p300 (Bernat et al., 2003), HPV16-E7 increased the binding of PKM2 with p300 (Figure 4B), providing a molecular basis for E7-stimulated K433 acetylation. Associated with its activity in promoting K433 acetylation, E7 expression caused a significant shift of wild-type PKM2, but not K433R mutant, from tetramer to dimer (Figure 4C). Importantly, unlike high-risk HPV16-E7, expression of E7 encoded by the low-risk type 11 HPV (HPV11-E7), which is known to have substantially reduced tumorigenic activity (Barbosa et al., 1991; Halbert et al., 1992), had virtually no effect in promoting K433 acetylation of PKM2 (Figure 4D).
Figure 4. Transformation-Competent Oncoprotein HPV16-E7 Inhibits PKM2 Tetramerization and Promotes K433 Acetylation.
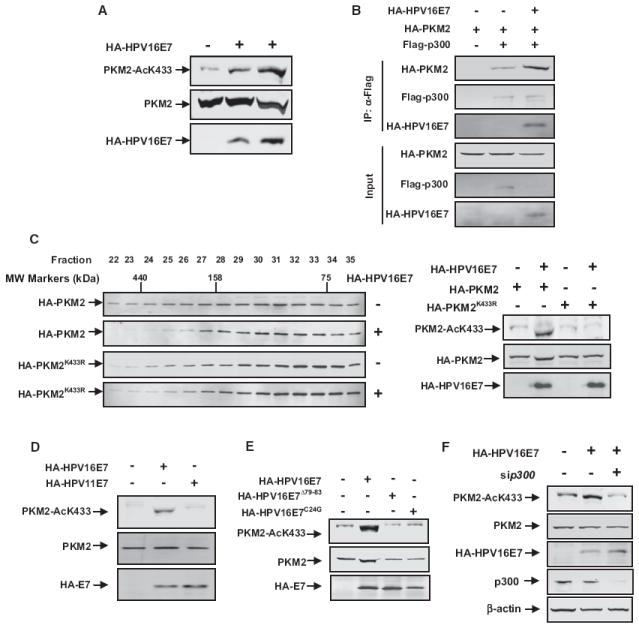
(A) HPV16-E7 promotes K433 acetylation of PKM2. Cell lysates were prepared from 3T3 cells transfected with empty vector or increasing amounts of HPV16-E7, followed by western analysis of total and K433-acetylated PKM2.
(B) HPV16-E7 promotes the interaction between PKM2 and p300. 3T3 cells were transfected with the indicated plasmids. p300-PKM2 interaction was examined by IP-western analysis.
(C) HPV16-E7 prevents tetramerization of wild-type, but not K433R mutant PKM2. Cell lysates were prepared from 1 × 107 cells cotransfected with empty vector or HPV16-E7 with either wild-type or K433R mutant PKM2, separated by gel filtration, followed by western analysis for PKM2 protein and K433 acetylation.
(D) High-risk HPV16-E7, but not low-risk HPV11-E7, promotes K433 acetylation. 3T3 cells were transfected with the indicated plasmids. The expression of individual proteins and K433 acetylation of PKM2 were determined by western analysis.
(E) Transformation-deficient HPV16-E7Δ79-83 and HPV16-E7C24G mutants are unable to promote K433 acetylation. 3T3 cells were transfected with the indicated plasmids. The expression of individual proteins and K433 acetylation of PKM2 were determined by western analysis.
(F) Small interfering p300 (si-p300) blocks HPV16-E7-induced K433 acetylation of PKM2. 3T3 cells were transfected with the indicated plasmids and oligos, and PKM2 acetylation at K433 was determined by western blot. See also Figure S3.
We have also examined two additional transformation-deficient E7 mutants, HPV16-E7C24G and HPV16-E7Δ79–83. C24G mutation reduces p300 binding (Halbert et al., 1992) and annuls E7’s transforming activity by disrupting the LXCXE motif that is conserved in E1A and SV40 transforming T oncoproteins (Figure S3A). HPV16-E7 Δ79–83 lost its ability to bind with PKM2 and cooperate with oncogenic ras in cell transformation (Mazurek et al., 2001; Zwerschke et al., 1999) (Figure S3B). We found that both transformation incompetent E7 mutants were also incapable of promoting K433 acetylation of PKM2 (Figure 4E). Knocking down p300 blocked HPV16-E7-induced PKM2 K433 acetylation, demonstrating that E7 promoting K433 acetylation was p300 dependent (Figure 4F). Together, these results indicate that the acetylation of PKM2 at K433 is stimulated by an oncogenic signal and uncover an activity of E7 oncoprotein in cell transformation.
K433 Acetylation Promotes EGF-Induced Nuclear Translocation of PKM2
Pyruvate kinase normally functions in the cytoplasm as a glycolytic enzyme, but several recent reports have discovered a nonmetabolic function of PKM2 in the nucleus as a protein kinase involved in transcriptional regulation (Gao et al., 2012; Luo et al., 2011; Yang et al., 2011; 2012). The mechanisms regulating the nuclear translocation of PKM2 and converting PKM2 into a protein kinase are both unclear. We carried out cell fractionation and immunofluorescence staining experiments and found that K433-acetylated PKM2 is highly enriched in the nucleus (Figures 5A and 5B).
Figure 5. K433 Acetylation Promotes EGF-Induced Nuclear Translocation of PKM2.
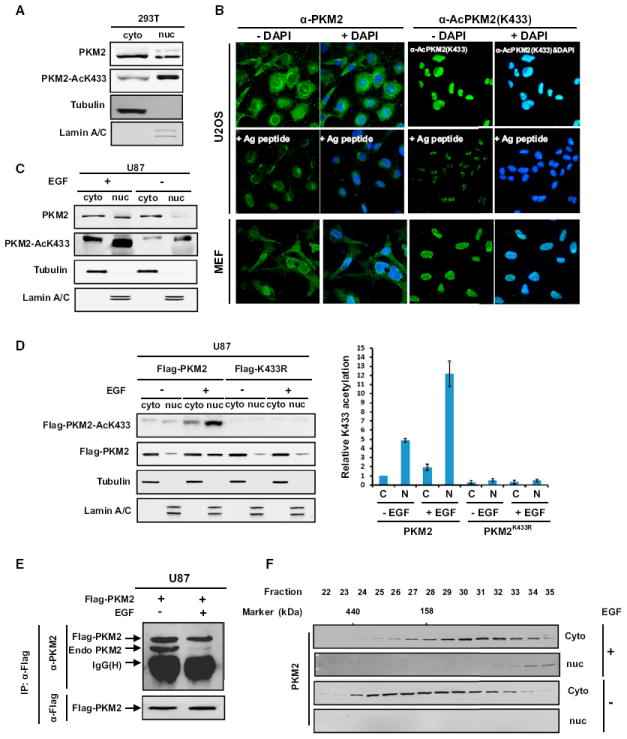
(A) K433-acetylated PKM2 is enriched in the nucleus. Cytosolic and nuclear fractions of lysates derived from 293T cells were separated, and total and K433-acetylated PKM2 were determined by western analysis.
(B) Subcellular localization of total PKM2 and K433-acetylated PKM2 was examined by immunofluorescence microscopy in both human U2OS and mouse embryo fibroblasts (MEFs). Antigen peptides were included to confirm the specificity of both antibodies.
(C) EGF promotes K433 acetylation and nuclear accumulation of PKM2. Serum-starved U87 cells were treated with EGF (100 ng/ml) for 6 hr. Cytosolic and nuclear fractions were separated, and total and K433-acetylated PKM2 were determined by western analysis.
(D) K433 mutation blocks EGF-stimulated PKM2 nuclear translocation. U87 cells were transfected with indicated plasmids for 24 hr, starved for another 24 hr with 0.5% serum + Dulbecco’s modified Eagle’s medium (DMEM), and followed by treatment with EGF (100 ng/ml) for 6 hr. Cells were harvested, cytosolic and nuclear fractions were separated, and Flag-PKM2 protein level and K433 acetylation level were determined by western analysis. Error bars represent ± SD for triplicate experiments.
(E) EGF treatment promotes PKM2 tetramer-dimer transition. U87 cells were transfected with Flag-PKM2 plasmids, followed by starvation for 24 hr and treatment with EGF for 6 hr. The expression of Flag-PKM2 and the binding amount of endogenous PKM2 were determined by western analysis.
(F) EGF treatment promotes PKM2 tetramer-dimer transition and nuclear accumulation. Nuclear and cytoplasmic cell extracts from U87 cells untreated or treated with EGF were fractionated by gel filtration, followed by western blotting analysis to determine the total and K433-acetylated PKM2. See also Figure S4.
To test the model that K433 acetylation promotes PKM2 nuclear translocation in response to mitogenic and oncogenic signals, we treated U87 cells with EGF or transfected 3T3 cells with HPV-16E7 and found that both EGF and E7 significantly promoted nuclear translocation of PKM2 (Figures 5C and S4A). Acetylation-mimetic mutant K433Q accumulated more in the nucleus compared with either wild-type or K433R mutant, which demonstrates the importance of acetylation for this change in localization (Figure S4B). K433R mutation can block EGF-induced PKM2 nuclear translocation (Figures 5D and S4C). Ectopically expressed Flag-PKM2 can bind well with endogenous PKM2 in U87 cells, but not in U87 cells treated by EGF (Figure 5E), suggesting that EGF-stimulated K433 acetylation and disruption of tetramerization of PKM2 is linked to PKM2 nuclear translocation. To directly test this notion, we carried out gel filtration experiments to determine the distributions of different forms of PKM2 in cells with and without EGF treatment. We found that in U87 cells, PKM2 localized predominantly in the cytoplasm mainly as tetramer and was hardly detected in the nucleus (Figure 5F, lower panel). Notably, EGF treatment caused not only a shift of PKM2 from tetramer to dimer and monomer in the cytoplasm, but also the accumulation of PKM2 in the nucleus with nearly all nuclear PKM2 being of the dimeric and monomeric forms (Figure 5F, upper panel). These results support a model that EGF stimulates K433 acetylation to prevent FBP-mediated dimer-to-tetramer transition and promotes PKM2 nuclear translocation.
K433 Acetylation Promotes Protein Kinase Activity of PKM2
It was recently reported that dimeric, but not tetrameric, PKM2 is an active protein kinase that stimulates transcription in the nucleus to phosphorylate Y705 of STAT3 and T11 of histone H3 (Gao et al., 2012; Yang et al., 2012). To determine if K433 acetylation regulates PKM2 tyrosine kinase activity, we first purified recombinant wild-type and K433Q mutant PKM2 proteins and assayed their kinase activity in vitro. We found that the acetylation-mimetic K433Q mutant, but not the wild-type PKM2, displayed very strong tyrosine kinase activity toward Y705 of STAT3 (Figure 6A). Furthermore, nuclear PKM2 that is almost predominantly enriched for K433-acetylated PKM2 exhibited a significantly higher level of tyrosine kinase activity than the cytoplasmic PKM2 (Figure 6B). Consistently, EGF treatment increased K433 acetylation, and this increase is correlated with the increase of protein tyrosine kinase activity of PKM2 toward STAT3 (Figure 6C).
Figure 6. K433 Acetylation Promotes Protein Kinase Activity of PKM2.
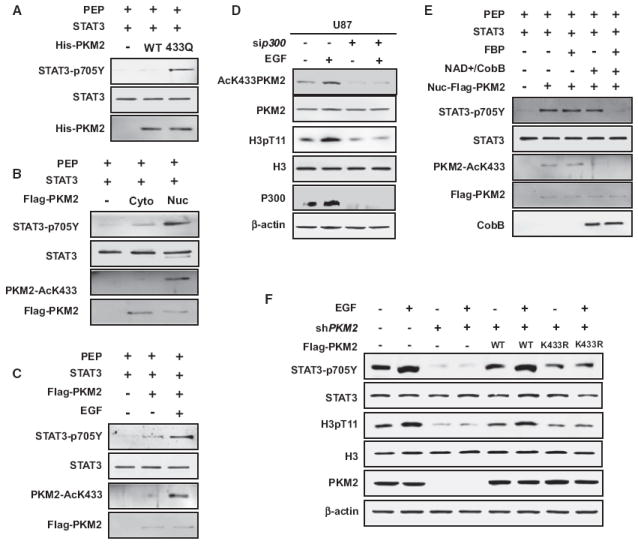
(A) Acetylation-mimetic K433Q mutation stimulates the protein kinase activity of PKM2. The recombinant wild-type and K433Q mutant PKM2 were incubated with purified STAT3, and protein kinase activity was determined by western blotting using the antibody recognizing Y705-phosphorylated STAT3.
(B) Nuclear PKM2 has a higher K433 acetylation level and protein kinase activity. U87 cells were transfected with Flag-PKM2 plasmid and starved for 24 hr with 0.5% serum, followed by treatment with EGF (100 ng/ml) for 6 hr. Cytosolic and nuclear PKM2 were immunopurified and incubated with STAT3. K433 acetylation level and protein kinase activity were determined by western analysis with the indicated antibodies.
(C) EGF promotes K433 acetylation and protein kinase activity of PKM2. U87 cells were transfected with Flag-PKM2 plasmid and serum starved for 24 hr (0.5% FBS), followed by EGF treatment (100 ng/ml) for 6 hr. Flag-PKM2 was immunopurified and incubated with purified STAT3, followed by the assay for protein kinase activity and K433 acetylation by western blotting.
(D) Knocking down p300 blocks EGF-stimulated K433 acetylation and histone H3 T11 phosphorylation. U87 cells were transfected with p300 siRNA or control siRNA. At 24 hr after transfection, cells were starved for another 24 hr (DMEM + 0.5% serum), followed by treatment with EGF (100 ng/ml) for 6 hr. Protein levels were determined by direct immunoblotting with the indicated antibodies.
(E) Acetylation is essential for the protein kinase activity of PKM2. U87 cells were transfected with Flag-PKM2 plasmid and starved for 24 hr with 0.5% serum, followed by treatment with EGF (100 ng/ml) for 6 hr. Cytosolic and nuclear PKM2 were immunopurified and treated with or without CobB in vitro, followed by incubation with purified recombinant STAT3 protein (10 μg/ml) in the presence or absence of FBP (5 mM) and the assay of protein kinase activity.
(F) PKM2 is required for EGF-dependent phosphorylation of STAT3 and H3. The U87/Flag-PKM2 stable cell lines described above were seeded for 24 hr and starved for another 24 hr with 0.5% serum + DMEM, followed by treatment with EGF (100 ng/ml) for 6 hr. Total cell lysates were analyzed by western blotting as indicated.
We then examined the effect of K433 acetylation on the protein threonine kinase activity of PKM2 toward T11 of histone H3. We found that EGF stimulation of serum-starved U87 cells resulted in a clear increase of K433 acetylation of PKM2 and that this increase was correlated with an increase of T11 phosphorylation of H3 (Figure 6D). Notably, knocking down p300 substantially reduced both the K433 acetylation and T11 phosphorylation of H3 (Figure 6D), linking the K433 acetylation to H3 T11 phosphorylation. To directly demonstrate that acetylation is essential for the protein kinase activity of PKM2, we incubated immunopurified nuclear PKM2 with recombinant bacterial deacetylase CobB, followed by the assay of its protein kinase activity toward STAT3 Y705 in the presence and absence of FBP. This experiment demonstrated that deacetylation almost completely abolished the tyrosine kinase activity of PKM2 in a FBP-dependent manner (Figure 6E). This result also supports the model that although K433 acetylation blocks FBP binding and thereby converts PKM2 into a dimeric form that is an active protein kinase, dimeric PKM2 cannot be converted to tetramer form by deacetylation alone in the absence of FBP and thus retains its protein kinase activity. Knocking down of PKM2 ablated EGF-induced STAT3 Y705 and H3 T11 phosphorylation, and the defect caused by PKM2 ablation can be rescued by putting back wild-type PKM2, but not nonacetylatable K433R mutant (Figure 6F). These results demonstrate that K433 acetylation promotes the protein kinase activity of PKM2.
K433 Acetylation Promotes Cell Proliferation and Tumorigenesis
That K433 acetylation of PKM2 is stimulated by both mitogenic and oncogenic signals led us to determine how K433 acetylation affects cell proliferation and tumorigenesis. To this end, we established three H1299 stable lines that knocked down endogenous PKM2 and expressed a Flag-tagged wild-type, K433R or K433Q mutants PKM2 (Figure 7A), followed by the analysis of cell proliferation in culture and tumorigenesis in nude mice. We found that cells expressing acetylation-mimetic PKM2(K433Q) have the highest proliferation rate, followed by wild-type PKM2 and nonacetylatable PKM2(K433R) (Figure 7B), demonstrating a proliferation-promoting function of K433 acetylation.
Figure 7. Increased K433 Acetylation Is Accumulated in the Nucleus during Tumorigenesis.
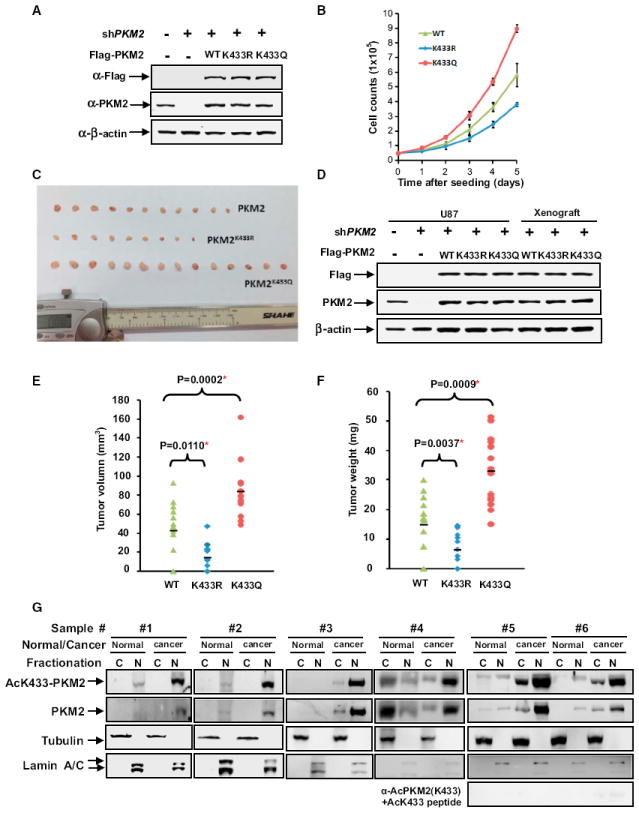
(A) Identification of PKM2 knocking-down and putting-back cell lines. Whole-cell extracts were prepared from PKM2 knocking-down and putting-back cell pools, and PKM2 knockdown efficiency and reexpression were determined by western blot.
(B) Acetylation-mimetic mutant K433Q promotes cell proliferation. Previously identified cells were seeded in each well (5 × 104/well). Cell numbers were counted every 24 hr. Error bars represent ± SD for triplicate experiments.
(C) Acetylation-mimetic mutant K433Q promotes xenograft tumor growth. Nude mice were injected with U87/PKM2 cells, U87/PKM2K433Q cells, and U87/PKM2K433R cells, respectively. The xenograft tumors were dissected and measured after 9 weeks and are shown here.
(D) The expression of PKM2, PKM2 K433Q, and PKM2 K433R in xenograft. Whole-cell extracts were prepared from either original, stable U87/PKM2, U87/PKM2K433Q, and U87/PKM2K433R pools or xenograft tumors, followed by western blot.
(E and F) The quantification of average volume (E) and weight (F) of xenograft tumors is shown. Error bars represent ± SD for 14 tumors.
(G) The K433 acetylation level is higher in the nucleus of breast cancer than in the normal tissue. Fresh breast cancer and matched surrounding normal tissues from the same patients were homogenated, cytosolic and nuclear fractions were separated, and total and K433-acetylated PKM2 was determined by western analysis.
We then established similar U87 stable cell lines and performed a xenograft assay. We found that PKM2(K433Q) cells develop tumors more rapidly than cells expressing wild-type PKM2, while cells expressing PKM2(K433R) exhibited the lowest tumorigenicity (Figures 7C). Sustained and similar levels of expression of putback wild-type, K433Q, and K433R mutants in parental U87 stable cells and xenograft tumors were verified by western blot analysis (Figure 7D). Measurement of the volume and weight of tumors demonstrated that U87/PKM2(K433Q) cells developed tumors significantly larger than those of the U87/PKM2 cells, while U87/PKM2(K433R) cells developed smallest tumors (Figures 7E and 7F). Taken together, these results demonstrated that substitution of K433 with an acetylation-mimetic residue confers tumor cell growth advantage in vivo.
Finally, we determined the expression of PKM2 in human tumors. Total extracts were prepared from six pairs of fresh breast cancer samples and their matched normal tissues from the same patient and were separated into nuclear and cytosolic fractions, followed by western analysis of total and K433-acetylated PKM2. In five out of six samples, the levels of total PKM2 was substantially higher in the tumors when compared to their matched normal tissues. K433-acetylated PKM2 was also expressed at significantly higher levels in these five tumor samples than in their matched normal surrounding tissues and, notably, was predominantly localized in the nucleus (Figure 7G). Collectively, these results demonstrate that K433-acetylated PKM2 is highly expressed in tumors and predominantly localized in the nucleus of breast tumor cells.
DISCUSSION
The key difference between PKM1 and PKM2 is that while PKM1 presents in tetrameric form, possesses a high level of constitutive activity, and is not allosterically regulated, PKM2 requires the binding of FBP for allosteric activation from a dimeric form with low basal activity to active tetrameric form. There are currently two different models explaining how the M2 isoform benefits the actively proliferating cells. One holds that the intrinsic lower pyruvate kinase activity of M2 reduces the rate of glycolysis and thus allows proliferating cells to accumulate various glycolytic intermediates needed for biosynthesis. The other proposes that PKM2 has a previously unrecognized, non-metabolic activity and can enter the nucleus to function as a protein kinase in the regulation of transcription. The mechanisms by which cells regulate the FBP-mediated allosteric activation of PKM2 and the conversion of PKM2 from a cytoplasmic metabolite kinase into a nuclear protein kinase are not known. The findings reported in this study reconcile both views and reveal a unique mechanism, acetylation of K433, that allows cells to regulate both cytoplasmic metabolic function and nuclear protein kinase activity of PKM2. More importantly, we demonstrate that K433 acetylation is linked to both mitogenic and oncogenic signaling pathways. It was recently reported that feeding cells with small molecules that bind to and activate PKM2, but not PKM1, converted a FBP-sensitive allosteric PKM2 into a constitutively active PKM1-like enzyme, resulting in increased pyruvate kinase activity and reduced tumor growth (Anastasiou et al., 2012). These results and our findings demonstrate biologically opposite effects after either promoting or blocking the conversion of FBP-sensitive to a constitutively active enzyme and support a mechanistically consistent notion that a reduced pyruvate kinase activity benefits the tumor cells.
Acetylation of K433 Is Unique to PKM2 and Dominates over FBP Regulation
Several covalent modifications of PKM2 have been previously reported to regulate PKM2, including phosphorylation of Y105 and oxidation of C358, which inhibits the pyruvate kinase activity of PKM2 (Hitosugi et al., 2009; Lv et al., 2011), acetylation of K305, which targets PKM2 for degradation through chaperone-mediated autophagy (Lv et al., 2011), and hydroxylation at P403 and P408, which enhances the ability of PKM2 to stimulate HIF-1 transactivation (Luo et al., 2011). All of these modification sites are either present in PKM1 (K305, C358) or in the other two PK isoforms (K305, C358, P403, and P408). K433 acetylation represents a unique covalent modification that is only specific to PKM2.
Of the 22 amino acid residues that are different between M1-encoded exon 9 and M2-encoded exon 10, K433 is the only residue involved in FBP binding (Dombrauckas et al., 2005; Jurica et al., 1998). We demonstrate that K433 is acetylated by p300 acetyltransferase and that acetylation of K433 directly and effectively blocks FBP binding, leading to the blockage of tetrameric forms, accumulation of dimers, and reduced pyruvate kinase activity of PKM2. The crystal structural analysis showed that the backbone amide of K433 binds directly to the 6′-phosphate oxygens of FBP and plays a critical role in FBP-mediated allosteric activation of PKM2. The electrostatic properties of the side group that occupies position 433 play prominent roles in eliciting allosteric activation by FBP, with positively charged amino acids resulting in FBP activation, and neutral amino acids, such as glutamine substitution or acetylated lysine, inhibit FBP activation. Binding of FBP not only causes a significant conformational change of the FBP-activating loop, but also promotes dimer-dimer intersubunit interface contacts (Dombrauckas et al., 2005). FBP binds PKM2 very tightly and cannot be dissociated from PKM2 by affinity purification, dialysis, or column chromatography (Christofk et al., 2008a; 2008b). Such a tight binding and the pivotal role of the positive charge at position 433 for binding of PKM2 to FBP may explain why cells have evolved acetylation of K433, which not only can effectively block noncovalent FBP binding, but also significantly reduced the positive charge of K433, as a mechanism to regulate the allosteric activation and dimer-tetramer conversion of PKM2.
K433 Acetylation Promotes Nuclear Translocation and Protein Kinase Activity of PKM2
In addition, to reduce the pyruvate kinase activity, acetylation of K433 promotes the translocation into the nucleus and protein kinase activity of PKM2. In vitro deacetylation almost completely abolished the tyrosine kinase activity of PKM2 in a FBP-dependent manner, providing direct evidence supporting the model that K433 acetylation converts the PKM2 form of a metabolite kinase into a protein kinase by blocking FBP-mediated dimer-to-tetramer conversion.
While the basis of how K433 acetylation reduces the pyruvate kinase activity of PKM2–by blocking FBP binding and allosteric activation–is apparent, it is currently not obvious how acetylation of K433 promotes nuclear translocation and protein kinase activity of PKM2. In multiple conditions that we have examined, nuclear translocation and protein kinase activity of PKM2 are always concurrently linked to K433 acetylation, including serum and EGF activation, expression of E7, cell-cell contact inhibition, and tumorigenesis. Such a tight correlation suggests that these two events are most likely controlled by a single change, as opposed to being regulated separately. We speculate that dimeric PKM2, which is induced and maintained by the K433 acetylation, possesses a different conformation from the FBP-bound tetrameric form that allows PKM2 to translocate to the nucleus by either opening a nuclear localization signal (NLS) concealed in the tetramer or allowing the binding with a nuclear protein that shuttles PKM2 into the nucleus. Likewise, the conformational change from tetramer to dimer could also allow PKM2 to bind with amino acid residues in proteins that would be inaccessible to the catalytic site in the tetramer.
K433 Acetylation Is Activated by Both Mitogenic and Oncogenic Signals and Promotes Cell Proliferation and Tumorigensis
While much attention on recent PKM2 research has been focused on its reexpression in tumors, PKM2 has long been known for its selective expression in actively proliferating cells and developing embryos. How both normal mitogenic and oncogenic signals regulate the activity of PKM2 is not known. The results presented here demonstrate that K433 acetylation of PKM2 is directly regulated by both mitogenic and oncogenic signals and promotes cell proliferation and tumorigenesis. First, K433 acetylation is downregulated in quiescent or contact-inhibited cells and activated when quiescent cells are stimulated to enter the cell cycle by either serum or EGF. Second, acetylation of K433 is stimulated by high-risk wild-type E7 oncoprotein, but not low-risk or transformation-deficient E7. Third, cells expressing acetylation-mimetic PKM2(K433Q) proliferate faster in culture and develop tumors more rapidly in mice. Finally, K433-acetylated PKM2 protein presents itself in breast cancer cells at levels substantially higher than those in surrounding normal breast tissues. Notably, during all of these regulations, an increase of K433-acetylated PKM2 is associated with the accumulation of PKM2 in the nucleus and the increases of both tyrosine and threonine kinase activities.
We propose that during active cell proliferation or oncogenic transformation, PKM2 is acetylated at K433 by p300 acetyltransferase, which blocks FBP binding and thereby prevents the conversion of PKM2 into the tetrameric form to become an active glycolytic enzyme in the cytoplasm, leading to the translocation of the dimeric form of PKM2 into the nucleus, where it acts as a protein kinase to regulate gene expression (Figure S5).
EXPERIMENTAL PROCEDURES
Measurement of Pyruvate Kinase Activity
Pyruvate kinase activity was measured by a continuous assay coupled to lactate dehydrogenase (LDH). The change in absorbance resulting from nicotinamide adenine dinucleotide (NADH) oxidation was measured using an F-4600 Fluorescence Spectrophotometer (HITACHI). A total of 5 mg of pCMV2B-Flag-PKM2 plasmids was transfected to HEK293T cells for 24 hr, followed by cell lysing, immunoprecipitation using Flag beads (Sigma), and elution by Flag peptide (100 μg/ml; Sigma). Assays for PK activity were carried out in a buffer containing purified Flag-PKM2 protein (100–200 ng), Tris-HCI (pH 7.6), imidazole (50 mM), KCI (120 mM), MgSO4 (60 mM), ADP (1.5 mM), PEP (1.5 mM), and NADH (25 μM), with or without FBP (500 μM) and LDH (10 units). Flag-PKM2 protein was added last. ADP (A2754), PEP (P0564), NADH (N8129), FBP (47810), and LDH (L2500) were all purchased from Sigma.
Gel Filtration
Cells were transfected with the indicated plasmids for 24 hr and then extracted in a lysis buffer containing 50 mM Tris (pH 7.5), 150 mM NaCI, 0.3% Nonidet P-40, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride (PMSF) for 30 min and centrifuged (15 min at 13,000 rpm) to remove cell debris. The gel filtration column (Superdex 200; Amersham Biosciences) was washed and equilibrated by cold PBS (4°C). Extracts were passed over the gel filtration column. The speed rate of flow is 0.5 ml/min. Fractions were collected every 0.5 ml per tube and analyzed by western blot. Molecular mass was determined by Gel Filtration Calibration Kit HMW (GE Healthcare).
In Vitro Protein Kinase Assay
The recombinant Flag-PKM2(pCMV2B-Flag-PKM2) and His-PKM2(pSJ3-His-PKM2) proteins (10 μg/ml) were purified from U87 cells and bacteria by immunoprecipitation using Flag beads (Sigma) and Ni-NTA beads (GE Healthcare), respectively, and incubated with purified STAT3 (10 μg/ml, OriGene China) under different conditions as previously described (Gao et al., 2012). Protein kinase activity was analyzed by western blotting for the level of total and Y705-phosphorylated STAT3.
Cell Proliferation and Xenograft Studies
For cell proliferation assay, 5 × 104 H1299 cells were seeded in triplicate in 6-well plates, and cell numbers were counted every 24 hr over a 5 day period. For xenograft assay, nude mice (nu/nu, 6- to 8-week-old males) were injected subcutaneously with 4 × 106 U87 cells. Around 9 weeks after injection, the tumors were dissected, and the volume and weight of tumors were measured. All animal-related procedures were performed under the Division of Laboratory Animal Medicine regulations of Fudan University.
Detailed experimental procedures can be found in the Supplemental Information.
Supplementary Material
Acknowledgments
We thank the members of the Fudan Molecular and Cell Biology laboratory for discussions throughout this study. This work was supported by 973 (2011CB910600, 2009CB918401), NSFC (31071192, 81225016, 81201540), NCET-09-0315, Shanghai Key basic research program (12JC1401100), “100 Talents” Program of Shanghai Health (XBR2011041), Scholar of “Dawn” Program of Shanghai Education Commission, and Shanghai Outstanding Academic Leader (13XD1400600) to Q.-Y.L. This work was also supported by the 985 Program, the Shanghai Leading Academic Discipline Project (project number B110), and NIH grants (to Y.X. and K.-L.G.). This work is dedicated to the memory of Zhen Yu, who prepared the K433 acetylation antibody.
Footnotes
AUTHOR CONTRIBUTIONS
L.L. and Y.-P.X. performed molecular cell biology experiments. D.Z. assayed enzyme activity. F.-L.L. established the stable cell lines. W.W. performed the FBP-PKM2 binding assay. N.S. characterized the anti-AcPKM2(K433) antibody and performed the IF experiment. Y.J. characterized and provided breast tumor samples. X.Z. and T.-T.L. assisted the xenograft experiment. K.-L.G., Q.-Y.L., and Y.X. conceived the study, designed the experiments, analyzed the data, and wrote the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2013.09.004.
References
- Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashizawa K, McPhie P, Lin KH, Cheng SY. An in vitro novel mechanism of regulating the activity of pyruvate kinase M2 by thyroid hormone and fructose 1, 6-bisphosphate. Biochemistry. 1991;30:7105–7111. doi: 10.1021/bi00243a010. [DOI] [PubMed] [Google Scholar]
- Barbosa MS, Vass WC, Lowy DR, Schiller JT. In vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. J Virol. 1991;65:292–298. doi: 10.1128/jvi.65.1.292-298.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernat A, Awakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22:7871–7881. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- Bond CJ, Jurica MS, Mesecar A, Stoddard BL. Determinants of allosteric activation of yeast pyruvate kinase and identification of novel effectors using computational screening. Biochemistry. 2000;39:15333–15343. doi: 10.1021/bi001443i. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008a;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008b;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 1998;6:195–210. doi: 10.1016/s0969-2126(98)00021-5. [DOI] [PubMed] [Google Scholar]
- Kato H, Fukuda T, Parkison C, McPhie P, Cheng SY. Cytosolic thyroid hormone-binding protein is a monomer of pyruvate kinase. Proc Natl Acad Sci USA. 1989;86:7861–7865. doi: 10.1073/pnas.86.20.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Mazurek S, Zwerschke W, Jansen-Durr P, Eigenbrodt E. Metabolic cooperation between different oncogenes during cell transformation: interaction between activated ras and HPV-16 E7. Oncogene. 2001;20:6891–6898. doi: 10.1038/sj.onc.1204792. [DOI] [PubMed] [Google Scholar]
- Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986;261:13807–13812. [PubMed] [Google Scholar]
- Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. The L-and R-type isozymes of rat pyruvate kinase are produced from a single gene by use of different promoters. J Biol Chem. 1987;262:14366–14371. [PubMed] [Google Scholar]
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Noguchi T. Alteration of isozyme gene expression during cell differentiation and oncogenesis. Nippon Rinsho. 1995;53:1112–1118. [PubMed] [Google Scholar]
- Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerschke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen-Dürr P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci USA. 1999;96:1291–1296. doi: 10.1073/pnas.96.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.