

Abstract
Both genetic and environmental factors play important roles in the pathophysiology of schizophrenia. Although prenatal hypoxia is a potential environmental factor implicated in schizophrenia, very little is known about the consequences of combining models of genetic risk factor with prenatal hypoxia. Heterozygous reeler (haploinsufficient for reelin; HRM) and wild-type (WT) mice were exposed to prenatal hypoxia (9% oxygen for two hr) or normoxia at embryonic day 17 (E17). Behavioral (Prepulse inhibition, Y-maze and Open field) and functional (regional volume in frontal cortex and hippocampus as well as hippocampal blood flow) tests were performed at 3 months of age. The levels of hypoxia and stress-related molecules such as hypoxia-inducible factor-1 α (HIF-1α), vascular endothelial factor (VEGF), VEGF receptor-2 (VEGFR2/Flk1) and glucocorticoid receptor (GR) were examined in frontal cortex and hippocampus at E18, 1 month and 3 months of age. In addition, serum VEGF and corticosterone levels were also examined. Prenatal hypoxia induced anxiety-like behavior in both HRM and WT mice. A significant reduction in hippocampal blood flow, but no change in brain regional volume was observed following prenatal hypoxia. Significant age and region-dependent changes in HIF-1α, VEGF, Flk1 and GR were found following prenatal hypoxia. Serum VEGF and corticosterone levels were found decreased following prenatal hypoxia. None of the above prenatal hypoxia-induced changes were either diminished or exacerbated due to reelin deficiency. These results argue against any gene-environment interaction between hypoxia and reelin deficiency.
Keywords: reelin, prenatal hypoxia, mice, schizophrenia, VEGF
1. Introduction
A history of maternal obstetric complications has been implicated in the pathophysiology of schizophrenia (Haukvik et al., 2010). Meta-analyses have shown that hypoxia, or low oxygen, is the most highly correlated insult documented for birth complications in schizophrenia (Van Erp et al., 2002; Demjaha et al., 2011). Moreover, the responses of blood vessels following hypoxia, which cause fluctuations in cerebral blood flow and metabolic demand, have been suggested to contribute to the protection and vulnerability in the developing brain (Baburamani et al., 2012). Studies in rodents have shown that chronic hypoxia leads to anatomical changes that strongly resemble volume loss, decreased myelination, and enlarged ventricles often observed in preterm babies (Mayoral et al., 2009). Interestingly, the many of above anatomical abnormalities are also observed in schizophrenia subjects (Asami et al., 2012; Andreasen et al., 2013).
Hypoxia inducible factor-1 (HIF-1) is the primary transcription factor that plays an integral role in the body's response to hypoxia (Semenza, 2012). HIF-1 is a key regulator responsible for the induction of genes that facilitate adaptation and survival of cells from normoxia (∼21% O2) to hypoxia (∼1% O2) (Semenza, 1998). Although a large number of genes have been shown to be regulated by hypoxia challenge, the vascular endothelial cell growth factor (VEGF) is the most potent HIF-1 regulated gene (Levy et al., 1995; Bunn and Poyton, 1996; Forsythe et al., 1996; Berra et al., 2000; Giordano and Johnson, 2001). VEGF is a neuroprotective, angiogenic and neurotrophic molecule (Zachary, 2005). VEGF is known to mediate its biological functions via activation of the protein tyrosine kinase receptors, VEGF receptor 1 (VEGFR-1/Flt1) and VEGFR-2 (KDR/Flk1) (Quinn et al., 1993). VEGF and its receptors are expressed on neurons and astrocytes, and VEGF induces neuronal outgrowth. Flk1 has been shown to mediate VEGF action in neuronal functions (Jin et al., 2002). Interestingly, subjects with schizophrenia have deficits in blood flow, altered regional brain volume, impaired myelination and reduced cortical VEGF mRNA levels (Hanson and Gottesman, 2005; Fulzele and Pillai, 2009).
In the present study, we investigated whether prenatal hypoxia exacerbates neurochemical, functional and behavioral deficits in adolescence when combined with a genetic risk factor of schizophrenia. We have used heterozygous reeler mice haploinsufficient for reelin (HRM), which have many behavioral and neurochemical deficits similar to schizophrenia (Costa et al., 2002). Reelin plays a central role in brain development and synaptic plasticity (Nullmeier et al., 2011) and significant reductions in reelin mRNA and protein expression have been reported in prefrontal cortex and hippocampus of schizophrenia subjects (Impagnatiello et al., 1998; Fatemi et al., 2000). Interestingly, a recent study has shown that prenatal hypoxia decreases reelin expression in the hippocampus of adult offspring mice when hypoxia (9% oxygen) was induced for 2 hours (Golan et al., 2009). We have utilized the above methodology for hypoxia induction on embryonic day 17 (E17) as this time point corresponds to the second trimester in humans (80-110 days post conception), which is most vulnerable to prenatal stress (Oliver and Davies, 2009).
We have analyzed the effect of prenatal hypoxia on behavioral (Prepulse inhibition, Y-maze and Open field) and functional (regional volume in frontal cortex and hippocampus as well as hippocampal blood flow) tests at 3 months of age. In neurochemical studies, the protein levels of hypoxia-related proteins such as HIF-1α, VEGF and Flk1, were examined in frontal cortex and hippocampus at E18, 1 month and 3 months of age. In addition, the effects of prenatal hypoxia on glucocorticoid receptor (GR) and corticosterone levels were also examined.
2. Experimental Procedures
Animals
All animal experiments were performed in accordance to Georgia Regents University's Committee on Animal Use for Research. Two original mating pairs of HRM were obtained from Jackson Laboratories (Bar Habor, ME, USA), and an in-house HRM colony was created. All the experiments were performed in HRM and wild-type mice (WT) littermates. Pregnant dams exposed to hypoxia were not used for subsequent breeding.
Serum Corticosterone
Mice were killed by decapitation in the early afternoon, and trunk blood was harvested and serum collected for analyses. Serum was assayed for corticosterone using a quantitative competitive enzyme immunoassay kit according to manufacturer's instructions (KGE008, R&D Systems). The reading absorbance was 450nm and corrected at 570nm, with a minimal detectable dose of 0.071ng/mL. The intra/inter-assay variation was 5.4%-21.2%.
Serum VEGF
VEGF protein levels were determined using a quantitative sandwich enzyme immunoassay kit (MMV00, R&D Systems). The reading absorbance was 450nm, and corrected at 570nm with a minimal detectable dose of 3 pg/mL. The intra/inter-assay variation was 4.3%-8.4%.
Genotyping
Animals were genotyped using the DNA extraction HOT SHOT method (Truett et al., 2000). The following Reelin primers used were: Wildtype Reelin RP 5′ACA GTT GAC ATA CCT TAA TC 3′; Reelin FP 5′TAA TCT GTC CTC ACT CTG CC 3′; Reelin RP 5′TGC ATT AAT GTG CAG TGT TG 3′. Samples were analyzed by PCR and run on a 2% agarose gel for UV determination of bands using ethidium bromide.
In Vivo Hypoxia
Timed pregnant mice were allowed to gestate to E17, at which time the pregnant dams underwent exposure to a controlled 9% Oxygen environment in a hypoxia chamber (BioSpherix, NY, USA) for two hours as previously described (Golan et al., 2009).
Sample Preparation
Frontal cortex and hippocampus samples were separately dissected at different time points and homogenized in ice-cold RIPA buffer (Teknova, Cat #R3792) contained additives such as EDTA-Free Halt Protease Inhibitor Cocktail (Thermo, Cat #78245; 100uL/10mL), sodium fluoride (200mM; 250uL/10mL), sodium orthovanadate (200mM; 100uL/10mL) and Phenylmethylsulfonyl fluoride (PMSF) (Sigma, P-7626, 100mM in ethanol; 100uL/10mL). After 15-min incubation period on ice, the extracts were clarified by centrifugation at 10000 g for 15 min at 4°C and stored at -70°C. Protein concentrations were determined by the bicinchoninic acid (BCA Protein Assay Kit, Sigma).
Western Blotting
Equal amounts of protein were resolved in SDS-polyacrylamide gels and transferred electrophoretically onto a 0.45-micron nitrocellulose membrane (Bio-Rad). Membranes were blocked for 1 h in TBST (10mM Tris-HCl, pH 8.0, 138mM NaCl, 2.7mM KCl, and 0.05% Tween-20) and 5% non-fat milk or bovine serum albumin (BSA) and incubated overnight with the indicated antibodies. After washing with TBST, the membranes were incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies in TBST and 5% non-fat milk. Secondary antibodies were washed off with TBST, and proteins were visualized by enhanced chemiluminescence. The densitometric values for the proteins of interest were corrected using β-actin or β-tubulin. Image J software (Wayne Rasband, NIH, USA) was used for densitometric analysis. The primary antibodies used in this study were: Reelin (1:1000; MAB5364), VEGF (1:6000; ABS82) (both were from Millipore, Billerica, MA, USA); Flk1 (1:1000; SC-504), HIF1-α (1:1000; SC-10790) and GR (1:1000; SC-1003) (Santa Cruz, Santa Cruz, CA, USA); β-actin (1:10,000; A4700; Sigma, St Louis, MO, USA) and β-tubulin (1:10,000; CS2146, Cell Signaling, Danvers, MA, USA). The secondary antibodies used were anti-Rabbit IgG (SC-2317; Santa Cruz) and anti-Mouse IgG (CS7076; Cell Signaling).
Prepulse Inhibition (PPI) test
Mice at 3 months of age were tested for PPI. On the first day, mice were allowed to habituate to the testing chamber apparatus (San Diego Instruments, San Diego, CA) for 20 minutes. The second day consisted of the mice being exposed to the various frequency auditory stimuli, randomized for a total of 24 trials to acclimate the mice to the startle and prepulse stimuli. The third day the mice underwent testing for a total of 60 psuedorandomized trials. These trials were set up such that no prepulse amplitude repeated itself consecutively or have the three prepulse stimuli in consecutive order (i.e. 75, 80, 85 dB). A continuous background noise of 70 dB was used. The nulstim background noise was 70 dB with duration of 40ms. Pre-pulse values were 75, 80, and 85 dB at 20ms durations. Startle amplitude of 120 dB at 40ms was used. The inter-stimulus interval between the prepulse/startle presentations ranged from 10 to 30 secs and the total experimental session lasted about 23 min.
Open Field test
Mice at 3 months of age were tested for habituation, anxiety and baseline activity behaviors using the Open Field apparatus (10.75″ × 10.75″ × 8″, Med Associates, St Albans, VT). Briefly, the mice were allowed to freely roam for 30 minutes. The center zone was defined as the center 6 inches. Movement was determined by laser beam breaks on horizontal and vertical axes. Data of interest included total distance traveled, distances traveled in the center zone, as well as time spent in the center zone. Total distance traveled gave an indication for baseline activity. Mice that spent more time in the center zone were considered less anxious than those who spent most of their testing time in the peripheral zone.
Y-maze test
The mice were tested sequentially so that each mouse underwent the testing at equal time lapses (i.e. mice were not tested repeatedly for the total number of trials before moving to the next mouse). The mice were allowed to feed for 3 hours/day for two days before habituation trials began. Subsequent food deprivation days allowed the mice to feed for 1 hour after training trials were complete. The mice were allowed to habituate to the testing apparatus for a total of 3 trials, 2 minutes each, for two days (total of 6 exploration trials). Training trials consisted of 4 trials per day for 3 days. Probe days allowed the mice 3 more training trials and then a probe/delay trial. The trials were pseudo-randomized, having each arm closed equally per day. The subject was held at the base of the Y in a box for 30 seconds. Habituation trials had both arms baited and open. During training and testing trials, both arms are baited, however one arm was blocked, forcing the subject to explore and retrieve the reward in the open arm. The subject was coerced back to the base of the maze and held again for 30 seconds, during which both arms are opened and baited. The subject was then released and allowed to choose which arm to explore. However, if the mouse went down the incorrect arm (the one previously visited), it was stopped before reaching the reward. If the mouse failed to complete the task in the allotted time (2 minutes), it was removed from the Y maze apparatus. The mice were scored on a 0 (incorrect choice or failure to complete task) or 100 (correct choice) basis.
MRI
All images were acquired on a 7.0 T, 20 cm bore MRI Spectrometer with a micro-gradient insert (950 mT/m) and a 35 mm volume coil to transmit and receive at 1H frequency. The T1-weighter images were generated using a MDEFT pulse sequence: matrix=128×182; TE/TR=3.5/3500 ms; slice thickness=0.45 mm; FOV=1.8×1.8 cm; NA=2. The perfusion images were generated using a FAIR-RARE pulse sequence: matrix=96×96; TE/TR =35.8/1200 ms; FOV=1.92×1.92 cm; NA=4. The perfusion images were analyzed with arterial spin labeling macro in the ParaVision 5.1 software package. Volumetric analysis was obtained using Image J software.
Statistical Analysis
Data are expressed as mean ± SE. A three-way ANOVA with post hoc Bonferroni's test was used for the PPI analysis. A Two-way ANOVA or one-way ANOVA with post hoc Bonferroni's multiple comparison procedure was used for other statistical analyses. Probability (P) values of less than 5% were considered significant.
3. Results
We did not find any significant effect of hypoxia or genotype on weight gain of the dams during pregnancy, litter size or sex ratio (data not shown).
Effect of prenatal hypoxia on sensorimotor gating in PPI
PPI scores were analyzed using three-way ANOVA with prepulse intensities as a within-subject factor, and genotype and treatment as between-subject factors. We found a significant effect of treatment [F(1,40)=19.03; p<0.0001], prepulse intensity [F(2,80)=163.45; p<0.0001] and treatment × genotype × prepulse intensity interaction [F(2,80)=5.687; p=0.004]. Genotype, treatment × genotype interaction, treatment × prepulse intensity interaction or genotype × prepulse intensity interaction was not significant. Further post-hoc analysis showed a significant increase in %PPI at 75 dB in hypoxia treated WT mice (Fig 1A; p=0.019). We found a small, but non-significant increase in %PPI at 80 dB in both HRM and WT following hypoxia as compared to normoxia treated WT mice (Fig 1A). Moreover, a marked increase in %PPI at 85 dB was found in HRM following hypoxia as compared to normoxia treated HRM.
Figure 1.
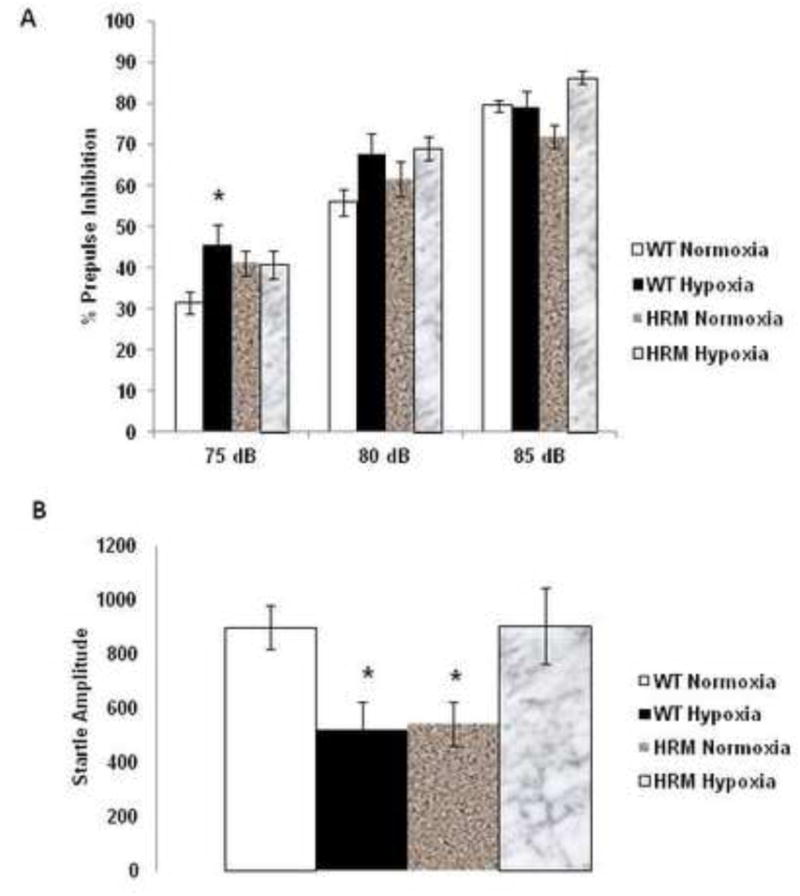
Effects of prenatal hypoxia on Prepulse inhibition (PPI) of the auditory startle response in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 and behavioral tests were performed at 3 months of age. Data represent mean±SE expressed as (A) %PPI and (B) startle response. * P < 0.05 vs WT Normoxia; Bonferroni's test.
Two-way ANOVA on startle amplitude showed a significant main effect of treatment × genotype interaction [F(1,42)=15.407; p<0.0001], but no effect of genotype [F(1,42)=0.065; p=0.799] or treatment [F(1,42)=0.033; p=0.857]. Subsequent post-hoc analysis indicated a significant decrease in startle amplitude in HRM normoxia group, which is in agreement with a previous report (Qiu et al., 2006) (Fig 1B; p<0.05). Furthermore, hypoxia resulted in a significant reduction in startle amplitude in WT, whereas it normalized the reduction in startle amplitude observed in HRM (Fig 1B; p<0.05).
Effect of prenatal hypoxia on working spatial memory in Y-maze
Training trials consisted of 4 trials per day for 3 days using 30 second delays (Fig 2A). Two-way ANOVA revealed a significant main effect of genotype [F(1,42)=8.04; p=0.005] and treatment [F(1,42)=4.081; p=0.045], but no effect of treatment × genotype interaction [F(1,42)=0.226; p=0.635] on the percentage of correct choices. We found that both normoxia and hypoxia treated HRM performed significantly better than the normoxia treated WT in the 30 second interval (Fig 2B; p<0.05). However, we did not find any significant difference in percentage of correct choices between groups in the 7 minute probe trial (Fig 2B).
Figure 2.
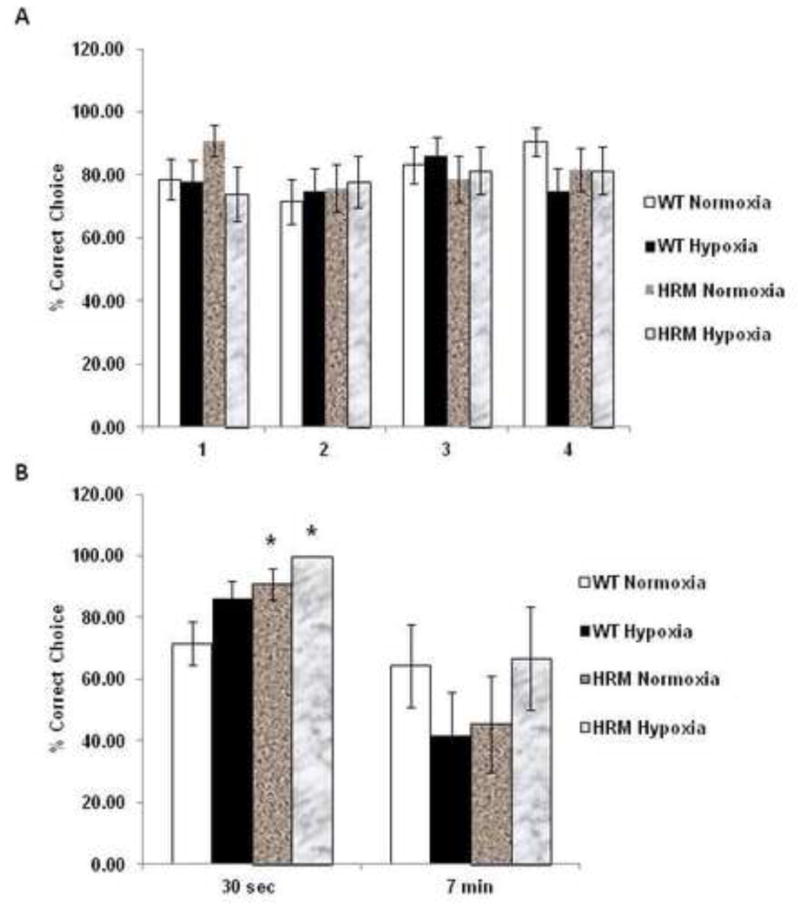
Effects of prenatal hypoxia on working memory (Y-maze) in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 and behavioral tests were performed at 3 months of age. Data represent mean±SE expressed as % correct choice in (A) Training Trials consist of 4 trials per day for 3 days using 30 second delay times and (B) Probe Trials consist of three 30 second trials followed by one 7 min delay probe trial. * P < 0.05 vs WT Normoxia; Bonferroni's test.
Effect of prenatal hypoxia on anxiety-like behavior in open field test
We did not find any significant difference in baseline activity (measured as total distance traveled) between WT and HRM at 3 months of age (Fig 3A). Statistical analysis of the data on time spent in center by two-way ANOVA with hypoxia/normoxia and wild-type/heterozygous as main effects revealed a significant main effect of treatment [F(1,42)=20.38; p<0.0001], but no significant effect of genotype [F(1,42)=0.025; p=0.874] and treatment × genotype interaction [F(1,42)=0.025; p=0.875]. Subsequent post-hoc analysis showed that both WT and HRM hypoxia-treated mice spent significantly less time in the center than their corresponding normoxia treated mice (Fig 3B; p<0.05). Similarly, the data on the distance traveled in the center showed a significant main effect of treatment [F(1,42)=10.86; p=0.002], but no significant effect of genotype [F(1,42)=0.169; p=0.683] and treatment × genotype interaction [F(1,42)=0.238; p=0.628]. We observed that hypoxia-treated mice traveled less distance in the center zone as compared to normoxia-treated mice (Fig 3C; p<0.05). These data suggest that hypoxia may induce an anxiety behavioral phenotype during late adolescent/early adulthood.
Figure 3.
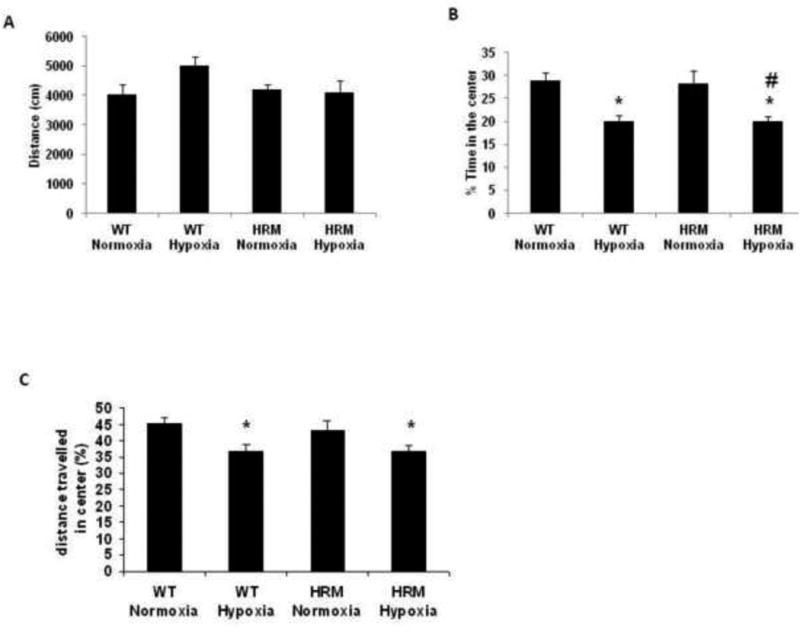
Effects of prenatal hypoxia on anxiety-like behavior (Open field Test) in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 and behavioral tests were performed at 3 months of age. (A) The total ambulatory distance, (B) the % time-spent in the center for the entire session and (C) the ambulatory distance in the center. Data represent mean±SE. * P < 0.05 vs WT Normoxia and P < 0.05 vs HRM Normoxia; Bonferroni's test.
Effect of prenatal hypoxia on blood flow and brain region volume
Two-way ANOVA on hippocampal blood flow revealed a significant main effect of treatment [F(1,9)=25.59; p=0.001] and treatment × genotype interaction [F(1,9)=7.087; p=0.028], but no significant effect of genotype [F(1,9)=0.028; p=0.869]. No significant difference in hippocampal blood flow was observed between normoxia-treated HRM and WT (Fig 4A). However, significant reductions in hippocampal blood flow were found in WT and HRM following hypoxia (p<0.05). We did not find any treatment or genotype effect on volume in frontal cortex or hippocampus (Fig 4B).
Figure 4.
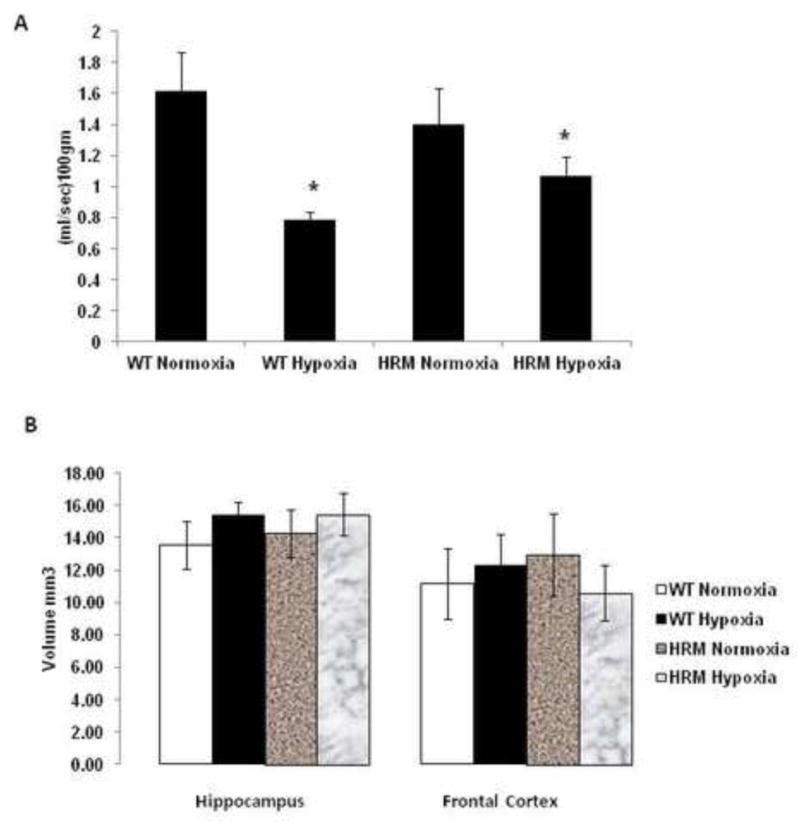
Effect of prenatal hypoxia on blood flow and brain region volume in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 and functional tests were performed at 3 months of age. (A) Hippocampal blood flow and (B) Hippocampus or frontal cortex volume based on MRI analysis. Data represent mean±SE. * P < 0.05 vs WT Normoxia; Bonferroni's test.
Effect of prenatal hypoxia on reelin protein levels
At E18, the two-way ANOVA showed a significant main effect of treatment [F(1,10)=30.97; p=0.0002] and genotype [F(1,10)=238.9; p<0.0001], but no effect of treatment × genotype interaction [F(1,10)=0.001; p=0.972] on reelin protein levels. Post-hoc analysis indicated that hypoxia significantly reduced reelin levels in forebrain of HRM and WT at 24 h following hypoxia (Fig 5A; p<0.05). The data on reelin levels in frontal cortex at 1 month following hypoxia showed significant main effect of treatment [F(1,9)=32.12; p=0.0003], genotype [F(1,9)=33.80; p=0.0003] and treatment × genotype interaction [F(1,9)=39.44; p=0.0001]. Further analysis using Bonferroni's test indicated a significant reduction in reelin levels in the frontal cortex of WT following hypoxia (Fig 5B; p<0.05). In hippocampus, we found a significant main effect of treatment [F(1,9)=14.38; p=0.004] and genotype [F(1,9)=23.61; p=0.0009], but no significant effect of treatment × genotype interaction [F(1,9)=1.394; p=0.268]. A significant reduction in reelin protein levels was observed in hippocampus of both WT and HRM at 1 month following hypoxia (Fig 5C; p<0.05). Two-way ANOVA showed significant main effect of genotype [F(1,9)=31.05; p=0.005], but no significant effect of treatment [F(1,9)=4.708; p=0.062] and treatment × genotype interaction [F(1,9)=2.724; p=0.137] on reelin protein levels in the frontal cortex at 3 month following normoxia/hypoxia treatment. The reelin levels were found significantly lower in the frontal cortex of HRM as compared to WT (Fig 5D; p<0.05). However, hypoxia did not have any significant effects on reelin levels in the frontal cortex of above mice. In hippocampus, no significant effect of genotype [F(1,9)=4.495; p=0.066], treatment [F(1,9)=0.669; p=0.437] or treatment × genotype interaction [F(1,9)=0.187; p=0.676] was found on reelin levels at 3 months following hypoxia. A significant reduction in reelin protein levels was found in the hippocampus of HRM at 3 months following normoxia/hypoxia treatment (Fig 5E; p<0.05).
Figure 5.
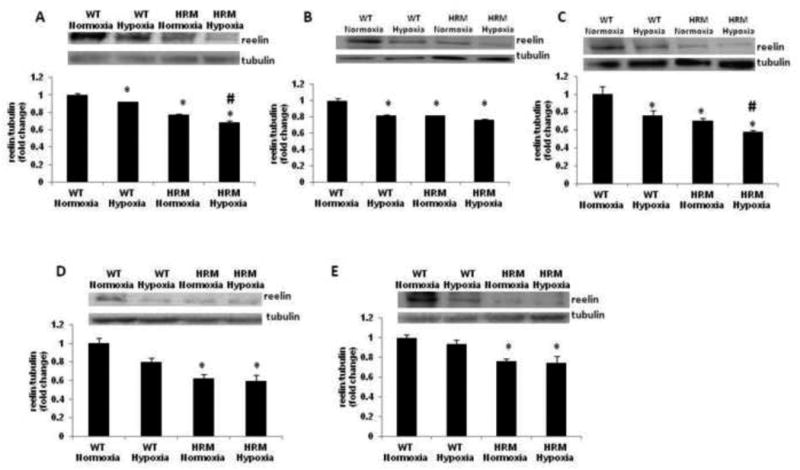
Effect of prenatal hypoxia on reelin protein levels in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 (E17) and reelin protein levels were measured in (A) forebrain at E18, (B) frontal cortex at 1 month, (C) hippocampus at 1 month, (D) frontal cortex at 3 months, and (E) hippocampus at 3 months of age. Data represent mean±SE. * P < 0.05 vs WT Normoxia and P < 0.05 vs HRM Normoxia; Bonferroni's test.
Effect of prenatal hypoxia on HIF-1 α, VEGF and Flk1 protein levels
We found a significant main effect of genotype [F(1,9)=16.58; p=0.0028], but no significant effect of treatment F(1,9)=1.765; p=0.216] and treatment × genotype interaction [F(1,9)=1.946; p = 0.196] on HIF-1α protein levels in the forebrain samples from HRM and WT at 24 h following normoxia/hypoxia treatment. In addition, significantly higher levels of HIF-1α was found in the forebrain of HRM at E18 as compared to WT littermates (Fig 6A; p<0.05).We did not find any significant effect of genotype, treatment or treatment × genotype interaction on HIF-1α levels in frontal cortex and hippocampus of HRM or WT following hypoxia at 1 month or 3 months following hypoxia (Fig 6B-E).
Figure 6.
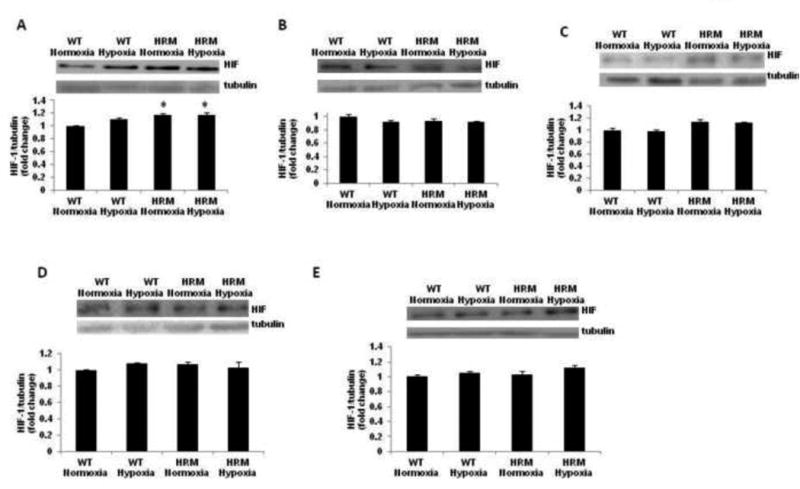
Effect of prenatal hypoxia on HIF-1α protein levels in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 (E17) and HIF-1α protein levels were measured in (A) forebrain at E18, (B) frontal cortex at 1 month, (C) hippocampus at 1 month, (D) frontal cortex at 3 months, and (E) hippocampus at 3 months of age. Data represent mean±SE. * P < 0.05 vs WT Normoxia; Bonferroni's test.
Data on VEGF protein levels at E18 as measured by immunoblot analysis showed a significant main effect of genotype [F(1,9)=12.10; p=0.007] and treatment × genotype interaction [F(1,9)=8.053; p=0.019], but no effect of treatment [F(1,9)=2.483; p=0.149]. Post-hoc analysis showed higher VEGF levels in the forebrain of HRM as compared to WT (Fig 7A; p<0.05). Moreover, hypoxia significantly increased VEGF levels in the forebrain of WT, but not in HRM as compared to their normoxia groups (p<0.05). We did not find any significant change in VEGF levels in frontal cortex of HRM or WT at 1 month following normoxia/hypoxia treatment (Fig 7B). We observed a significant main effect of treatment [F(1,9)=6.233; p=0.034], but no effect of genotype [F(1,9)=0.697; p=0.425] or treatment × genotype interaction [F(1,9)=2.579; p=0.143] on VEGF levels in the hippocampus at 1 month following normoxia/hypoxia treatment. Moreover, VEGF levels in the hippocampus of hypoxia-treated HRM were significantly lower than their normoxia groups (Fig 7C; p<0.05). The data on VEGF protein levels in frontal cortex at 3 months following hypoxia showed significant main effect of treatment [F(1,9)=14.52; p=0.0052] and treatment × genotype interaction [F(1,9)=27.76; p = 0.0008], but no effect of genotype [F(1,9)=1.286; p=0.289]. Further analysis using Bonferroni's test indicated no significant effect of hypoxia on VEGF levels in the frontal cortex of HRM and WT (Fig 7D). In hippocampus, no significant effect of genotype [F(1,9)=4.556; p=0.063], treatment [F(1,9)=2.847; p=0.13] or treatment × genotype interaction [F(1,9)=0.337; p=0.577] on VEGF protein levels was found in WT or HRM at 3 month following normoxia/hypoxia treatment. A significant increase in VEGF protein levels was found in the hippocampus of HRM and WT following hypoxia as compared to WT normoxia groups (Fig 7E; p<0.05). Next, we examined serum VEGF levels in HRM and WT at 3 months of age following hypoxia. We found a significant main effect of treatment [F(1,26)=18.84; p=0.0002], but no significant effect of genotype [F(1,26)=0.0075; p=0.931], or treatment × genotype interaction [F(1,26)=0.045; p=0.832] on serum VEGF levels. Moreover, hypoxia induced significant reductions in serum VEGF levels in both HRM and WT (Fig 7F; p<0.05).
Figure 7.
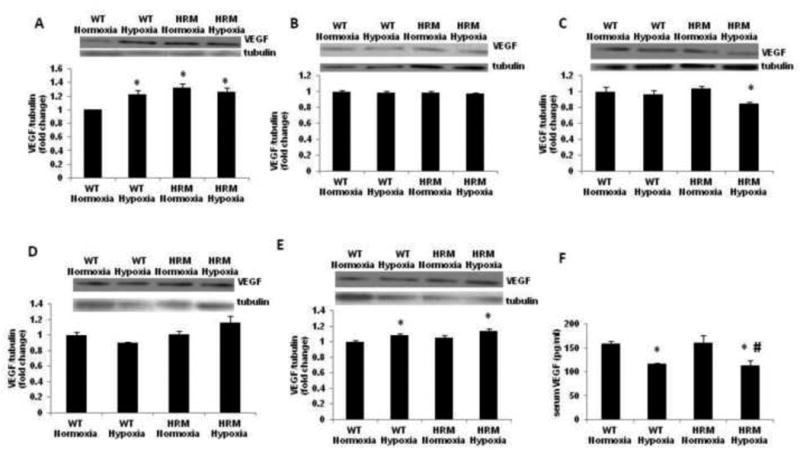
Effect of prenatal hypoxia on VEGF protein levels in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 (E17) and VEGF protein levels were measured in (A) forebrain at E18, (B) frontal cortex at 1 month, (C) hippocampus at 1 month, (D) frontal cortex at 3 months, (E) hippocampus at 3 months and (F) serum at 3 months of age. Data represent mean±SE. * P < 0.05 vs WT Normoxia; Bonferroni's test.
Two-way ANOVA showed a significant main effect of genotype [F(1,11)=15.12; p=0.0025] and treatment × genotype interaction [F(1,11)=9.22; p=0.011], but no significant effect of treatment [F(1,11)=2.168; p=0.168] on Flk1 protein levels in the forebrain samples from HRM and WT at 24 h following normoxia/hypoxia treatment. Subsequent analysis showed a significant reduction in Flk1 protein levels in HRM mice following hypoxia (Fig 8A; p<0.05). The data on Flk1 protein levels in frontal cortex at 1 month following hypoxia showed significant main effect of genotype [F(1,9)=8.784; p=0.010], but no effect of treatment [F(1,9)=2.692; p=0.135] and treatment × genotype interaction [F(1,9)=0.002; p = 0.958]. Further analysis using Bonferroni's test did not indicate any significant change in Flk1 levels following hypoxia in HRM and WT (Fig 8B). In hippocampus, we found a significant main effect of treatment [F(1,9)=5.802; p=0.039], but no effect of genotype [F(1,9)=0.061; p=0.809] and treatment × genotype interaction [F(1,9)=1.175; p=0.306] on Flk1 levels. Moreover, a significant reduction in Flk1 levels was found in the hippocampus of HRM at 1 month following hypoxia as compared to WT normoxia group (Fig 8C; p<0.05). At 3 months, the two-way ANOVA showed significant main effect of treatment [F(1,9)=10.66; p=0.011], but no effect of genotype [F(1,9)=4.179; p=0.075] and treatment × genotype interaction [F(1,9)=3.302; p=0.107] on Flk1 protein levels in the frontal cortex of HRM and WT. Post-hoc analysis showed a significant reduction in Flk1 levels in the frontal cortex of HRM at 3 months following hypoxia as compared to WT normoxia (Fig 8D; p<0.05). A significant main effect of genotype [F(1,9)=7.815; p=0.023], but no effect of treatment [F(1,9)=0.54; p=0.483] and treatment × genotype interaction [F(1,9)=2.453; p=0.156] was found on Flk1 levels in the hippocampus of HRM and WT at 3 month following normoxia/hypoxia treatment. Further analysis showed no significant difference in Flk1 levels in hypoxia treated mice as compared to normoxia groups.
Figure 8.
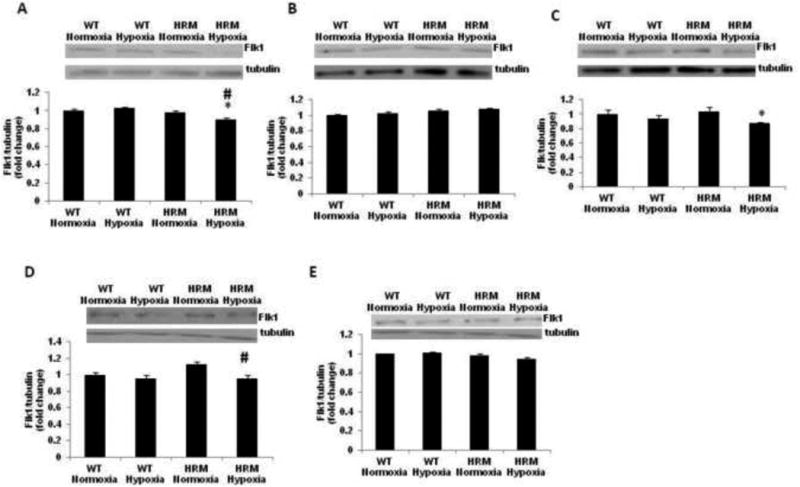
Effect of prenatal hypoxia on Flk1 protein levels in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 (E17) and Flk1 protein levels were measured in (A) forebrain at E18, (B) frontal cortex at 1 month, (C) hippocampus at 1 month, (D) frontal cortex at 3 months, and (E) hippocampus at 3 months of age. Data represent mean±SE. * P < 0.05 vs WT Normoxia and # P < 0.05 vs HRM Normoxia; Bonferroni's test.
Effect of prenatal hypoxia on GR and corticosterone levels
Two-way ANOVA revealed a significant main effect of genotype [F(1,9)=19.38; p=0.002], treatment [F(1,9)=21.92; p=0.001] and treatment × genotype interaction [F(1,9)=15.29; p = 0.0036] on GR levels in the forebrain samples from HRM and WT at 24 h following normoxia/hypoxia treatment. We found significantly higher GR protein levels in the forebrain of HRM at E18 as compared to WT littermates (Fig 9A; p<0.05). Moreover, hypoxia significantly increased GR levels in the forebrain of WT, but not in HRM as compared to their normoxia groups (p<0.05). A significant effect of treatment [F(1,9)=8.701; p=0.014], and treatment × genotype interaction [F(1,9)=5.622; p=0.039], but no effect of genotype [F(1,9)=0.644; p=0.441] on GR protein levels in frontal cortex of WT or HRM at 1 month following normoxia/hypoxia treatment. Post-hoc analysis showed a significant increase in GR levels in the frontal cortex of HRM and WT following hypoxia as compared to WT normoxia groups (Fig 9B; p<0.05). In hippocampus, a significant effect of genotype [F(1,9)=15.67; p=0.003], and treatment × genotype interaction [F(1,9)=29.82; p=0.0004], but no effect of treatment [F(1,9)=0.296; p=0.599] was found on GR protein levels at 1 month following normoxia/hypoxia treatment. Furthermore, a significant increase in GR levels was observed in the hippocampus of HRM and WT following hypoxia as compared to WT normoxia groups (Fig 9C; p<0.05). The data on GR protein levels in frontal cortex at 3 months following hypoxia showed significant main effect of genotype [F(1,9)=6.709; p=0.032], but no effect of treatment [F(1,9)=0.469; p=0.512] and treatment × genotype interaction [F(1,9)=2.594; p=0.146]. Further analysis using Bonferroni's test revealed a significant increase in GR levels in HRM and WT following hypoxia as compared to WT normoxia groups (Fig 9D; p<0.05). In hippocampus, we found a significant main effect of genotype [F(1,9)=11.44; p=0.009], but no effect of treatment [F(1,9)=2.445; p=0.156] and treatment × genotype interaction [F(1,9)=1.780; p=0.219] on GR levels. Moreover, a significant increase in GR levels was found in the hippocampus of HRM and WT at 3 month following hypoxia as compared to their normoxia groups (Fig 9E; p<0.05). Next, we examined serum corticosterone levels in HRM and WT at 3 months of age following hypoxia. We found a significant main effect of treatment [F(1,12)=15.30; p=0.0021], but no effect of genotype [F(1,12)=0.223; p=0.645] and treatment × genotype interaction [F(1,12)=0.687; p=0.420] on serum corticosterone levels. Bonferroni's test showed a significant decrease in serum corticosterone levels in HRM and WT following hypoxia as compared to WT normoxia groups (Fig 9F; p<0.05).
Figure 9.
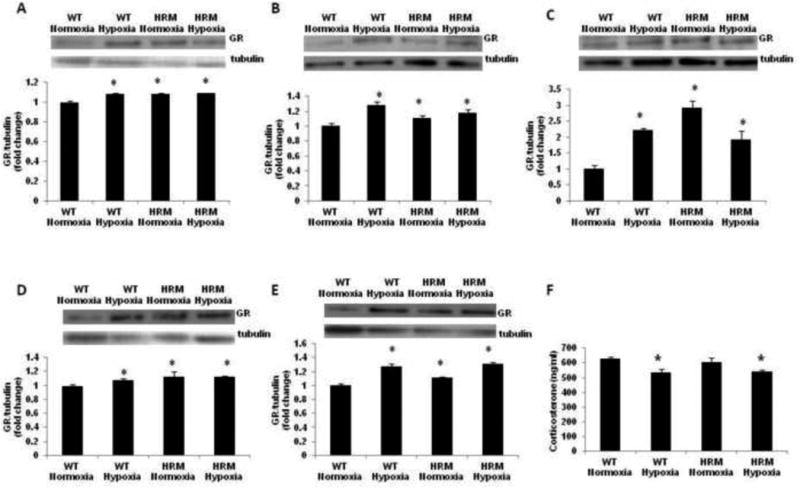
Effect of prenatal hypoxia on GR and corticosterone levels in heterozygous reeler mice (HRM). HRM and wild-type (WT) mice underwent prenatal hypoxia or normoxia at embryonic day 17 (E17) and GR protein levels were measured in (A) forebrain at E18, (B) frontal cortex at 1 month, (C) hippocampus at 1 month, (D) frontal cortex at 3 months, and (E) hippocampus at 3 months of age. (F) serum corticosterone levels at 3 months of age. Data represent mean±SE. * P < 0.05 vs WT Normoxia; Bonferroni's test.
4. Discussion
The main findings of the present study are as follows: First, prenatal hypoxia induced anxiety-like behavior in both HRM and WT. Second, a significant reduction in blood flow was observed in hippocampus of both WT and HRM following prenatal hypoxia. Finally, prenatal hypoxia induced brain region specific as well as age-dependent changes in the protein levels of HIF-1, VEGF, Flk1 and GR.
Hypoxia during pregnancy has been considered as a potential environmental risk factor for schizophrenia (Van Erp et al., 2002; Demjaha et al., 2011). Moreover, serious obstetric complications interacting with specific genetic risk factors may increase the risk for schizophrenia (Nicodemus et al., 2008). We hypothesized that prenatal hypoxia would exacerbate a reelin haploinsufficiency-induced schizophrenia endophenotype in mice. While neurochemical analyses were performed at E18, 1 month and 3 months following normoxia/hypoxia treatment, behavioral testing was carried out only at 3 months since HRM do not exhibit behavioral deficits until after puberty (Costa et al., 2002).
Loss of normal PPI is widely accepted as an endophenotype of schizophrenia and considered indicative of disrupted sensorimotor gating (Braff et al., 1992). We did not find any significant difference in PPI between WT and HRM. The available literature show contradictory data on PPI deficits in HRM (Tueting et al., 1999; Salinger et al., 2003; Podhorna and Didriksen, 2004; Kutiyanawalla et al., 2012). A detailed behavioral analyses showed no significant difference in behavior tests including spatial working memory, novel-object detection, fear conditioning, and sensorimotor reflex modulation in HRM as compared to WT (Salinger et al., 2003) suggesting that HRM might be more tolerant than humans to reelin synthesis deficits, and as a consequence, different insults (such as prenatal hypoxia) might also not produce any significant behavioral or molecular alterations in HRM. Although we did not find genotype effects on PPI, there appears to be a treatment effect of hypoxia on PPI indicating a small, but no-significant increase in %PPI following prenatal hypoxia. Moreover, we observed a diminished startle in prenatal hypoxia-treated WT mice, similar to normoxia treated HRM. We found differential effects of prenatal hypoxia on startle amplitude in WT and HRM. A difference in the nature of complexity in cellular mechanisms has been suggested between the startle response and PPI (Geyer et al., 2002). It has been shown that increased startle reactivity is not necessarily associated with decreased PPI (Paylor and Crawley, 1997). The differential regulation of startle amplitude and PPI has been reported in many rodent studies (Acri et al., 1994; Schreiber et al., 2002; Duncan et al., 2003). Together, the above data suggest that there does not appear to be a predictable relationship between startle amplitude and PPI.
We have used a modified Y maze protocol that tests both spatial and working memory. The traditional Y maze for spatial memory without food restriction tests the mouse's ability to remember based on visual cues only, and are scored on which arm they enter spontaneously in a single trial of 10 minutes for exploration. A ratio of correct to incorrect is used to determine if spatial memory is intact (i.e. A, B, C is correct as none were repeated before all were 3 arms are explored while B, C, B, A would indicate a possible memory deficit if incorrect choices are prevalent). In our model, each arm has distinct cues for the spatial memory aspect, which allows the mice to remember which arm they initially entered with delays of 30 sec. Due to using a forced choice with food reward setup, we decided to run several discrete trials over several days to establish a learning curve and used a retention delay interval to assess task learning On the last day of testing, three 30 sec delay trials similar to the training trials were performed and then we introduced the long delay of 7 minutes to test their working memory. This allows for strengthened confidence in accuracy versus a one day trial to test their learning and memory capabilities. Our data from Y maze indicate that HRM have better working spatial memory at shorter time delays. The probe of 7 minutes did not show any significant difference in treatment or genotype. Interestingly, the above probe trial data is similar to the PPI startle profile. The hippocampus is sensitive to hypoxic insults and studies have shown additional reductions in hippocampal volume and/or brain abnormalities in subjects with schizophrenia and their siblings who experienced obstetric complications as compared to controls (Cannon et al., 2002; Nicodemus et al., 2008). We found a significant decrease in hippocampal blood flow following prenatal hypoxia in both WT and HRM suggesting a potential impact of hypoxia during pregnancy on vascular function in adolescence.
To find out the possible biological mediators of prenatal hypoxia-induced changes in behavior and blood flow, we examined the profile of molecules associated with hypoxia in frontal cortex and hippocampus. HIF-1 is considered as the main regulatory protein of VEGF (Semenza, 2012). A significant interaction between putative hypoxia regulated genes and serious obstetric complications has been found to influence the risk for schizophrenia (Nicodemus et al., 2008). Although we did not find any significant effect of prenatal hypoxia on HIF-1α and VEGF levels in frontal cortex of WT and HRM at 1 month and 3 months of age, HRM with prenatal hypoxia insult showed robust deficits in hippocampal VEGF levels at 1 month of age. However, the above reduction in VEGF levels did not last for 3 months of age. Instead, there was a significant increase in hippocampal VEGF levels in HRM at 3 months of age following prenatal hypoxia. Moreover, we found a significant increase in VEGF levels without any change in HIF-1 levels in hippocampus at 3 months following prenatal hypoxia. The above data suggest whether VEGF expression might be regulated by prenatal hypoxia via HIF-1 independent pathway. A recent study has indicated that peroxisome-proliferator-activated receptor-gamma coactivator-1alpha (PGC-1α) regulates VEGF via HIF-1 independent pathway (Arany et al., 2008). Further studies need to be performed to understand the role of PGC-1 α in VEGF regulation following prenatal hypoxia.
It is important to note that prenatal hypoxia induced a significant reduction in both serum VEGF levels and hippocampal blood flow at 3 months of age. VEGF is an angiogenic factor involved in the regulation of blood flow and has neurotrophic and neuroprotective properties (Greenberg and Jin, 2005). Moreover, alterations in brain cellular energy metabolism and blood flow are also observed in schizophrenia subjects (Hanson and Gottesman, 2005). Studies have shown that the various positive and negative symptom dimensions observed in subjects with schizophrenia are associated with specific patterns of the regional blood flow (Andreasen et al., 1997). Although the data on peripheral VEGF levels in schizophrenia are lacking, one study has found a significant reduction in VEGF mRNA in prefrontal cortex of schizophrenia subjects (Fulzele and Pillai, 2009). Our earlier study has found a significant increase in VEGF levels in frontal cortex, but reduction in serum VEGF levels following chronic corticosterone exposure in mice (Howell et al., 2011). Together, these data suggest that a feedback mechanism may operate to increase VEGF production in CNS in response to inhibition of the VEGF signaling pathway. However, the possibility of increase in VEGF in hippocampus as a response to reduction in peripheral VEGF levels following hypoxia cannot be ruled out. Further studies are warranted to understand the above possible mechanisms.
A number of studies have suggested that corticosteroids are involved in the anxiety-like symptoms (de Kloet et al., 2005). Our data also indicate that prenatal hypoxia induces anxietylike phenotype as well as increases in GR levels following prenatal hypoxia. Moreover, studies have shown that chronic corticosteroid exposure, but not acute exposure manifests an anxietylike phenotype in rodents (David et al., 2009). While our data found increases in anxiety, without deficits in prepulse inhibition, further studies are warranted to test whether such behavioral differences are due to differential effects of hypoxia on specific brain regions (i.e. frontal cortex vs hippocampus). It is plausible that subjects with schizophrenia have altered hypothalamic–pituitary–adrenal (HPA) axes and fail to respond to stress conditions appropriately. The observed increase in GR levels in frontal cortex and hippocampus in our study could be a result of diminished corticosterone levels in the periphery following prenatal hypoxia. Thus, modulating GR function may be a possible way to manage the disease progress in stress-related neuropsychiatric disorders such as schizophrenia. It has been shown that short-term use of Mifepristone, a GR antagonist (RU38486), is effective in the treatment of psychotic major depression via re-regulating the HPA axis (Flores et al., 2006). It would be important to investigate whether blocking GR function could restore prenatal hypoxia-induced changes in anxiety-like behavior and hippocampal blood flow in mice.
In summary, the data from our studies indicate that prenatal hypoxia has differential effects on different domains of behavioral parameters related to schizophrenia. It is important to note that when taking into account the “two-hit” hypothesis of schizophrenia, the second hit occurs usually during adolescence or pubertal stages, when the brain is developing connections enabling executive higher functions, a time highly susceptible to stress. A couple of previous studies have found a lack of change in PPI when two stressors in neonatal and young-adult life in animals were combined (Ellenbroek and Cools, 2002; Choy and van den Buuse, 2008). In one study, the apomorphine treatment was found to disrupt PPI in controls and in rats that had undergone either one of the two stressors (maternal deprivation or prolonged periadolescent corticosterone treatment) whereas no apomorphine-induced PPI-disruptions were observed in the group that had experienced both the stressors (Choy and van den Buuse, 2008). In another study, a subsequent stressor (isolation rearing) could reverse PPI-disruptive effects of the primary stressor (maternal separation) (Ellenbroek and Cools, 2002). These studies suggest an inhibitory interaction, instead of an augmentation, of early and later developmental stressors in rodents. Our study combined an early stressor (prenatal hypoxia) with a candidate gene (reelin) since several studies have successfully identified candidate genes that could contribute to the induction of schizophrenia-like phenotypes in interaction with stress (Duncan et al., 2004; Eells et al., 2006; Oliver and Davies, 2009). However, the behavioral changes following prenatal hypoxia do not seem to be influenced by reelin deficiency suggesting that the change in behavioral, functional or neurochemical phenotypes resulting from gene-environment interaction depends on the specificity of the susceptibility gene. Our studies indicate that corticosterone may be a possible mediator of prenatal hypoxia-induced changes in behavioral and/or functional changes. Our ongoing studies will explore the role of GR in mediating prenatal hypoxia-induced changes in anxiety-like phenotype and hippocampal blood flow in mice. Our data also have broader implications for finding association between the peripheral marker and functional changes in schizophrenia. Further studies should explore the possibility whether peripheral VEGF can correlate with changes in hippocampal blood flow in schizophrenia subjects.
Acknowledgments
The authors would like to thank the members of Small Animal Behavior Core Facility, Georgia Regents University for training KRH for behavioral studies. The authors are thankful to Core Imaging Facility, Small Animals of Georgia Regents University for imaging analysis.
Role of funding source: This work was supported by grant from NIH/NIMH to A.P.
Footnotes
Conflict of interest: All authors declare no conflict of interest.
Contributors: AP designed the study. KRH acquired the data and performed data analysis. KRH and AP wrote the manuscript. All authors approved the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acri JB, Morse DE, Popke EJ, Grunberg NE. Nicotine increases sensory gating measured as inhibition of the acoustic startle reflex in rats. Psychopharmacology (Berl) 1994 Mar;114(2):369–374. doi: 10.1007/BF02244861. [DOI] [PubMed] [Google Scholar]
- Andreasen N, O'Leary D, Flaum M, Nopoulos P, Watkins G, Ponto L, Hichwa R. Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naive patients. Lancet. 1997;349:1730–1734. doi: 10.1016/s0140-6736(96)08258-x. [DOI] [PubMed] [Google Scholar]
- Andreasen NC, Liu D, Ziebell S, Vora A, Ho BC. Relapse duration, treatment intensity, and brain tissue loss in schizophrenia: a prospective longitudinal MRI study. Am J Psychiatry. 2013;170(6):609–615. doi: 10.1176/appi.ajp.2013.12050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Asami T, Bouix S, Whitford TJ, Shenton ME, Salisbury DF, McCarley RW. Longitudinal loss of gray matter volume in patients with first-episode schizophrenia: DARTEL automated analysis and ROI validation. Neuroimage. 2012;59(2):986–996. doi: 10.1016/j.neuroimage.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baburamani AA, Ek CJ, Walker DW, Castillo-Melendez M. Vulnerability of the developing brain to hypoxic-ischemic damage: contribution of the cerebral vasculature to injury and repair? Front Physiol. 2012;3:424. doi: 10.3389/fphys.2012.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra E, Pagès G, Pouysségur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19(1-2):139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and Habituation of the Startle Reflex in Schizophrenic Patients. Arch Gen Psychiatry. 1992;49(3):206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996 Jul;76(3):839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002 Jul;159(7):1080–1092. doi: 10.1176/appi.ajp.159.7.1080. [DOI] [PubMed] [Google Scholar]
- Choy KH, van den Buuse M. Attenuated disruption of prepulse inhibition by dopaminergic stimulation after maternal deprivation and adolescent corticosterone treatment in rats. Eur Neuropsychopharmacol. 2008 Jan;18(1):1–13. doi: 10.1016/j.euroneuro.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Costa E, Davis J, Pesold C, Tueting P, Guidotti A. The heterozygote reeler mouse as a model for the development of a new generation of antipsychotics. Curr Opin Pharmacol. 2002 Feb;2(1):56–62. doi: 10.1016/s1471-4892(01)00121-7. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Sibug RM, Helmerhorst FM, Schmidt MV. Stress, genes and the mechanism of programming the brain for later life. Neurosci Biobehav Rev. 2005 Apr;29(2):271–281. doi: 10.1016/j.neubiorev.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Demjaha A, MacCabe JH, Murray RM. How Genes and Environmental Factors Determine the Different Neurodevelopmental Trajectories of Schizophrenia and Bipolar Disorder. Schizophrenia Bulletin. 2011;38(2):209–14. doi: 10.1093/schbul/sbr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan EJ, Szilagyi S, Efferen TR, Schwartz MP, Parwani A, Chakravorty S, Madonick SH, Kunzova A, Harmon JW, Angrist B, et al. Effect of treatment status on prepulse inhibition of acoustic startle in schizophrenia. Psychopharmacology (Berl) 2003 Apr;167(1):63–71. doi: 10.1007/s00213-002-1372-z. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Koller BH. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004 Aug 31;153(2):507–19. doi: 10.1016/j.bbr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Eells JB, Misler JA, Nikodem VM. Early postnatal isolation reduces dopamine levels, elevates dopamine turnover and specifically disrupts prepulse inhibition in Nurr1-null heterozygous mice. Neuroscience. 2006 Jul 21;140(4):1117–26. doi: 10.1016/j.neuroscience.2005.12.065. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Cools AR. Early maternal deprivation and prepulse inhibition: the role of the postdeprivation environment. Pharmacol Biochem Behav. 2002 Aug;73(1):177–84. doi: 10.1016/s0091-3057(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle JA, McMenomy T. Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry. 2000 Nov;5(6):654–663. doi: 10.1038/sj.mp.4000783. [DOI] [PubMed] [Google Scholar]
- Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology. 2006 Mar;31(3):628–636. doi: 10.1038/sj.npp.1300884. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulzele S, Pillai A. Decreased VEGF mRNA expression in the dorsolateral prefrontal cortex of schizophrenia subjects. Schizophr Res. 2009 Dec;115(2-3):372–373. doi: 10.1016/j.schres.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry. 2002;7(10):1039–1053. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- Giordano FJ, Johnson RS. Angiogenesis: the role of the microenvironment in flipping the switch. Curr Opin Genet Dev. 2001 Feb;11(1):35–40. doi: 10.1016/s0959-437x(00)00153-2. [DOI] [PubMed] [Google Scholar]
- Golan MH, Mane R, Molczadzki G, Zuckerman M, Kaplan-Louson V, Huleihel M, Perez-Polo JR. Impaired migration signaling in the hippocampus following prenatal hypoxia. Neuropharmacology. 2009 Oct-Nov;57(5-6):511–522. doi: 10.1016/j.neuropharm.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Hanson DR, Gottesman II. Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet. 2005:6–7. doi: 10.1186/1471-2350-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haukvik UK, Saetre P, McNeil T, Bjerkan PS, Andreassen OA, Werge T, Jönsson EG, Agartz I. An exploratory model for G × E interaction on hippocampal volume in schizophrenia; obstetric complications and hypoxia-related genes. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1259–1265. doi: 10.1016/j.pnpbp.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Howell KR, Kutiyanawalla A, Pillai A. Long-term continuous corticosterone treatment decreases VEGF receptor-2 expression in frontal cortex. PLoS One. 2011;6(5):e20198. doi: 10.1371/journal.pone.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P, Sharma RP, Costa E. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutiyanawalla A, Promsote W, Terry A, Pillai A. Cysteamine treatment ameliorates alterations in GAD67 expression and spatial memory in heterozygous reeler mice. Int J Neuropsychopharmacol. 2012 Sep;15(8):1073–1086. doi: 10.1017/S1461145711001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270(22):13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- Mayoral SR, Omar G, Penn AA. Sex differences in a hypoxia model of preterm brain damage. Pediatr Res. 2009;66(3):248–253. doi: 10.1203/PDR.0b013e3181b1bc34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodemus KK, Marenco S, Batten AJ, Vakkalanka R, Egan MF, Straub RE, Weinberger DR. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry. 2008 Sep;13(9):873–877. doi: 10.1038/sj.mp.4002153. [DOI] [PubMed] [Google Scholar]
- Nullmeier S, Panther P, Dobrowolny H, Frotscher M, Zhao S, Schwegler H, Wolf R. Region-specific alteration of GABAergic markers in the brain of heterozygous reeler mice. Eur J Neurosci. 2011 Feb;33(4):689–698. doi: 10.1111/j.1460-9568.2010.07563.x. [DOI] [PubMed] [Google Scholar]
- Oliver PL, Davies KE. Interaction between environmental and genetic factors modulates schizophrenic endophenotypes in the Snap-25 mouse mutant blind-drunk. Hum Mol Genet. 2009 Dec 1;18(23):4576–89. doi: 10.1093/hmg/ddp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997 Jul;132(2):169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Podhorna J, Didriksen M. The heterozygous reeler mouse: behavioural phenotype. Behav Brain Res. 2004 Aug 12;(1):153. 43–54. doi: 10.1016/j.bbr.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Qiu S, Korwek KM, Pratt-Davis AR, Peters M, Bergman MY, Weeber EJ. Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol Learn Mem. 2006 May;85(3):228–242. doi: 10.1016/j.nlm.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Quinn TP, Peters KG, De Vries C, Ferrara N, Williams LT. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci U S A. 1993;90:7533–7537. doi: 10.1073/pnas.90.16.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinger WL, Ladrow P, Wheeler C. Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav Neurosci. 2003 Dec;117(6):1257–1275. doi: 10.1037/0735-7044.117.6.1257. [DOI] [PubMed] [Google Scholar]
- Schreiber R, Dalmus M, De Vry J. Effects of alpha 4/beta 2- and alpha 7-nicotine acetylcholine receptor agonists on prepulse inhibition of the acoustic startle response in rats and mice. Psychopharmacology (Berl) 2002 Jan;159(3):248–257. doi: 10.1007/s00213-001-0927-8. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet. 1998 Dev;8(5):588–594. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med. 2012;18(9):534–543. doi: 10.1016/j.molmed.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tueting P, Costa E, Dwivedi Y, Guidotti A, Impagnatiello F, Manev R, Pesold C. The phenotypic characteristics of heterozygous reeler mouse. Neuroreport. 1999 Apr 26;10(6):1329–1334. doi: 10.1097/00001756-199904260-00032. [DOI] [PubMed] [Google Scholar]
- Van Erp TG, Saleh PA, Rosso IM, Huttunen M, Lönnqvist J, Pirkola T, Salonen O, Valanne L, Poutanen VP, Standertskjöld-Nordenstam CG, Cannon TD. Contributions of Genetic Risk and Fetal Hypoxia to Hippocampal Volume in Patients With Schizophrenia or Schizoaffective Disorder, Their Unaffected Siblings, and Healthy Unrelated Volunteers. Am J Psychiatry. 2002;159:1514–1520. doi: 10.1176/appi.ajp.159.9.1514. [DOI] [PubMed] [Google Scholar]
- Zachary I. Neuroprotective role of vascular endothelial growth factor: signalling mechanisms, biological function, and therapeutic potential. Neurosignals. 2005;14:207–221. doi: 10.1159/000088637. [DOI] [PubMed] [Google Scholar]