

Abstract
Cadmium (Cd), a toxic environmental contaminant, induces neurodegenerative diseases. Celastrol, a plant-derived triterpene, has shown neuroprotective effects in various disease models. However, little is known regarding the effect of celastrol on Cd-induced neurotoxicity. Here, we show that celastrol protected against Cd-induced apoptotic cell death in neuronal cells. This is supported by the findings that celastrol strikingly attenuated Cd-induced viability reduction, morphological change, nuclear fragmentation, and condensation, as well as activation of caspase-3 in neuronal cells. Concurrently, celastrol remarkably blocked Cd-induced phosphorylation of c-Jun N-terminal kinase (JNK), but not extracellular signal-regulated kinases 1/2 and p38, in neuronal cells. Inhibition of JNK by SP600125 or over-expression of dominant negative c-Jun potentiated celastrol protection against Cd-induced cell death. Furthermore, pre-treatment with celastrol prevented Cd down-regulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and activation of phosphoinositide 3′-kinase/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling in neuronal cells. Over-expression of wild-type PTEN enhanced celastrol inhibition of Cd-activated Akt/mTOR signaling and cell death in neuronal cells. The findings indicate that celastrol prevents Cd-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. Our results strongly suggest that celastrol may be exploited for the prevention of Cd-induced neurodegenerative disorders.
Keywords: apoptosis, cadmium, Celastrol, c-Jun N-terminal kinase, mammalian target of rapamycin, phosphatase and tensin homolog on chromosome 10
Cadmium, a toxic transition metal, is also an environmental contaminant, which is mainly released from smelting and refining of metals, burning of chemical fuels and municipal wastes, and cigarette smoking. Its very long biological half-life (15–20 years) is responsible for accumulation in many human organs, including kidney (Johri et al. 2010), liver (Koyu et al. 2006), lung (Jiang et al. 2008), testis (Thompson and Bannigan 2008), bone (Akesson et al. 2006), blood system (Kocak and Akcil 2006), which leads to their structural and functional damages. As cadmium (Cd) has high blood–brain barrier permeability, clinical data have shown that chronic Cd exposure affects the nervous system such as learning disabilities and hyperactivity in children (Pihl and Parkes 1977; Wright et al. 2006), olfactory dysfunction, and neurobehavioral defects in attention, psychomotor speed, and memory in workers (Jarup et al. 1993). Besides that, Parkinsonism after acute Cd poisoning has been reported (Okuda et al. 1997). Accumulated evidence implicates that Cd intoxication is a possible etiological factor of neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS) (Okuda et al. 1997; Panayi et al. 2002; Chen et al. 2011c).
Mounting studies have demonstrated that sustained activation of c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase 1/2 (Erk1/2), and/or p38 MAPK contribute to Cd-induced apoptosis in various cells, including neuronal cells (Rockwell et al. 2004; Kim et al. 2005). Recently, we have found that all three MAPK members can be activated by Cd in neuronal cells, and identified that Cd-induced neuronal apoptosis is partially associated with activation of JNK and Erk1/2, but not p38 (Chen et al. 2008). A number of studies have revealed that mammalian target of rapamycin (mTOR) activity, which plays an important role in differentiation, development and survival of neurons, is modified in various pathologic states of the nervous system such as neurodegenerative disorders and brain tumors (Ravikumar et al. 2004; Swiech et al. 2008). Our group has demonstrated that Cd-induced neuronal apoptosis is also partially involved in activation of mTOR pathway in neuronal cells, and Cd activates mTOR pathway by inhibiting negative regulator phosphatase and tensin homolog on chromosome 10 (PTEN), a dual-specificity protein and lipid phosphatase (Chen et al. 2008, 2011b). Thus, we deduce that a compound that can inhibit MAPK and mTOR signaling network may be protective against Cd-poisoning.
Celastrol, a quinone methide triterpene, is a pharmacologically active compound extracted from the root of the plant Tripterygium wilfordii (Thunder of God vine). Celastrol has been shown to possess a wide variety of biological and pharmacological effects, including antioxidant, anti-apoptotic, anti-inflammatory, and anti-carcinogenic properties (Salminen et al. 2010; Kannaiyan et al. 2011). Celastrol inhibits growth and induces apoptotic cell death by activating JNK or p38 MAPK in cancer cells (Chen et al. 2011a; Kannaiyan et al. 2011) and microglial cells (Boridy et al. 2012), and by suppressing phosphoinositide 3′-kinase/protein kinase B (Akt) signaling in wide variety of human tumor cells (Kannaiyan et al. 2011; Lee et al. 2012). Anti-carcinogenic ability of celastrol is partially attributed to effectively suppressing activated mTOR signaling pathway (Pang et al. 2010; Kannaiyan et al. 2011; Li et al. 2012). Interestingly, celastrol also has neuroprotective effects in the models of neurodegenerative disorders, such as PD, AD, and ALS (Allison et al. 2001; Cleren et al. 2005; Kiaei et al. 2005). However, whether and how celastrol protects against Cd neurotoxicity is largely unknown. Here, for the first time, we show that celastrol prevented Cd-induced neuronal apoptosis via inhibiting activation of JNK and mTOR signaling pathways. Furthermore, celastrol suppressed Cd-induced activation of mTOR signaling pathway and neuronal apoptosis by elevating PTEN activity.
Materials and methods
Materials
Cadmium chloride was purchased from Sigma (St. Louis, MO, USA) and dissolved in sterile distilled water to prepare the stock solutions (10 and 20 mM), aliquoted, and stored at 23°C for all experiments. Dulbecco’s modified Eagle’s medium, 0.05% Trypsin-EDTA, NEUROBASAL™ Media, and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum were supplied by Hyclone (Logan, UT, USA). Enhanced chemiluminescence solution was from Millipore (Billerica, MA, USA). The MAPK inhibitors, SP600125, U0126, and PD169136 were obtained from LC Laboratories (Woburn, MA, USA). The following antibodies were used: phospho-Akt (Ser473), phospho-S6 kinase 1 (S6K1) (Thr389), phospho-Erk1/2 (Thr202/Tyr204), phospho-p38 (Thr180/Tyr182), phospho-4E-BP1 (Thr70), 4E-BP1, caspase-3 (all from Cell Signaling Technology, Beverly, MA, USA), Akt, S6K1, Erk2, JNK1, phospho-JNK (Thr183/Tyr185), c-Jun, phospho-c-Jun (Ser63), p38 (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA), β-tubulin (Sigma), goat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP (Pierce, Rockford, IL, USA). 4′,6-diamidino-2-phenylindole (DAPI), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), poly-d-lysine (PDL), and celastrol were from Sigma. Other chemicals were purchased from local commercial sources and were of analytical grade.
Cell culture
Rat pheochromocytoma (PC12) cell line was from American Type Culture Collection (ATCC) (Manassas, VA, USA), which was used for no more than 10 passages, and maintained in antibiotic-free Dulbecco’s modified Eagle’s medium supplemented with 10% horse serum and 5% fetal bovine serum. Cells were incubated at 37°C in a humidified incubator containing 5% CO2.
To isolate primary neurons, female ICR mice were purchased from the Laboratory Animal Center, Nanjing Medical University (Nanjing, China). Animals were handled in accordance with the ARRIVE guidelines and the guidelines of the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals. Fetal mouse cerebral cortexes of 14–18 days of gestation were used and primary murine neurons were isolated as described (Chen et al. 2010). Next, the cells were seeded at a density of 2 × 106 cells/well in a 6-well plate coated with 10 μg/mL PDL in NEUROBASAL™ Media (Invitrogen) supplemented with 2% B27 Supplement (Invitrogen), 2 mM glutamine (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 5 μg/mL insulin (Sigma), and 40 μg/mL of gentamicin (Invitrogen), and grown in a humid incubator (37°C, 5% CO2). Fresh medium was replaced every 3 days. The cells were used for experiments after 6 days of culture.
Recombinant adenoviral constructs and infection of cells
The recombinant adenoviral vectors encoding FLAG-tagged dominant negative c-Jun (FLAG-Δ169) (Ad-dn-c-Jun), wild-type human PTEN (Ad-PTEN), and green fluorescence protein (Ad-GFP) were described previously (Chen et al. 2008, 2011b). The viruses were amplified, titrated, and used as described (Huang et al. 2003; Liu et al. 2008). For experiments, PC12 cells were grown in the growth medium and infected with the individual adenovirus for 24 h at five of multiplicity of infection (MOI = 5). Subsequently, cells were used for experiments. Ad-GFP served as a control. Expression of PTEN and FLAG-tagged dn-c-Jun was determined by western blot analysis with antibodies to PTEN and FLAG, respectively.
Cell viability evaluation and morphological analysis
PC12 cells were seeded in a 96-well plate (1 × 104 cells/well) or 6-well plate (5 × 105 cells/well), pre-coated with PDL (0.2 μg/mL). Next day, cells were treated with different concentration of celastrol (0–10 μM) for 24 h, with/without Cd (10 and 20 μM) for 24 h following pre-incubation with/without celastrol (0.1, 0.5, and 1 μM) for 1 h, or with/without Cd (10 μM) for 24 h following pre-incubation with/without celastrol (1 μM) in the presence or absence of SP600125 (20 μM), U0126 (5 μM), or PD169136 (20 μM) for 1 h with five replicates of each treatment. In some cases, after infection with Ad-PTEN, Ad-dn-c-Jun, or Ad-GFP, the cells were pre-treated with 1 μM celastrol for 1 h and then exposed to Cd (10 μM) for 24 h. Subsequently, cell viability was evaluated by MTT assay as described (Chen et al. 2011c). The images for morphological analysis were taken with a Nikon Eclipse TE2000-U inverted phase-contrast microscope (Nikon, Tokyo, Japan) (200×) equipped with a digital camera.
LDH release assay
Cytotoxicity was determined by measuring cell membrane damage through the release of lactate dehydrogenase (LDH). PC12 cells were seeded in PDL-coated 96-well plates and incubated overnight in the complete growth medium, and then treated with celastrol at the indicated concentrations (0–10 μM) for 24 h. LDH activities in culture medium were evaluated using a LDH assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the protocol suggested by the supplier.
DAPI staining
Cells were seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. Next day, cells were exposed to Cd (10 and 20 μM) following pre-incubation with/without celastrol (0.1, 0.5 and 1 μM) for 1 h. After treatment with Cd for 24 h, the cells with fragmented and condensed nuclei were determined using DAPI staining as described (Chen et al. 2008). Photographs were taken with a fluorescence microscope (Nikon 80i, Japan) equipped with a digital camera.
Western blot analysis
PC12 cells and/or primary neurons were seeded at a density of 2 × 106 cells/well in a PDL-coated 6-well plate. Next day, cells were treated with/without Cd (10 and 20 μM) for 12 h following pre-incubation with/without celastrol (0.1, 0.5, and 1 μM) for 1 h, or with/without Cd (10 μM) for 12 h following pre-incubation with/without celastrol (1 μM) in the presence or absence of SP600125 (20 μM), U0126 (5 μM), or PD169136 (20 μM) for 1 h. In addition, after infection with Ad-PTEN, Ad-dn-c-Jun or Ad-GFP, PC12 cells were pre-treated with celastrol (1 μM) for 1 h and then exposed to Cd (10 μM) for 12 h. Finally, western blotting was performed as described (Chen et al. 2008). The blots for detected proteins were semi-quantified using NIH Image J software (http://rsb.info.nih.gov/nih-image/) and were normalized using β-tubulin as an internal control.
Statistical analysis
Results were expressed as Mean ± SE. The Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one-way or two-way anova followed by Bonferroni’s post-tests to compare replicate means. Significance was accepted at p < 0.05.
Results
Celastrol attenuates Cd-induced cell viability reduction and morphological change in neuronal cells
To find an appropriate concentration of celastrol for the studies, we first performed cell viability assay for PC12 cells treated with celastrol. As shown in Fig. 1a, at low concentrations (0.1–1 μM), treatment of PC12 cells with celastrol for 24 h did not affect cell viability significantly. However, at high concentrations (> 1.5 μM), celastrol reduced the cell viability significantly and in a concentration-dependent manner (Fig. 1a). This is consistent with the notion that celastrol displays cytotoxicity when its concentration exceeds cell toleration (Sun et al. 2010). Our LDH release assay further showed that significant cytotoxic effects triggered by celastrol appeared at high concentrations (2–10 μM), but not at low concentrations (0.1–1.5 μM) (Fig. 1b). Therefore, the data indicate that 0.1–1 μM celastrol is not toxic to PC12 cells, which can be used to study its protective effect on Cd-induced neurotoxicity.
Fig. 1.
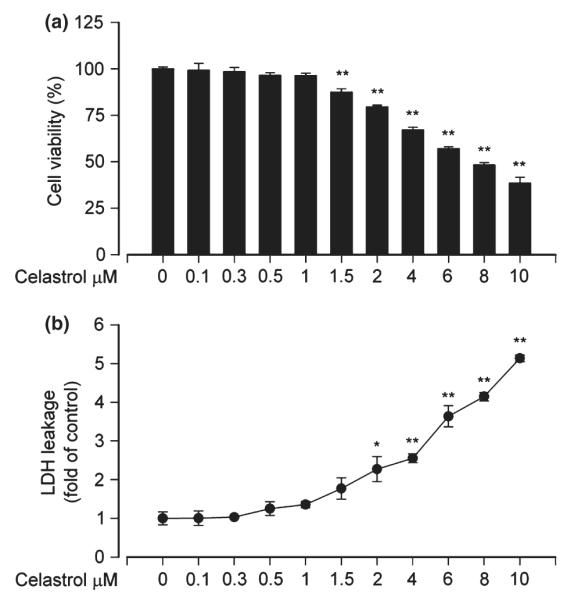
Celastrol does not have cytotoxic effects on PC12 cells at low concentrations. Pheochromocytoma (PC12) cells were treated with 0–10 μM celastrol for 24 h. (a) Cell viability was evaluated using 3-(4,5-dimethylazol-2-yl)-2,5- diphenyltetrazolim bromide (MTT) assay, and (b) lactate dehydrogenase (LDH) activities in culture medium were determined by LDH release assay, showing that 0.1–1 μM celastrol had no cytotoxic effects on PC12 cells in culture. Results are presented as mean ± SE, n = 5. *p < 0.05, **p < 0.01, difference with control group.
To test whether celastrol exerts a protective effect on Cd-induced neuronal cell death, PC12 cells were pre-incubated with celastrol (0–1 μM) for 1 h, followed by treatment with/without Cd (10 and 20 μM) for 24 h. By phase-contrast microscopic observation, many PC12 cells, because of exposure to Cd alone, became round or shrunken. Celastrol alone did not apparently alter cell morphology, but markedly prevented Cd-induced morphological change in the cells (Fig. 2a). Consistently, we observed that cell viability in the Cd plus celastrol group was significantly higher than that in the Cd alone group (Fig. 2b). At 1 μM, celastrol exhibited a best protection against Cd-reduced viability or altered morphology in the cells (Fig. 2a and b). The results suggest that celastrol may prevent Cd-induced neuronal cell death.
Fig. 2.

Celastrol attenuates cadmium (Cd)-reduced viability and altered morphology in neuronal cells. Pheochromocytoma (PC12) cells were pre-treated with celastrol (0–1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 24 h. (a) Morphology of PC12 cells was visualized under a Nikon Eclipse TE2000-U inverted phase-contrast microscope (200×) equipped with a digital camera. Scale bar: 100 μm. (b) Cell viability was evaluated by 3-(4,5-dimethylazol-2-yl)-2,5- diphenyltetrazolim bromide (MTT) assay. Results are presented as mean ± SE, n = 5. ap < 0.05, difference with control group; bp < 0.05, difference with 10 μM Cd group; cp < 0.05, difference with 20 μM Cd group.
Celastrol prevents Cd-induced apoptosis in neuronal cells
Earlier studies have consistently shown that Cd mediates neurotoxicity, which is attributed to its induction of neuronal cell apoptosis (Lopez et al. 2003; Mendez-Armenta and Rios 2007). To evaluate the effect of celastrol on Cd-induced neuronal apoptosis, PC12 cells were treated with Cd (10 and 20 μM) for 24 h following pre-incubation with celastrol for 1 h. Subsequently, DAPI staining was used to assess nuclear fragmentation and condensation, a hallmark of apoptosis (Hao et al. 2013). We found that treatment with Cd for 24 h significantly increased nuclear fragmentation and condensation, which was markedly attenuated by celastrol in a concentration-dependent fashion (Fig. 3a and b), revealing that celastrol has an obvious protective effect on Cd-induced neuronal apoptosis.
Fig. 3.

Celastrol prevents cadmium (Cd)-induced apoptosis of neuronal cells. Indicated cells were pre-treated with celastrol (0–1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 12 h (for western blotting) or 24 h (for 4′, 6-diamidino-2-phenylindole (DAPI) staining). (a) DAPI staining displayed nuclear fragmentation and condensation (arrows) in pheochromocytoma (PC12) cells. Scale bar: 20 μm. (b) Celastrol markedly attenuated Cd-increased percentage of cells with fragmented nuclei in PC12 cells in a concentration-dependent manner. (c) Indicated cell lysates were subjected to western blot analysis using antibodies to cleaved-caspase-3, and (d) blots for cleaved-caspase-3 were semi-quantified using NIH image J. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. Results are presented as mean ± SE; n = 3–5. ap < 0.05, difference with control group; bp < 0.05, difference with 10 μM Cd group; cp < 0.05, difference with 20 μM Cd group.
To gain more insights into in the event that celastrol possesses neuroprotection by reversing Cd-induced neuronal apoptosis, we determined proteolytic cleavages of caspase-3 in PC12 cells and primary neurons. Our western blot results showed that treatment with Cd resulted in robust activation of caspase-3 in PC12 cells and primary neurons, as detected by increased cleavages of caspase-3 (Fig. 3c and d). However, celastrol strikingly blocked the event dose-dependently, consistent with the result from DAPI staining.
Celastrol inhibits Cd-induced neuronal cell death by blocking JNK pathway
Studies have shown that celastrol may alter the activity of MAPKs including JNK, Erk1/2, and/or p38 under different conditions (Yu et al. 2010; Zhu et al. 2010; Chen et al. 2011a; Kannaiyan et al. 2011; Boridy et al. 2012). We have demonstrated that Cd activates JNK, Erk1/2, and p38, but only JNK and Erk1/2 participate in Cd-induced apoptosis in neuronal cells (Chen et al. 2008). Therefore, we hypothesized that celastrol might prevent Cd-induced neuronal apoptosis by inhibiting MAPK pathway. To this end, PC12 cells and primary neurons were pre-treated with celastrol (0–1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 12 h, followed by western blot analysis. We found that celastrol slightly activated the basal level of phosphorylation of JNK, Erk1/2, and p38 in PC12 cells and primary neurons (Fig. 4a–d). However, the effects of celastrol on Cd-activated MAPKs were complex. It appeared that celastrol remarkably inhibited Cd-induced phosphorylation of JNK, particularly protein expression and phosphorylation of c-Jun, a substrate of JNK (Fig. 4a and b). Celastrol slightly enhanced Cd-induced phosphorylation of Erk1/2, but did not obviously affect Cd-induced phosphorylation of p38 (Fig. 4c and d). Our previous studies have demonstrated that Cd activates JNK, Erk1/2, and p38, but only JNK and Erk1/2 participate in Cd-induced apoptosis of neuronal cells (Chen et al. 2008). To determine how JNK, Erk1/2 and p38 are involved in celastrol prevention against Cd-induced cell death, SP600125 (JNK inhibitor), U0126 (MEK1/2 inhibitor), and PD169136 (p38 inhibitor) were employed. When PC12 cells were treated with celastrol (1 μM), SP600125 (20 μM), U0126 (5 μM), or PD169136 (20 μM) alone, or co-treated with celastrol (1 μM)/SP600125 (20 μM), U0126 (5 μM), or PD169136 (20 μM) for 1 h, and then exposed to Cd (10 μM) for 12 h, we found that celastrol or SP600125 alone remarkably attenuated Cd-induced activation of JNK/c-Jun and caspase-3 in PC12 cells (Fig. 4e and f). Furthermore, co-treatment with celastrol/SP600125 exhibited a stronger inhibitory effect on Cd-induced JNK and caspase activation (Fig. 4e and f). In line with this, co-treatment with celastrol/SP600125 also rescued cells from Cd-induced death more potently than celastrol or SP600125 alone (Fig. 4g). In addition, consistent with our previous findings (Chen et al. 2008), although U0126 (5 μM) and PD169136 (20 μM) blocked Cd-induced phosphorylation of Erk1/2 and p38, respectively, only U0126 partially attenuated Cd-induced cell death. However, neither celastrol/U0126 nor celastrol/PD169136 co-treatment enhanced the protective effect of celastrol on Cd-induced cytotoxicity (data not shown). Taken together, our data suggest that celastrol may prevent Cd-induced neuronal cell death partially by blocking JNK pathway.
Fig. 4.

Celastrol inhibits cadmium (Cd)-induced neuronal cell death by blocking c-Jun N-terminal kinase (JNK) pathway. (a, c) Pheochromocytoma (PC12) cells and primary neurons were pre-treated with celastrol (0–1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 12 h, followed by western blot analysis with antibodies against the indicated proteins. Celastrol partially blocked Cd-induced phosphorylation of JNK/c-Jun (a, b), but not extracellular signal-regulated kinase 1/2 (Erk1/2) and p38 (c, d). (e, g) PC12 cells were pre-treated with or without celastrol (1 μM) and/or SP600125 (20 μM) for 1 h, and then exposed to Cd (10 μM) for 12 h (for western blotting) or 24 h [for 3-(4,5-dimethylazol-2-yl)-2,5- diphenyltetrazolim bromide (MTT) assay]. Cells were harvested and total lysates were subjected to western blot analysis using indicated antibodies (e), or cell viability was evaluated by MTT assay (g). For (a), (c), and (e), similar results were observed in at least three independent experiments, and blots for p-JNK, p-c-Jun, p-Erk1/2, p-p38, cleaved-caspase-3 were semi-quantified (b, d, f). Results are presented as mean ± SE; n = 3–5. ap < 0.05, difference with control group; bp < 0.05, difference with 10 μM Cd group; cp < 0.05, difference with 20 μM Cd group; dp < 0.05, difference with Cd/SP600125 group or Cd/Celastrol group.
Expression of dominant negative c-Jun strengthened celastrol inhibition of Cd-induced neuronal apoptosis
To confirm the finding that celastrol inhibits Cd-activation of JNK cascade, preventing cell death, PC12 cells, infected with recombinant adenoviral vectors expressing dominant negative (dn) c-Jun (Ad-dn-c-Jun) and GFP (Ad-GFP) (as control), respectively, were pre-treated with celastrol (1 μM) for 1 h, and then exposed to Cd (10 μM) for 12 h. Western blot analysis revealed that ectopic expression of dn-c-Jun obviously blocked Cd-induced phosphorylation of c-Jun (Fig. 5a). Consistently, Cd activation of caspase-3 was apparently attenuated by expression of dn-c-Jun (Fig. 5a and b). Furthermore, our cell viability assay revealed that expression of dn-c-Jun also partially protected PC12 cells from death induced by Cd (Fig. 5c). Moreover, addition of celastrol exhibited more inhibitory effect on Cd-activated c-Jun and cell death (Fig. 5a–c). Therefore, our results indicate that celastrol inhibits Cd-induced cell death in neuronal cells at least in part through blocking JNK cascade.
Fig. 5.

Expression of dominant negative c-Jun strengthens celastrol inhibition of cadmium (Cd)-induced neuronal cell death. Over-expression of FLAG-tagged dominant negative c-Jun by infection of pheochromocytoma (PC12) cells with Ad-dn-c-Jun, as detected by western blotting with antibodies to FLAG, obviously attenuated Cd-induced phosphorylation of c-Jun and expression of cleaved-caspase-3 protein (a), and blots for cleaved-caspase-3 were semi-quantified (b). Ectopic expression of dn-c-Jun also partially rescued cells from death induced by Cd (c). Addition of celastrol exhibited more significant inhibition on Cd-induced activation of c-Jun and cell death in Ad-dn-c-Jun group than in Ad-GFP group (a–c). Results are presented as mean ± SE; n = 3–5. ap < 0.05, difference with control group; bp < 0.05, Ad-dn-c-Jun group versus Ad-GFP group.
Celastrol blocks Cd-induced neuronal cell death by preventing Cd inactivation of PTEN and activation of Akt/mTOR
Akt/mTOR signaling is crucial not only for cell proliferation/growth but also for cell survival (Bai and Jiang 2010; Don et al. 2012). Studies have implicated that celastrol inhibits proliferation and survival by inhibiting Akt/mTOR pathway in tumor cells (Pang et al. 2010; Kannaiyan et al. 2011; Li et al. 2012). Our previous studies have identified that Cd activates Akt/mTOR pathway leading to apoptosis of the neuronal cells (Chen et al. 2008). Therefore, we asked whether celastrol inhibits Cd-induced neuronal apoptosis by blocking Cd-activated Akt/mTOR signaling. To this end, the effect of celastrol on Cd-induced phosphorylation of Akt, S6K, and 4E-BP1 was detected. We found that pre-treatment with celastrol remarkably inhibited Cd-induced phosphorylation of Akt, S6K, and 4E-BP1 in PC12 cells and primary neurons (Fig. 6a and b). At 1 μM, celastrol almost completely blocked the events (Fig. 6a and b). Consistent with our previous finding (Chen et al. 2011b), we found that Cd-activated Akt/mTOR was associated with down-regulation of PTEN, a negative regulator of Akt-mTOR pathway (Fig. 6a and b). Interestingly, treatment with celastrol was able to strikingly prevent Cd-induced decrease in PTEN expression dose-dependently in PC12 cells and primary neurons (Fig. 6a and b), suggesting that celastrol inhibits Cd-activation of Akt/mTOR probably by preventing Cd from reducing PTEN expression.
Fig. 6.

Celastrol blocks Cd-induced neuronal cell death by preventing cadmium (Cd) from reducing phosphatase and tensin homolog deleted on chromosome 10 (PTEN) expression and activating Akt/mammalian target of rapamycin (mTOR) pathway. (a) Pheochromocytoma (PC12) cells and primary neurons were pre-treated with celastrol (0–1 μM) for 1 h, and then exposed to Cd (10 and 20 μM) for 12 h, following by western blot analysis with antibodies against the indicated proteins. Celastrol inhibited Cd-induced phosphorylation of Akt, S6K, and 4E-BP1, with a concomitant restoring of Cd-down-regulated PTEN dose-dependently. (c, e, f) PC12 cells, infected with Ad-PTEN-wt or Ad-GFP (as control), were pre-treated with celastrol (1 μM) for 1 h, and then exposed to Cd (10 μM) for 12 h (for western blotting) or 24 h [for 3-(4,5-dimethylazol-2-yl)-2,5- diphenyltetrazolim bromide (MTT) assay or morphological analysis], followed by (c) western blot analysis using the indicated antibodies, (e) cell viability evaluation using MTT assay, or (f) morphological analysis using a Nikon Eclipse TE2000-U inverted phase-contrast microscope (200×) equipped with a digital camera. Scale bar: 100 μm. For (a) and (c), similar results were observed in at least three independent experiments, and blots for PTEN, p-Akt, p-S6 kinase 1 (S6K1), p-4E-BP1, cleaved-caspase-3 were semi-quantified (b, d). Results are presented as mean ± SE; n = 3–5. ap < 0.05, difference with control group; bp < 0.05, difference with 10 μM Cd group; cp < 0.05, difference with 20 μM Cd group; dp < 0.05, Ad-PTEN group versus Ad-GFP group.
To corroborate the above finding, PC12 cells were infected with recombinant adenoviral vector (Ad-PTEN) encoding wild-type human PTEN or Ad-GFP (as control) and then exposed to Cd (10 μM) for 12 h post pre-incubation with/without celastrol (1 μM) for 1 h, followed by western blot analysis. We observed that the infection with Ad-PTEN increased the expression of PTEN and slightly inhibited the basal levels of phosphorylation of Akt and S6K1, compared to the infection with Ad-GFP (Fig. 6c and d). In agreement with the above result, treatment with Cd decreased PTEN expression, and correspondingly increased phosphorylation of Akt and S6K1 in the control cells infected with Ad-GFP (Lane 3 vs. Lane 1). Over-expression of PTEN blocked Cd-activated phosphorylation of Akt and S6K1 (Lane 7 vs. Lane 3) (Fig. 6c and d). Celastrol attenuated Cd-induced decrease in PTEN expression and increase in Akt/S6K1 phosphorylation (Lane 4 vs. Lane 3). Furthermore, over-expression of PTEN was able to potentiate the inhibitory effect of celastrol on Cd-induced phosphorylation of Akt/S6K1 (Lane 8 vs. Lane 4). Moreover, over-expression of PTEN also enhanced the protective effect of celastrol against Cd-induced cleavage of caspase-3 (Lane 8 vs. Lane 4) (Fig. 6c and d). By cell viability assay and morphological analysis, we observed that over-expression of PTEN alone partially prevented Cd-induced cell death in PC12 cells (Fig. 6e and f). Addition of celastrol elicited more significant protection against Cd-induced cell death (Fig. 6e and f). Collectively, the findings support the notion that celastrol inhibits Cd-activated Akt/mTOR pathway, as well as cell death in neuronal cells, by preventing Cd from reducing PTEN expression.
Discussion
Cadmium, as one of the most toxic environmental and industrial pollutants, targets several organs and tissues such as kidney (Johri et al. 2010), blood (Kocak and Akcil 2006), bones (Akesson et al. 2006), testis (Thompson and Bannigan 2008), and brain (Okuda et al. 1997; Lopez et al. 2003; Mendez-Armenta and Rios 2007), resulting in nephrotoxicity, immunotoxicity, osteotoxicity, genotoxicity, neurotoxicity, and tumors following either acute or chronic exposure. Current effective therapy for acute Cd- poisoning is mainly utilizing chelating agents to increase the excretion of Cd, but this method is not suitable for long-term exposure to Cd because of the side effects of the chelators (Nordberg 1984; Sinicropi et al. 2010). Celastrol, a natural compound extracted from the plant Tripterygium wilfordii, has been recognized as an effective agent for neurodegenerative diseases because of its neuroprotective effects on PD, AD, and ALS (Allison et al. 2001; Cleren et al. 2005; Kiaei et al. 2005). However, whether it has a protective effect on Cd-induced neurotoxicity remains unknown. Here, for the first time, we present evidence that celastrol protects against Cd-induced apoptotic cell death in neuronal cells. This is supported by the findings that celastrol attenuated Cd-reduced viability and altered morphology of neuronal cells, and strikingly blocked nuclear fragmentation and condensation, and activation of caspase-3 in neuronal cells induced by Cd exposure. These results are in line with the recent studies on natural compound inhibiting Cd toxicity. Abib et al. have demonstrated that epigallocatechin 3-gallate plays a potent protective role in Cd-induced rat brain mitochondrial dysfunction (Abib et al. 2011). Similarly, curcumin and resveratrol also individually prevent Cd-induced oxidative damage (Eybl et al. 2006). Our data suggest that celastrol has an ability to detoxicate Cd in neuronal cells as well.
In the studies, we found that celastrol inhibited Cd-induced phosphorylation of JNK including its downstream molecule, protein expression and phosphorylation of c-Jun, but not Cd-induced phosphorylation of Erk1/2 and p38 (Fig. 4b) in PC12 cells and primary neurons. Our previous studies have demonstrated that Cd activates JNK, Erk1/2, and p38, but only JNK and Erk1/2 participate in Cd-induced apoptosis of neuronal cells (Chen et al. 2008). To confirm the role of celastrol in inhibition of Cd-induced activation of JNK pathway and neuronal apoptosis, pharmacological inhibitor for JNK activity or genetic manipulation for c-Jun activity was utilized. Co-treatment with celastrol and JNK inhibitor SP600125 attenuated Cd-induced activation of JNK/c-Jun and caspase-3, as well as apoptosis of neuronal cells more potently than single treatment with celastrol or SP600125. Furthermore, ectopic expression of dn-c-Jun also strengthened celastrol inhibition of Cd-induced neuronal apoptosis. Our finding is in consistence with a recent report that celastrol has protective effect in rat cerebral ischemia model through down-regulation of phospho-JNK and phospho-c-Jun (Li et al. 2012). Therefore, our data support the notion that celastrol protects against Cd-induced neuronal apoptosis partially by blocking JNK pathway.
It is worth mentioning that the effects of celastrol on Cd-activated MAPKs in neuronal cells were unexpectedly complicated. In this study, we found that celastrol slightly activated the basal level of phosphorylation of JNK, Erk1/2, and p38. However, celastrol inhibited Cd-induced phosphorylation of JNK, slightly activated Cd-induced Erk1/2, and did not obviously affect Cd-induced activation of p38 in PC12 cells. Our results are is in contrast to the findings in non-neuronal cells. For instance, celastrol exerts its anti-inflammatory activity by inhibiting Erk1/2 in basophilic leukemia (RBL-2H3) cells (Kim et al. 2009), and attenuates hypertension-induced inflammation and oxidative stress partially by inhibiting Erk1/2 in vascular smooth muscle cells (Yu et al. 2010). Celastrol inhibits cell adhesion and β1 integrin function partially by activating p38 MAPK in mouse melanoma (B16F10) and human lung cancer (95-D) cells (Zhu et al. 2010), and induces apoptosis and autophagy by activating p38, JNK and Erk1/2 in cervix (HeLa), lung (A549), and prostate (PC-3 cancer cells) (Wang et al. 2012). Celastrol inhibits lipopolysaccharide-induced inflammatory signaling and cytokine release from microglial cells by inhibiting p38 (Boridy et al. 2012). The above findings suggest that the effects of celastrol on MAPKs are dependent on cell lines or experimental conditions (e.g., stimuli) used.
Akt/mTOR signaling is crucial for cell growth, differentiation, and survival (Bai and Jiang 2010; Don et al. 2012). Studies have shown that celastrol suppresses proliferation and promotes apoptosis in many kinds of tumor cells via blocking Akt/mTOR pathway (Pang et al. 2010; Yu et al. 2010; Kannaiyan et al. 2011; Lee et al. 2012). Our previous evidence has demonstrated that Cd activates Akt/mTOR signaling pathway, promoting neuronal apoptosis (Chen et al. 2008). This prompted us to test whether celastrol exerts a protective effect through inhibition of Cd-activated Akt/mTOR signaling in neuronal cells. As expected, we found that pre-treatment with celastrol did obviously attenuate Cd-induced phosphorylation of Akt, S6K, and 4E-BP1, as well as cell death in PC12 cells and primary neurons. Recently, we have further observed that Cd inactivation of PTEN results in activation of Akt/mTOR signaling and apoptosis of neuronal cells, implying that loss of PTEN function may promote development of neurodegenerative disorders (Chen et al. 2011b). In this study, we noticed that celastrol suppressed Cd-activated Akt/mTOR by preventing Cd from reducing PTEN expression. Of interest, over-expression of wild-type PTEN was able to further enhance celastrol blockage of Cd-induced activation of Akt/mTOR signaling and apoptosis of neuronal cells. Therefore, our findings strongly suggest that celastrol inhibits Cd-induced activation of Akt/mTOR signaling pathway and apoptosis in neuronal cells, at least in part by elevating the negative regulator PTEN.
A new question that arises from this work is how celastrol suppresses Cd-induced JNK activation and PTEN reduction. It is well known that celastrol belongs to triterpene family. Studies have shown the natural triterpenoids have a potentiality for attenuating Cd toxicity (Sunitha et al. 2001; Renugadevi and Prabu 2010a,b). For example, quercetin protects against oxidative stress-related renal dysfunction induced by Cd in rats (Renugadevi and Prabu 2010b). Cd-induced hepatotoxicity in rats is ameliorated by lupeol (Sunitha et al. 2001) and naringenin (Renugadevi and Prabu 2010a). Further investigation has demonstrated that the triterpenoids increase resistance to oxidative stress and act as agents with antioxidative bioactivity (Dumont et al. 2009). In response to oxidative stress, JNK can be activated (Choi et al. 2011; Yang et al. 2013), and PTEN can be down-regulated (Flaherty et al. 2006; Chen et al. 2011b; Kim et al. 2013). In addition, superoxide can also oxidize PTEN, leading to inactivation of PTEN (Lim and Clement 2007). Currently, we have no idea whether Cd affects oxidation of PTEN. However, recent studies have reported that celastrol’s oxidative stability is characterized by potently inhibiting NADPH oxidases (NOXs) activity (Jaquet et al. 2011). Celastrol specifically bound to p47phox and disrupted the binding of p22phox to the tandem SH3 domain of NOX organizer type 1 (NOXO1) and p47phox, even inhibited superoxide production by NOX2 and NOX5 (Jaquet et al. 2011). Our group has demonstrated that Cd induces the generation of reactive oxygen species by up-regulating the expression of NOX2 and its regulatory proteins (p22phox, p67phox, p40phox, p47phox, and Rac1) in PC12 and SH-SY5Y cells (Chen et al. 2011b). Therefore, we deduce that celastrol is likely to act by mechanisms that counteract Cd-induced oxidative stress, thereby not only preventing Cd-induced activation of JNK but also blocking Cd-induced down-regulation of PTEN. Celastrol may be involved in modulating macromolecular interactions and expression of genes associated with Cd-induced neurotoxicity. Undoubtedly, more studies are needed to address these issues.
In this study, we showed that celastrol, at high concentrations (> 1.5 μM), reduced cell viability in a concentration-dependent manner, which is consistent with a recent report (Deng et al. 2013). Therefore, much more attention should be paid for use of celastrol and its related preparations. Especially, detailed pharmacokinetic data of celastrol are in great demand to achieve the best therapeutic effect and least toxicity of celastrol. The pharmacokinetics of celastrol has been studied in rats for oral administration or intravenous injection of celastrol (Zhang et al. 2012), and in human for oral administration of celastrol tablets for more than 2 days (Xu et al. 2007). Celastrol’s bioavailability in brain has not been reported, but the neuroprotective effects of celastrol on neurodegenerative diseases (such as PD, AD, and ALS) imply that celastrol could cross the blood–brain barrier. These data provide valuable reference for further studying the effect of celastrol on in vivo Cd neurotoxicity.
In conclusion, we have identified that celastrol prevented Cd-induced neuronal apoptosis via inhibiting activation of JNK and Akt/mTOR signaling pathways. Celastrol suppressed Cd-activated Akt/mTOR signaling pathway by preventing Cd from reducing PTEN expression. Our results underscore a potential role for celastrol in the prevention of Cd-induced neurodegenerative disorders.
Acknowledgements
This study was supported in part by the grants from National Natural Science Foundation of China (30971486, 81271416; L.C.), NIH (CA115414; S.H.), American Cancer Society (RSG-08-135-01-CNE; S.H.), Project for the Priority Academic Program Development and Natural Science Foundation of Jiangsu Higher Education Institutions of China (10KJA180027; L.C.), Louisiana Board of Regents (NSF-2009-PFUND-144; S.H.), NSFC for Talents Training in Basic Science (J1103507, J1210025; C.G., L.C.), and Innovative Research Program of Jiangsu College Graduate of China (CXZZ11-0888; S.C.). The authors declare no conflict of interest.
Abbreviations used
- 4E-BP1
eukaryotic initiation factor 4E binding protein 1
- AD
Alzheimer disease
- Akt
protein kinase B (PKB)
- ALS
amyotrophic lateral sclerosis
- Cd
cadmium
- DAPI
4′, 6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- Erk1/2
extracellular signal-regulated kinase 1/2
- FBS
fetal bovine serum
- JNK
c-Jun N-terminal kinase
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- MEK
mitogen extracellular kinase
- mTOR
mammalian target of rapamycin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
phosphate buffered saline
- PDL
poly-d-lysine
- PD
Parkinson’s disease
- PI3K
phosphoinositide 3′-kinase
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- S6K1
S6 kinase 1
References
- Abib RT, Peres KC, Barbosa AM, et al. Epigallocatechin-3-gallate protects rat brain mitochondria against cadmium-induced damage. Food Chem. Toxicol. 2011;49:2618–2623. doi: 10.1016/j.fct.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Akesson A, Bjellerup P, Lundh T, Lidfeldt J, Nerbrand C, Samsioe G, Skerfving S, Vahter M. Cadmium-induced effects on bone in a population-based study of women. Environ. Health Perspect. 2006;114:830–834. doi: 10.1289/ehp.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- Bai X, Jiang Y. Key factors in mTOR regulation. Cell. Mol. Life Sci. 2010;67:239–253. doi: 10.1007/s00018-009-0163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boridy S, Soliman GM, Maysinger D. Modulation of inflammatory signaling and cytokine release from microglia by celastrol incorporated into dendrimer nanocarriers. Nanomedicine. 2012;7:1149–1165. doi: 10.2217/nnm.12.16. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Luo Y, Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J. Neurochem. 2008;105:251–261. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab. Invest. 2010;90:762–773. doi: 10.1038/labinvest.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Zhang X, Zhao M, Wang Y, Cheng X, Wang D, Xu Y, Du Z, Yu X. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer. 2011a;11:170. doi: 10.1186/1471-2407-11-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic. Biol. Med. 2011b;50:624–632. doi: 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu Y, Xu B, et al. CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J. Neurochem. 2011c;119:1108–1118. doi: 10.1111/j.1471-4159.2011.07493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Kang KS, Fukui M, Zhu BT. CriticalroleoftheJNK-p53-GADD45alpha apoptotic cascade in mediating oxidative cytotoxicity in hippocampal neurons. Br. J. Pharmacol. 2011;162:175–192. doi: 10.1111/j.1476-5381.2010.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005;94:995–1004. doi: 10.1111/j.1471-4159.2005.03253.x. [DOI] [PubMed] [Google Scholar]
- Deng YN, Shi J, Liu J, Qu QM. Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem. Int. 2013;63:1–9. doi: 10.1016/j.neuint.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Don AS, Tsang CK, Kazdoba TM, D’Arcangelo G, Young W, Zheng XF. Targeting mTOR as a novel therapeutic strategy for traumatic CNS injuries. Drug Discov. Today. 2012;17:861–868. doi: 10.1016/j.drudis.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M, Wille E, Calingasan NY, et al. Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2009;109:502–512. doi: 10.1111/j.1471-4159.2009.05970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eybl V, Kotyzova D, Koutensky J. Comparative study of natural antioxidants - curcumin, resveratrol and melatonin - in cadmium-induced oxidative damage in mice. Toxicology. 2006;225:150–156. doi: 10.1016/j.tox.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Flaherty DM, Monick MM, Hinde SL. Human alveolar macrophages are deficient in PTEN. The role of endogenous oxidants. J. Biol. Chem. 2006;281:5058–5064. doi: 10.1074/jbc.M508997200. [DOI] [PubMed] [Google Scholar]
- Hao B, Cheng S, Clancy CJ, Nguyen MH. Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis. Antimicrob. Agents Chemother. 2013;57:326–332. doi: 10.1128/AAC.01366-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Shu L, Dilling M, Easton J, Harwood F, Ichijo H, Houghton P. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol. Cell. 2003;11:1491–1501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Jaquet V, Marcoux J, Forest E, et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011;164:507–520. doi: 10.1111/j.1476-5381.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarup L, Persson B, Edling C, Elinder CG. Renal function impairment in workers previously exposed to cadmium. Nephron. 1993;64:75–81. doi: 10.1159/000187282. [DOI] [PubMed] [Google Scholar]
- Jiang G, Xu L, Song S, Zhu C, Wu Q, Zhang L, Wu L. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology. 2008;244:49–55. doi: 10.1016/j.tox.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Johri N, Jacquillet G, Unwin R. Heavy metal poisoning: the effects of cadmium on the kidney. Biometals. 2010;23:783–792. doi: 10.1007/s10534-010-9328-y. [DOI] [PubMed] [Google Scholar]
- Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A, Lam P, Kumar AP, Sethi G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis. 2011;16:1028–1041. doi: 10.1007/s10495-011-0629-6. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 2005;2:246–254. doi: 10.1159/000090364. [DOI] [PubMed] [Google Scholar]
- Kim SD, Moon CK, Eun SY, Ryu PD, Jo SA. Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochem. Biophys. Res. Commun. 2005;328:326–334. doi: 10.1016/j.bbrc.2004.11.173. [DOI] [PubMed] [Google Scholar]
- Kim Y, Kim K, Lee H, Han S, Lee YS, Choe J, Kim YM, Hahn JH, Ro JY, Jeoung D. Celastrol binds to ERK and inhibits FcepsilonRI signaling to exert an anti-allergic effect. Eur. J. Pharmacol. 2009;612:131–142. doi: 10.1016/j.ejphar.2009.03.071. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Jung HJ, Lim CJ. reactive oxygen species-dependent down-regulation of tumor suppressor genes PTEN, USP28, DRAM, TIGAR, and CYLD under oxidative stress. Biochem. Genet. 2013 doi: 10.1007/s10528-013-9616-7. doi:10.1007/s10528-013-9616-7. [DOI] [PubMed] [Google Scholar]
- Kocak M, Akcil E. The effects of chronic cadmium toxicity on the hemostatic system. Pathophysiol. Haemost. Thromb. 2006;35:411–416. doi: 10.1159/000102047. [DOI] [PubMed] [Google Scholar]
- Koyu A, Gokcimen A, Ozguner F, Bayram DS, Kocak A. Evaluation of the effects of cadmium on rat liver. Mol. Cell. Biochem. 2006;284:81–85. doi: 10.1007/s11010-005-9017-2. [DOI] [PubMed] [Google Scholar]
- Lee JH, Won YS, Park KH, Lee MK, Tachibana H, Yamada K, Seo KI. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis. 2012;17:1275–1286. doi: 10.1007/s10495-012-0767-5. [DOI] [PubMed] [Google Scholar]
- Li Y, He D, Zhang X, et al. Protective effect of celastrol in rat cerebral ischemia model: down-regulating p-JNK, p-c-Jun and NF-kappaB. Brain Res. 2012;1464:8–13. doi: 10.1016/j.brainres.2012.04.054. [DOI] [PubMed] [Google Scholar]
- Lim S, Clement MV. Phosphorylation of the survival kinase Akt by superoxide is dependent on an ascorbate-reversible oxidation of PTEN. Free Radic. Biol. Med. 2007;42:1178–1192. doi: 10.1016/j.freeradbiomed.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27:4998–5010. doi: 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP. Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br. J. Pharmacol. 2003;138:901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Armenta M, Rios C. Cadmium neurotoxicity. Environ. Toxicol. Pharmacol. 2007;23:350–358. doi: 10.1016/j.etap.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Nordberg GF. Chelating agents and cadmium toxicity: problems and prospects. Environ. Health Perspect. 1984;54:213–218. doi: 10.1289/ehp.8454213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda B, Iwamoto Y, Tachibana H, Sugita M. Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg. 1997;99:263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- Panayi AE, Spyrou NM, Iversen BS, White MA, Part P. Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J. Neurol. Sci. 2002;195:1–10. doi: 10.1016/s0022-510x(01)00672-4. [DOI] [PubMed] [Google Scholar]
- Pang X, Yi Z, Zhang J, Lu B, Sung B, Qu W, Aggarwal BB, Liu M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70:1951–1959. doi: 10.1158/0008-5472.CAN-09-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihl RO, Parkes M. Hair element content in learning disabled children. Science. 1977;198:204–206. doi: 10.1126/science.905825. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, et al. InhibitionofmTORinduces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Renugadevi J, Prabu SM. Cadmium-induced hepatotoxicity in rats and the protective effect of naringenin. Exp. Toxicol. Pathol. 2010a;62:171–181. doi: 10.1016/j.etp.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Renugadevi J, Prabu SM. Quercetin protects against oxidative stress-related renal dysfunction by cadmium in rats. Exp. Toxicol. Pathol. 2010b;62:471–481. doi: 10.1016/j.etp.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Rockwell P, Martinez J, Papa L, Gomes E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell. Signal. 2004;16:343–353. doi: 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: Molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun. 2010;394:439–442. doi: 10.1016/j.bbrc.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Sinicropi MS, Amantea D, Caruso A, Saturnino C. Chemical and biological properties of toxic metals and use of chelating agents for the pharmacological treatment of metal poisoning. Arch. Toxicol. 2010;84:501–520. doi: 10.1007/s00204-010-0544-6. [DOI] [PubMed] [Google Scholar]
- Sun H, Xu L, Yu P, Jiang J, Zhang G, Wang Y. Synthesis and preliminary evaluation of neuroprotection of celastrol analogues in PC12 cells. Bioorg. Med. Chem. Lett. 2010;20:3844–3847. doi: 10.1016/j.bmcl.2010.05.066. [DOI] [PubMed] [Google Scholar]
- Sunitha S, Nagaraj M, Varalakshmi P. Hepatoprotective effect of lupeol and lupeol linoleate on tissue antioxidant defence system in cadmium-induced hepatotoxicity in rats. Fitoterapia. 2001;72:516–523. doi: 10.1016/s0367-326x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Thompson J, Bannigan J. Cadmium: toxic effects on the reproductive system and the embryo. Reprod. Toxicol. 2008;25:304–315. doi: 10.1016/j.reprotox.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH, Yang M, Liu X, Guo DA. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J. Cell. Physiol. 2012;227:2196–2206. doi: 10.1002/jcp.22956. [DOI] [PubMed] [Google Scholar]
- Wright RO, Amarasiriwardena C, Woolf AD, Jim R, Bellinger DC. Neuropsychological correlates of hair arsenic, manganese, and cadmium levels in school-age children residing near a hazardous waste site. Neurotoxicology. 2006;27:210–216. doi: 10.1016/j.neuro.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Xu Q, Huang M, Jin M, Ren Q. LC-APCI-MS-MS for the determination of celastrol in human whole blood. Chromatographia. 2007;66:735–739. [Google Scholar]
- Yang Y, Hou L, Li Y, Ni J, Liu L. Neuronal necrosis and spreadingdeathinaDrosophilageneticmodel. CellDeathDis. 2013;4:e723. doi: 10.1038/cddis.2013.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Tao W, Jiang F, Li C, Lin J, Liu C. Celastrol attenuates hypertension-induced inflammation and oxidative stress in vascular smooth muscle cells via induction of heme oxygenase-1. Am. J. Hypertens. 2010;23:895–903. doi: 10.1038/ajh.2010.75. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li CY, Xu MJ, Wu T, Chu JH, Liu SJ, Ju WZ. Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J. Ethnopharmacol. 2012;144:195–200. doi: 10.1016/j.jep.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Zhu H, Liu XW, Cai TY, Cao J, Tu CX, Lu W, He QJ, Yang B. Celastrol acts as a potent antimetastatic agent targeting beta1 integrin and inhibiting cell-extracellular matrix adhesion, in part via the p38 mitogen-activated protein kinase pathway. J. Pharmacol. Exp. Ther. 2010;334:489–499. doi: 10.1124/jpet.110.165654. [DOI] [PubMed] [Google Scholar]