

Significance
In 1953, Stanley Miller reported on the spontaneous formation of glycine when applying an electric discharge on a mixture of simple molecules, giving birth to modern research on the origins of life. The effect of electric fields on mixtures of simple molecules is presently studied in computer simulations at the quantum level, and Miller results are reproduced for the first time, to our knowledge, in atomistic simulations, as glycine forms spontaneously only in the presence of electric fields. However, this occurs through reaction pathways more complex than believed, identifying formamide as a key compound in prebiotic chemistry. Moreover, electric fields are naturally present at mineral surfaces, suggesting a potentially crucial role in the biogeochemistry of both the primordial and the modern Earth.
Abstract
The celebrated Miller experiments reported on the spontaneous formation of amino acids from a mixture of simple molecules reacting under an electric discharge, giving birth to the research field of prebiotic chemistry. However, the chemical reactions involved in those experiments have never been studied at the atomic level. Here we report on, to our knowledge, the first ab initio computer simulations of Miller-like experiments in the condensed phase. Our study, based on the recent method of treatment of aqueous systems under electric fields and on metadynamics analysis of chemical reactions, shows that glycine spontaneously forms from mixtures of simple molecules once an electric field is switched on and identifies formic acid and formamide as key intermediate products of the early steps of the Miller reactions, and the crucible of formation of complex biological molecules.
In 1953, Miller reported (1) the surprising results he had achieved by the application of an electric discharge on a mixture of the gases CH4, NH3, H2O, and H2 that simulated what was considered at the time as a model atmosphere for the primordial Earth. The result of this experiment was a substantial yield of a mixture of amino acids (2–4). Similarly, Orò demonstrated the spontaneous formation of nucleic acid bases from aqueous hydrogen cyanide subject to heating and/or spark discharges (5). Since then, a number of different hypotheses have been formulated to explain the synthesis, from simple molecules (water, ammonia, methane, carbon oxides), of simple organic molecules (formaldehyde, hydrogen cyanide, formic acid), and from the latter ones to biological monomers (amino acids, purines, pyrimidines) up the ladder of the complexity of life (6, 7). However, a “standard model” explaining the emergence of larger and larger molecular assemblies is lacking. Various sources of energy that might have initiated this synthesis have been suggested in the last 60 y, including UV irradiation both in extraterrestrial and terrestrial environments (8–11), hydrothermal energy from deep sea vents (12, 13), shock waves from meteorite impact (14–20), redox energy in the “iron−sulphur world” hypothesis (21), radioactive emission from uranium accumulation (22), and, of course, electric discharges originated by lightning, which motivated the Miller experiment, and are the inspiration of the present work. Despite the historical importance of Miller’s experiments, no attempts have been made to explore the role of the electric field, traditionally considered merely in terms of just another thermal activation.
A key aspect of that experiment was the formation of hydrogen cyanide, aldehydes, ketones, and amino acids, suggesting that the compounds were produced by the Strecker cyanohydrin reaction (23). Subsequent Miller-like experiments, run with other types and combinations of gases (24, 25) representing plausible geochemical conditions, produced other classes of basic building block molecules, namely sugars and nucleotides. To date, all of the major classes of organic compounds found in biological systems have been synthesized in experiments of this nature; in particular, in the most recent study of Miller and coworkers (26), it was shown that a CO-CO2-N2-H2O atmosphere can give a variety of bioorganic compounds with yields comparable to those obtained from a strongly reducing atmosphere, suggesting that atmospheres containing carbon monoxide might therefore have been conducive to prebiotic synthesis and perhaps the origin of life.
Theory and modeling have generally provided considerable insight (27) in prebiotic chemistry, particularly by studying the synthesis of simple organic molecules and the mechanisms and barriers of such reactions on substrates such as ice (28, 29) or minerals (30, 31). A remarkable theoretical study, based on ab initio molecular dynamics (AIMD), on prebiotic synthesis focused on the simulation of the effect of the pressure/temperature shock waves induced by the impact of bolides in the early Earth (17). Other high-quality AIMD works show that extreme static pressures, such as those present in the planetary interiors, can induce the synthesis of small organic molecules from simpler ones (32, 33). However, the full atomistic dynamical simulation of gas-phase Miller experiments is still well beyond the computational feasibility of modern quantum chemistry methods, and even computational studies on the bare effect of external electric fields on solutions of Miller molecules are lacking. As a consequence, the chemical mechanisms governing the reactivity of the C-H-O-N system under electric field are unknown. On one hand, accurate high-level quantum chemical methods are computationally too expensive to deal with the dynamics of relatively large systems containing more than 100 atoms. On the other hand, the fundamental difficulty of treating the electronic problem, within standard ab initio density functional theory (DFT) implementations, in the presence of an external electric field, was solved just a few years ago for solids (34), and was only recently applied by our group to the study of dissociation and proton conduction in water (35).
Ab Initio Miller-Like Experiments
We present here, to our knowledge, the first computer study of Miller-like experiments in the condensed-phase, i.e., the study of solutions of “Miller molecules” under external electric field. Ab initio Car−Parrinello (36) molecular dynamics simulations were performed within state-of-the-art DFT via the Quantum ESPRESSO code (37). Technical details and calculation strategies are provided in Methods. Several cubic supercells were set up to study and amplify the different reaction steps as long as they were successively observed. All systems were put under progressively stronger external electric fields, up to intensities of the order of ∼0.5 V/Å. Such strong field values are necessary to dissociate water within the picosecond timescale of AIMD (35), and are compatible both with the experimental values of water dissociation in the presence of electrical discharges (38), and thus with the likely conditions of Miller experiments, and with the naturally occurring short-ranged electric fields at the surface of minerals (39), also implied in the prebiotic geochemical reactions.
We set up three simulation boxes by decomposing the Miller−Strecker reaction into its elementary steps: (i) a reactants box, named MS01, containing 8 H2O, 8 NH3, 8 CH4, 10 CO, and 5 N2 molecules; (ii) the so far supposed intermediate MS02 box, containing nine molecules each of water, ammonia, formaldehyde, and hydrogen cyanide; and (iii) the supposed products MS03 supercell, containing nine glycine, plus nine ammonia molecules. These proportions were carefully chosen so as to correctly reproduce the intermediate and end products of the Strecker reaction, and to allow each box to have the exact same content of each atomic species as the other ones, i.e., 18:72:18:18 atoms of C:H:O:N, respectively. This choice allows a direct comparison of the potential energies of all three systems at all steps of their dynamical and chemical evolution under electric fields, and thus a reasonable description of the corresponding relative free energies, as all simulations are carried out in the canonical ensemble at the same volumes and temperatures. To this end, all three systems have been at first simulated for about 5 ps in the absence of external fields; we report in Table S1 their composition and potential energies relative to the end products.
The first important result is that the MS02 system, containing the supposedly intermediate reactants, has a higher potential energy than both the MS03 and, especially, the starting MS01 systems. In other words, the formation of formaldehyde and HCN from the Miller molecules is thermodynamically disfavored, at least at the DFT level, and thus very unlikely to occur in our small simulation boxes over short picosecond timescales. Interestingly, while no chemical reactions were observed in MS01 and MS03, at the end of the zero-field evolution of MS02, we observed that a H2CO and an ammonia molecule combined to yield a formamide molecule (HCONH2).
Once the zero-field energetics have been evaluated, we set up the actual in silico Miller experiment, by putting the MS01 system under a finite electric field, to possibly observe the spontaneous formation of formaldehyde and/or HCN, and thus make a chemical connection with the intermediate Strecker (MS02) system. However, once the electric field was switched on, up to 0.25 V/Å, the system only exhibited occasional water/ammonia proton dissociations/jumps/recombinations, analogously to the case of pure water (35). At fields around 0.35 V/Å, however, more interesting chemical reactions occurred in the MS01 system. In particular, we observed, within 2.1 ps of our 10-ps-long trajectory, the field-induced formation of two formic acid molecules, both achieved via the association of a hydroxide ion with a carbon monoxide, later neutralized by a proton diffusing via a typical Grotthuss mechanism (40). Even more interestingly, at about 2.4 ps, a formamide ion HCONH3+, later evolving into a neutral formamide molecule, formed via the simultaneous association of a “Grotthuss proton” and an ammonia molecule on the two available bonds of a carbon atom in a CO molecule, thus creating the simplest possible O−C−N backbone.
Metadynamics Analysis of the Formamide Reaction
We note that all those reactions, as the ones described in the following, were not simply due to the presence of reactive charged species in solution, and that the field is the indispensable driving force. We specifically checked this point in two ways. First, we checked whether the presence of ionic species in solution could explain the observed reactions under field by running a zero-field simulation of the same initial set of Miller molecules but replacing all water and ammonia molecules with their ionic OH− and NH4+ counterparts. Although the proportion of ionic species was thus much larger than any instantaneous one ever observed at finite field, no reactions other than proton jumps until the full neutralization of all molecules occurred, thus proving that the presence of ionic species is not sufficient to promote the Miller reactions without an external field.
Secondly, we carried out independent calculations, based on the metadynamics (MetD) approach (41), to quantitatively determine the effect of the electric field in the free energy of the reaction of CO and NH3 to form formamide in aqueous solution, and of the dissociation of the latter one into the starting reactants. To this end, we set up a metadynamics simulation box, named MetD01, containing 1 CO, 1 NH3, and 30 H2O molecules, and chose the C−N distance as the single reaction coordinate (RC). This choice, while being only a projection of the multidimensional space of the RCs, proved to be quite effective to yield a quantitative energetic analysis of the formamide formation reaction. All metadynamics technical details are provided in Methods.
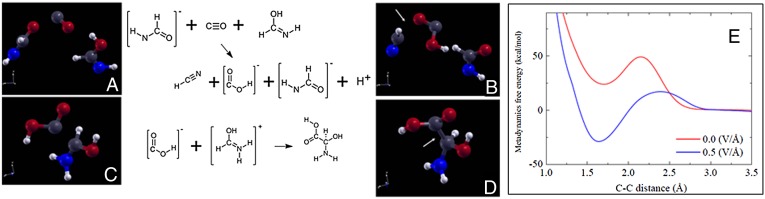
We report in Fig. 1B the free-energy curve as a function of the RC both without an external electric field and with an electric field of 0.5 V/Å, and a graphical representation of the MetD-based reaction analysis. We observe that the effect of the field is to strongly reduce the reaction barrier from about 58 to about 11 kcal/mol, and to improve both the kinetics and energetics. Moreover, the MetD trajectories allow decomposition of the chemical path between the reactants (CO, NH3) and the products (formamide), and identification of the statistically relevant intermediate states or potential transition states (TS): In fact, to obtain formamide, (i) one C–N bond must form (TS1), (ii) one C–H bond must form (TS2), and (iii) one N–H bond must break (TS3). In principle, these three steps, which do not qualify as transition states yet, must all be accomplished to yield formamide, but they can be permuted in any order to do so, thus passing through secondary intermediate steps (SIS), as shown in Fig. 1A.
Fig. 1.

Metadynamics-based analysis of the formation of formamide. (A) Representation of the chemical possible paths and intermediate/transition states. C–N, +H(C), and –H(N) indicate the formation of a CN bond, the formation of a CH bond, or the break of a NH bond, respectively. Secondary intermediate states are referred to in the text as SIS1, SIS2, and SIS3 from the top to the bottom, respectively. (B) Metadynamics-based free-energy landscape as function of the C-N distance reaction coordinate. (C) Statistical occurrence of the potential transition states (TS) with and without an external field, following the same color code as in A, and normalized with respect to the total occurrences in the fieldless case. (D) Energetics of the reaction with (blue line) and without (red line) the electric field, evaluated by identifying the “purple” (TS2) step as the reaction transition state as E(TS2)=EB+ln(P(TS2)/β, where EB is the MetD barrier, P(TSx) is the normalized statistical occurrence of a given TSx state, and β is the thermal term. Energy values are expressed in kilocalories per mole throughout the whole image.
However, our MetD analysis indicates that the three putative transition states (TS1, TS2, TS3) are not energetically equivalent, and that they occur in dramatically different proportions in the presence of an electric field, as we report in Fig. 1A. In fact, in neutral conditions, the C–N bond formation (TS1, “orange” species in Fig. 1A) has an occurrence of about 70% among all intermediate states along the MetD trajectories (Fig. 1C); the break of one N–H bond (TS3, “yellow” species) occurs about 29% of the times, while the C–H bond formation (TS2, “purple” species) has only a marginal occurrence (∼ 1%). The situation is radically different in the presence of the electric field (Table S2). First of all, the system visits the transition states about 20% more often than in the neutral case, indicating that it more easily explores its chemical landscape. More importantly, the relative presence of the potential transition states is reversed: The C–H bond formation (TS2) occurs 51% of the times, and the other two intermediate states about 23% (TS1) and 26% (TS3). Although one might be tempted to directly model from these results the reaction free energies, this is not a rigorous procedure within the MetD approach, as visited states do not necessarily correspond to actual transition states. To this aim, we follow the procedure described in ref. 42, i.e., the so-called committor analysis (see Methods), which reveals that, in the absence of an external electric field, the TS2 state is capable of yielding formamide in more than 50% of the independent simulations, while the TS1 state falls into the formamide basin about 1/3 of the time. The TS3 state, on the contrary, yields formamide only less than 5% of the time despite being by far the most likely intermediate state in the fieldless case. On the other hand, in the presence of the field, all three potential TSs fall the majority of the time (>75%) into the formamide basin. We can thus conclude that TS2, i.e., the formation of a C–H bond, is the actual transition state of this chemical reaction both with and without field, and its extremely low occurrence in the latter case explains its very large barrier at ambient conditions. This conclusion is further supported by the fact that, among the SIS shown in Fig. 1A, the uppermost one (SIS1), NH2 + CO, occurs, in the fieldless case, almost 62% of the time, although being the only SIS that cannot be achieved directly from TS3 (see Table S3). In other words, when formamide is about to dissociate into its reactants, it often takes the wrong chemical path in the neutral case. On the other hand, in the presence of the field, the most frequent secondary intermediate states (>83%) are precisely the ones chemically connected to TS2.
On the basis of all of the above results, we can provide its schematic energetic diagram (Fig. 1D) and suggest that the larger availability of protons in the presence of an external field is very likely the reason why the barriers and energetics are so different in the two cases. A deeper analysis of the fine details of this reaction is beyond the scope of the present work and will be carried out in future studies. We note in passing that the same behavior is observed in the subsequent breakdown reaction of formamide into water and hydrogen cyanide, which we also analyzed with the metadynamics tools. In fact, the formamide oxygen atom, in the presence of the electric field, more easily catches a proton to form, at first, formimidic acid, subsequently breaking its C bond and leaving behind a cyanide species. On the other hand, this intermediate formimidic acid step is observed about 40 times less often in the absence of the field. We report all these MetD data in Tables S2−S4.
Of course, at the DFT level, we cannot exclude that the presence of radicals in real systems could play a role and likely enhance the observed reactivity. On the other hand, the above results confirm that the electric field is not just a mere source of activation energy but has a more active role in favoring the assembling of larger chemical units from smaller ones, and thus complexity. This ordering effect is confirmed by experiments showing that charged electrodes in protein solutions favor crystallization (43).
Results: From Formamide to Amino Acids
On the basis of the above results and of the absence of Strecker intermediate products in the MS01 system and, on the contrary, of the spontaneous formation of formamide in the MS02 system even in the absence of field, we decided to explore a different path to explain the Miller experiments. We conceived another system, hereon named Miller−formamide (MF01), based on the MS01 one, but containing, from the start, eight molecules of each chemically relevant molecular species so far identified, i.e., formic acid, formamide, water, ammonia, and carbon monoxide, while the inert nitrogen and methane were discarded. This choice was also consistent with a century-old experiment of wet formamide under an electric discharge, capable of unexpectedly producing glycine (44). We set up the corresponding simulation box, and carried out the AIMD calculations along the same lines of the other systems. At a field of 0.35 V/Å, the only observed relevant reaction within 5 ps of trajectory, besides the typical proton hopping, concerned a formamide and a formic acid, which combined to form the smallest O=CH–N=CH–OH carboximide alcohol, whose pentagonal ring was closed by a H bond between the oxygen atoms. At a higher field of 0.5 V/Å, and along a 20-ps trajectory, we observed the most interesting reactions to occur. In particular, in agreement with a well-known industrial manufacture process (45), we observed all along the trajectory that ammonia and carbon monoxide often combined to yield formamide, thus confirming once more the previous analysis of the effect of the field in favoring this reaction. Once formed, formamide then either fueled the synthesis of larger and more complex molecules or broke down into water and hydrogen cyanide, as observed in the classical Strecker reaction (2–4) and shown in Table S5, possibly after converting into its formimidic acid tautomer, as follows:
A MetD analysis confirms that this latter reaction is also enhanced by the presence of the field (Table S4). At the same time, a similar yet less frequent reaction occurred between water/CO and formic acid:
After about 3.7 ps, as shown in Fig. 2, we observed that a carbon monoxide combined with a formamide molecule (Fig. 2A), a complex which dissociated into a hydrogen cyanide and a formate anion (Fig. 2B), which, within 0.05 ps, combined with a just-formed formamide-proton cation (i.e., a formamide whose carbonyl oxygen has bound a free proton), to yield a α-hydroxyglycine, as shown in Fig. 2 C and D.
Fig. 2.
Formation of hydroxyglycine. Trajectory snapshots showing, within 50 fs, (A) the bonding of a formamide molecule with a carbon monoxide one, followed by (B) the dissociation of a hydrogen cyanide, (C) leaving behind a formate ion that bonds another formamide (ion) molecule, (D) to yield a hydroxyglycine. Standard color codes are adopted: carbon atoms are displayed in dark gray, nitrogen atoms in blue, oxygen atoms in red, and hydrogen atoms in white. The relevant bond forming and breaking is highlighted by white arrows. The corresponding chemical reactions of A−B and C−D are sketched in between the corresponding configurations. (E) The corresponding metadynamics-determined free energy as function of the C−C distance reaction coordinate is reported.
This reaction is of course a crucial point of the whole study, since it shows that the presence of the electric field induces important chemical reactions, particularly the formation of an amino acid from smaller organic molecules, i.e., formamide and formic acid, in turn originated by the field-induced reactions of simpler and smaller Miller molecules such as water, ammonia, and carbon monoxide. As in the formamide case, we decided to explore this reaction with the MetD approach, using the C–C distance as the single reaction coordinate. To this end, we set up a specific supercell containing 30 water molecules and one formic acid and one formamide molecule. This MetD study, although less refined than the formamide one, allowed us a first approximation of the reaction energy landscape, reported in Fig. 2E, which qualitatively confirms, once again, the impact of the electric field in the reactivity of these species. A more detailed and quantitative analysis of this reaction would be significantly more complex, given the number of atomic and molecular species potentially involved.
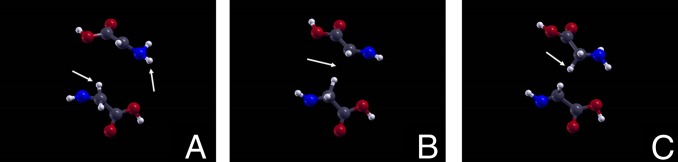
Interestingly, within a short time (0.03 ps), the carbonylic oxygen bonded to the central α carbon atom, after losing a proton, moved (in the opposite direction with respect to the field) onto the carboxylic β carbon (Figs. 3 A−C), to finally bond a proton moving along the field direction, and dissociated from the rest of the molecule, thus leaving behind a NH=CH–COOH molecule (Fig. 3 D−F). This latter molecule is known as dehydroglycine, an α-amino acid very similar to glycine, the only difference being the presence of a double bond (rather than a single one) between the nitrogen and the central carbon atoms.
Fig. 3.

Transformation of hydroxyglycine into dehydroglycine. Trajectory snapshots showing, at first, the α-carbonylic oxygen moving to the β-carbon atom (A−C, indicated by a white arrow), then, the dissociation as a hydroxide ion (D and E, indicated by a white arrow), quickly neutralizing into a water molecule by capturing a proton from formamide, which breaks into a hydrogen cyanide and another water molecule (F, indicated by the second white arrow). On the left of the figure, a dehydroglycine molecule remains. The atomic color code is the same as in Fig. 2. The chemical formulae and reactions at the bottom describe the events observed in D−F.
To attempt to find glycine at the end of the Miller chemical path, we first checked that the average energy of a simulation box containing glycine and water molecules (named Gly) is about 32 kcal/mol lower than the same box containing equal amounts of dehydroglycine, H2, and water molecules (named DHGW). We then set up a simulation box containing 10 dehydroglycine, 10 H2, and 10 formic acids molecules (labeled DHGF), the latter ones belonging to our family of “homemade” Miller molecules, preferred to water to improve the efficiency of the calculations and favor an acid attack on the double bond between the N and C atoms of dehydroglycine. The simulations were carried out at the usual conditions and in the presence of a ∼0.5 V/Å field. We observed, within less than a picosecond, one favorable and seemingly barrierless event, namely the simultaneous attack of two hydrogen atoms on an α-carbon atom of one dehydroglycine and a N atom of an adjacent one. Within about 100 fs the two α-carbons approached, and the hydrogen atom finally jumped from one to another, thus effectively yielding glycine, as shown in Fig. 4.
Fig. 4.
Formation of glycine. Trajectory snapshots showing, at first, two hydrogen atoms bonding with the α-carbon of one dehydroglycine molecule and with the nitrogen of a nearby one (A, shown by arrows). After about 90 fs, the two α-carbon of both molecules approach (B, indicated by the arrow), and the extra proton jumps from one of the two molecules onto the other one, yielding glycine (C, indicated by the arrow). The atomic color code is the same as in Fig. 2.
Discussion
This latter result is a significant one that represents the end of our quest. We can summarize it as follows: (i) a computer simulation Miller-like experiment has been hereby attempted via ab initio calculation methods that include external electric fields; (ii) the initial mixture of simple Miller molecules spontaneously gives birth, only under strong fields, to small intermediate organic compounds, such as formic acid and formamide; this latter reaction, one of the crucial results of this work, is driven by the presence of the electric field, as our detailed MetD analysis quantitatively shows; (iii) a mixture including more of those molecules, once put under a field stronger than 0.35 V/Å, is able to generate at least one proto-amino acid, α-hydroxyglycine, again confirmed by a metadynamics analysis, which, under the effect of the field, spontaneously transforms into α-dehydroglycine; (iv) a mixture containing the latter species, along with a subset of exclusively inbred molecules from previous steps, spontaneously forms glycine, the simplest (and most often found in real Miller experiments) standard amino acid; it is important to stress that all of the chemical reactions hereby observed only occur under strong electric fields. Moreover, we observe, even within the very short picosecond timescale of our simulations, that the chemical pathway leading to glycine is seemingly more complex, at least in the earliest stages of the reactions, than the standard interpretation of Miller experiments, i.e., that of a classical Strecker reaction, as we observe that formamide and formic acid form first, and formaldehyde and hydrogen cyanide are secondary intermediate steps to the formation of glycine. The presence of formamide and formic acid is consistent with recent ab initio simulations (17) and experiments (20) on the effect of bolide impact shockwaves in prebiotic chemistry, suggesting that very different physical conditions might drive similar reaction mechanisms. It is also worth noting that the possibility of new routes to make amino acids without formaldehyde intermediate is novel and gaining ground, especially in extraterrestrial contexts (46).
In our study, we provide new insights precisely on this point and, to our knowledge, for the first time at the quantum atomistic level, by showing that the first simple organic molecules that spontaneously form, in the presence of an electric field, are formic acid and formamide. We find in particular that the latter one is at the crucible of the Miller experiment, and most likely of many of the prebiotic chemical reactions leading to biological life, since it is continuously formed under electric fields, and it continuously fuels the formation of more complex organic molecules. It is in fact the combination of formamide and formate ion that spontaneously yields an amino acid, eventually evolving into glycine. Interestingly, our discovery of the role of formamide in building one major class of biochemical species necessary to biological life, amino acids, complements the findings of more and more studies (47–50) on its role in the formation of the other major class of biochemical species necessary to biological life, RNA bases. A recent work (51) shows in fact that formamide can be the crucial step in the formation of the so-called “missing G” of the nucleotide bases, i.e., guanine, once subject to UV light. This result is particularly interesting since another recent study (52) shows the presence of formamide in the environment of a solar-type protostar. In this context, the implications of our work on the spontaneous formation of formamide in the early pathway leading Miller molecules to evolve into amino acids go beyond the first in silico reproduction of Miller-like experiments, suggesting that the presence of formamide might be a most telling fingerprint of abiotic terrestrial and extraterrestrial amino acids.
We conclude by considering that the potential implications of our results on the understanding of the origins of life could go beyond the historical Miller experiments. In fact, one of the leading hypotheses in the field of the origins of life suggests that important early Earth prebiotic chemical reactions might have occurred at the surface of minerals (53–55); so far, this has been interpreted as due to catalytic effects originated on the steric concentration and/or on the chemical interaction of the reacting species with the active sites at the mineral surfaces. However, surface polymerization scenarios are still very far from being fully understood. In this regard, it is interesting to notice that the comprehensive review of Lambert (56) on prebiotic chemistry at mineral surfaces concludes with the following sentence: “Finally, a later step could involve the study of activation mechanisms different from simple heating.” We show in this study that the electric field acts as an order maker, promoting the spontaneous assembling of simple Miller molecules into more complex ones of biochemical interest. Our results suggest that the role of the electric field at the surface of minerals, even in the closest proximity to the surfaces, needs to be explored in full depth, as it could analogously be an important, and so far overlooked, component in driving the prebiotic chemical reactions that have made life possible.
Methods
Using the Car−Parrinello (CP) approach (36) we performed a series of AIMD simulations on several “Miller boxes”, under the effect of intense electric fields. All results were obtained using the software suite Quantum ESPRESSO (37). Our systems were initially set up as shown in Table S1. Our ab initio calculations were performed by using a plane−wave/pseudopotential scheme. We chose the Perdew−Burke−Ernzerhov (57) exchange and correlation functional, quite reliable for aqueous systems.
The electronic interaction with the nuclei was described through ultrasoft pseudopotential; we chose a kinetic energy cutoff of 35 rydberg (Ry) and a charge density cutoff of 280 Ry. The fictitious electronic mass was set at a value of 300 a.u.; the ion dynamics was carried out in the NVT ensemble using the Verlet algorithm and Nosé−Hoover thermostats at a frequency of 13.5 THz. We carried out the ab initio simulations at the nominal temperature of 400 K, which is considered a reasonable value of typical prebiotic chemistry temperatures (4). The typical time step was 0.1 fs, and the total simulations time was of the order of 250 ps, as shown at the bottom of Table S1, where we report additional figures on the accuracy and stability of our method.
Macroscopic polarization and finite electric fields are treated within the Quantum ESPRESSO package with the modern theory of polarization (Berry phases) (34), and have been used by our group to study dissociation and proton conduction in water (35). Based on this implementation, we studied the effect of electric field intensity in the range of 0.0–0.5 V/Å; the field direction was chosen parallel to the z axis of each of our cubic cells. The electronic kinetic energy typically does not exceed 0.8 eV, while the electronic gap of the system is at least 2–3 times larger along the simulations.
Several cubic supercells were set up to study and amplify the different reaction steps as long as they were successively observed, described in detail in Table S1, and labeled as follows: Original Miller (M00), Miller−Strecker first step (MS01), Miller−Strecker second step (MS02), Miller−Strecker third step (MS03), Miller−formamide (MF01), dehydroglycine + formic acid and H2 (DHGF), dehydroglycine + water and H2 (DHGW), and glycine + water (Gly). All systems had a cubic cell side chosen to obtain a density of 1.0 g/mL. All boxes were set up and equilibrated at first via a classical force field.
A preliminary test to all simulations reported in the main text was carried out on a system, named M00, based on the original Miller mixture and relative proportions of ammonia, methane, and hydrogen, containing 32 H2O, 8 NH3, 8 CH4, and 4 H2 molecules, i.e., 160 atoms. This system showed, once the electric field was switched on, a large number of events of proton dissociations/recombinations/jumps involving water and/or ammonia molecules. However, no other reactions were observed, due to the extremely high proportion of hydrogen atoms with respect to the other atomic species, making very likely the saturation of instantaneous dangling bonds with the many available protons regularly produced in the solution, and, as a consequence, more significant C-N-O reactions unlikely to occur within the relatively short simulation timescales. More generally, a calculation strategy expecting within a few tens of picoseconds the formation of glycine and other amino acids in simulation boxes containing very few Miller molecules appears quite inefficient. The full reaction mechanisms need instead to be decomposed in more elementary steps, including at each one of them the products of the previous one, as described in the text.
The energy landscape of the formamide reaction from CO and NH3 was calculated with the metadynamics approach (41), using the MetD01 supercell described above and the PLUMED 2.0 code (58). We chose the distance between the C and the N atom as the reaction coordinate of the system. Potential transition states have been tested with the committor analysis (42), i.e., by running 50 independent AIMD simulations for each state and zero or nonzero field. Each simulation started from the same configuration of each of the three TSs, but with a random distribution of initial velocities, transition states being defined if both the initial and the final states of the reaction occur a statistically significant fraction of times.
Supplementary Material
Acknowledgments
The authors thank Vivia Bruni for inspiration, support, and insights in prebiotic chemistry; F. Guyot for chemistry and geochemistry advice and insights and for a thorough critical reading of the manuscript; R. Vuilleumier for advice on transition state theory; F. Pietrucci for insights in metadynamics and committor analysis; and P. V. Giaquinta and F. M. Raymo for discussions and support. A.M.S. acknowledges the French Agence Nationale de la Recherche for support through Grant ANR-13-IS04-0006-01, and thanks the Grand Equipement National de Calcul Intensif (GENCI – project x2013091387 and following) and the Université Pierre et Marie Curie Computational Mesocenter for computer time.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402894111/-/DCSupplemental.
References
- 1.Miller SL. A production of amino acids under possible primitive Earth conditions. Science. 1953;117(3046):528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 2.Miller SL, Urey HC. Organic compound synthesis on the primitive Earth. Science. 1959;130(3370):245–251. doi: 10.1126/science.130.3370.245. [DOI] [PubMed] [Google Scholar]
- 3.Miller SL. The mechanism of synthesis of amino acids by electric discharges. Biochim Biophys Acta. 1957;23(3):480–489. doi: 10.1016/0006-3002(57)90366-9. [DOI] [PubMed] [Google Scholar]
- 4.Lazcano A, Bada JL. The 1953 Stanley L. Miller experiment: Fifty years of prebiotic organic chemistry. Orig Life Evol Biosph. 2003;33(3):235–242. doi: 10.1023/a:1024807125069. [DOI] [PubMed] [Google Scholar]
- 5.Oro J. Mechanism of synthesis of adenine from hydrogen cyanide under possible primitive Earth conditions. Nature. 1961;191:1193–1194. doi: 10.1038/1911193a0. [DOI] [PubMed] [Google Scholar]
- 6.Harada K, Fox SW. Thermal synthesis of natural amino-acids from a postulated primitive terrestrial atmosphere. Nature. 1964;201:335–336. doi: 10.1038/201335a0. [DOI] [PubMed] [Google Scholar]
- 7.Sutherland JD, Whitfield JN. Prebiotic chemistry: A bioorganic perspective. Tetrahedron. 1997;53(34):11493–11527. [Google Scholar]
- 8.Cnossen I, et al. Habitat of early life: Solar X-ray and UV radiation at Earth's surface 4−3.5 billion years ago. J Geophys Res. 2007;112(E2):E02008. [Google Scholar]
- 9.Dworkin J, Deamer D, Sandford S, Allamandola L. Self-assembling amphiphilic molecules: Synthesis in simulated interstellar/precometary ices. Proc Natl Acad Sci USA. 2001;98(3):815–819. doi: 10.1073/pnas.98.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muñoz Caro GMM, et al. Amino acids from ultraviolet irradiation of interstellar ice analogues. Nature. 2002;416(6879):403–406. doi: 10.1038/416403a. [DOI] [PubMed] [Google Scholar]
- 11.Raulin F, Brassé C, Poch O, Coll P. Prebiotic-like chemistry on Titan. Chem Soc Rev. 2012;41(16):5380–5393. doi: 10.1039/c2cs35014a. [DOI] [PubMed] [Google Scholar]
- 12.Foustoukos DI, Seyfried WE., Jr Hydrocarbons in hydrothermal vent fluids: The role of chromium-bearing catalysts. Science. 2004;304(5673):1002–1005. doi: 10.1126/science.1096033. [DOI] [PubMed] [Google Scholar]
- 13.Amend JP, Shock EL. Energetics of amino acid synthesis in hydrothermal ecosystems. Science. 1998;281(5383):1659–1662. doi: 10.1126/science.281.5383.1659. [DOI] [PubMed] [Google Scholar]
- 14.Bar-Nun A, Bar-Nun N, Bauer SH, Sagan C. Shock synthesis of amino acids in simulated primitive environments. Science. 1970;168(3930):470–473. doi: 10.1126/science.168.3930.470. [DOI] [PubMed] [Google Scholar]
- 15.Elsila JE, Glavin DP, Dworkin JP. Cometary glycine detected in samples returned by stardust. Meteorit Planet. 2009;44(9):1323–1330. [Google Scholar]
- 16.Fegley B, Jr, Prinn RG, Hartman H, Watkins GH. Chemical effects of large impacts on the Earth’s primitive atmosphere. Nature. 1986;319:305–308. doi: 10.1038/319305a0. [DOI] [PubMed] [Google Scholar]
- 17.Goldman N, Reed EJ, Fried LE, William Kuo IF, Maiti A. Synthesis of glycine-containing complexes in impacts of comets on early Earth. Nat Chem. 2010;2(11):949–954. doi: 10.1038/nchem.827. [DOI] [PubMed] [Google Scholar]
- 18.McKay CP, Borucki WJ. Organic synthesis in experimental impact shocks. Science. 1997;276(5311):390–392. doi: 10.1126/science.276.5311.390. [DOI] [PubMed] [Google Scholar]
- 19.Goldman N, Tamblyn I. Prebiotic chemistry within a simple impacting icy mixture. J Phys Chem A. 2013;117(24):5124–5131. doi: 10.1021/jp402976n. [DOI] [PubMed] [Google Scholar]
- 20.Martins Z, Price MC, Goldman N, Sephton MA, Burchell MJ. Shock synthesis of amino acids from impacting cometary and icy planet surface analogues. Nat Geosci. 2013;6(12):1045–1049. [Google Scholar]
- 21.Huber C, Wächtershäuser G. Peptides by activation of amino acids with CO on (Ni,Fe)S surfaces: Implications for the origin of life. Science. 1998;281(5377):670–672. doi: 10.1126/science.281.5377.670. [DOI] [PubMed] [Google Scholar]
- 22.Parnell J. Mineral radioactivity in sands as a mechanism for fixation of organic carbon on the early Earth. Orig Life Evol Biosph. 2004;34(6):533–547. doi: 10.1023/b:orig.0000043132.23966.a1. [DOI] [PubMed] [Google Scholar]
- 23.Bada JL. How life began on Earth: A status report. Earth Planet Sci Lett. 2004;226:1–15. [Google Scholar]
- 24.Miller SL. In: The endogenous synthesis of organic compounds. The Molecular Origins of Life, Assembling Pieces of the Puzzle. Brack A, editor. Cambridge, UK: Cambridge Univ Press; 1998. pp. 59–85. [Google Scholar]
- 25.Parker ET, et al. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc Natl Acad Sci USA. 2011;108(14):5526–5531. doi: 10.1073/pnas.1019191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyakawa S, Yamanashi H, Kobayashi K, Cleaves HJ, Miller SL. Prebiotic synthesis from CO atmospheres: Implications for the origins of life. Proc Natl Acad Sci USA. 2002;99(23):14628–14631. doi: 10.1073/pnas.192568299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coveney PV, Swadling JB, Wattis JAD, Greenwell HC. Theory, modelling and simulation in origins of life studies. Chem Soc Rev. 2012;41(16):5430–5446. doi: 10.1039/c2cs35018a. [DOI] [PubMed] [Google Scholar]
- 28.Menor-Salván C, Ruiz-Bermejo DM, Guzmán MI, Osuna-Esteban S, Veintemillas-Verdaguer S. Synthesis of pyrimidines and triazines in ice: Implications for the prebiotic chemistry of nucleobases. Chemistry. 2009;15(17):4411–4418. doi: 10.1002/chem.200802656. [DOI] [PubMed] [Google Scholar]
- 29.Rimola A, Sodupe M, Ugliengo P. Deep-space glycine formation via Strecker-type reactions activated by ice water dust mantles. A computational approach. Phys Chem Chem Phys. 2010;12(20):5285–5294. doi: 10.1039/b923439j. [DOI] [PubMed] [Google Scholar]
- 30.Rimola A, Sodupe M, Ugliengo P. Aluminosilicate surfaces as promoters for peptide bond formation: An assessment of Bernal’s hypothesis by ab initio methods. J Am Chem Soc. 2007;129(26):8333–8344. doi: 10.1021/ja070451k. [DOI] [PubMed] [Google Scholar]
- 31.Schreiner E, Nair NN, Wittekindt C, Marx D. Peptide synthesis in aqueous environments: The role of extreme conditions and pyrite mineral surfaces on formation and hydrolysis of peptides. J Am Chem Soc. 2011;133(21):8216–8226. doi: 10.1021/ja111503z. [DOI] [PubMed] [Google Scholar]
- 32.Chau R, Hamel S, Nellis WJ. Chemical processes in the deep interior of Uranus. Nat Commun. 2011;2:203. doi: 10.1038/ncomms1198. [DOI] [PubMed] [Google Scholar]
- 33.Lee MS, Scandolo S. Mixtures of planetary ices at extreme conditions. Nat Commun. 2011;2:185. doi: 10.1038/ncomms1184. [DOI] [PubMed] [Google Scholar]
- 34.Umari P, Pasquarello A. Ab initio molecular dynamics in a finite homogeneous electric field. Phys Rev Lett. 2002;89(15):157602. doi: 10.1103/PhysRevLett.89.157602. [DOI] [PubMed] [Google Scholar]
- 35.Saitta AM, Saija F, Giaquinta PV. Ab initio molecular dynamics study of dissociation of water under an electric field. Phys Rev Lett. 2012;108(20):207801. doi: 10.1103/PhysRevLett.108.207801. [DOI] [PubMed] [Google Scholar]
- 36.Car R, Parrinello M. Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett. 1985;55(22):2471–2474. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- 37.Giannozzi P, et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J Phys Condens Matter. 2009;21(39):395502. doi: 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- 38.Hammadi Z, Descoin M, Salancon E, Morin R. Proton and light ion nanobeams from field ionization of water. Appl Phys Lett. 2012;101(24):243110. [Google Scholar]
- 39.Vlcek L, et al. Electric double layer at metal oxide surfaces: Static properties of the cassiterite-water interface. Langmuir. 2007;23(9):4925–4937. doi: 10.1021/la063306d. [DOI] [PubMed] [Google Scholar]
- 40.Grotthuss CJT. Sur la decomposition de l’eau et des corps qu’elle tient en dissolution à l’aide de l’électricité galvanique. Ann. Chim. (Paris) 1806;58:54–74. [Google Scholar]
- 41.Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA. 2002;99(20):12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallet GA, Pietrucci F, Andreoni W. Bridging static and dynamical descriptions of chemical reactions: An ab initio study of CO2 interacting with water molecules. J Chem Theory Comput. 2012;8(11):4029–4039. doi: 10.1021/ct300581n. [DOI] [PubMed] [Google Scholar]
- 43.Hammadi Z, Astier JP, Morin R, Veesler S. Protein crystallization induced by a localized voltage. Cryst Growth Des. 2007;7(8):1472–1475. [Google Scholar]
- 44.Löb W. Uber das Verhalten des Formamids unter der Wirkung der stillen Entladung. Ein Beitrag zur Frage der Stickstoff-Assimilation. Ber Dtsch Chem Ges. 1913;46(1):684–697. [Google Scholar]
- 45.Hohn A. 1999. Formamide. Kirk-Othmer Concise Encyclopedia of Chemical Technology, eds Kroschwitz J, Kirk-Othmer I. (Wiley, New York), 4th Ed, pp 943−944.
- 46.Pilling S, Baptista L, Boechat-Roberty HM, Andrade DPP. Formation routes of interstellar glycine involving carboxylic acids: Possible favoritism between gas and solid phase. Astrobiology. 2011;11(9):883–893. doi: 10.1089/ast.2011.0650. [DOI] [PubMed] [Google Scholar]
- 47.Saladino R, Botta G, Pino S, Costanzo G, Di Mauro E. Genetics first or metabolism first? The formamide clue. Chem Soc Rev. 2012;41(16):5526–5565. doi: 10.1039/c2cs35066a. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen VS, Orlando TM, Leszczynski J, Nguyen MT. Theoretical study of the decomposition of formamide in the presence of water molecules. J Phys Chem A. 2013;117(12):2543–2555. doi: 10.1021/jp312853j. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Gu J, Nguyen MT, Springsteen G, Leszczynski J. From formamide to purine: An energetically viable mechanistic reaction pathway. J Phys Chem B. 2013;117(8):2314–2320. doi: 10.1021/jp311423q. [DOI] [PubMed] [Google Scholar]
- 50.Scott AM, Dawley MM, Orlando TM, Hill FC, Leszczynski J. Theoretical study of the roles of Na+ and water on the adsorption of formamide on kaolinite surfaces. J Phys Chem C. 2012;116(45):23992–24005. [Google Scholar]
- 51.Barks HL, et al. Guanine, adenine, and hypoxanthine production in UV-irradiated formamide solutions: Relaxation of the requirements for prebiotic purine nucleobase formation. ChemBioChem. 2010;11(9):1240–1243. doi: 10.1002/cbic.201000074. [DOI] [PubMed] [Google Scholar]
- 52.Kahane C, Ceccarelli C, Faure A, Caux E. Detection of formamide, the simplest but crucial amide in a solar type protostar. Astr J Lett. 2013;763(2):L38. [Google Scholar]
- 53.Bernal JD. The Physical Basis of Life. London: Routledge; 1951. [Google Scholar]
- 54.Orgel LE. Polymerization on the rocks: Theoretical introduction. Orig Life Evol Biosph. 1998;28(3):227–234. doi: 10.1023/a:1006595411403. [DOI] [PubMed] [Google Scholar]
- 55.Holland G, et al. Deep fracture fluids isolated in the crust since the Precambrian era. Nature. 2013;497(7449):357–360. doi: 10.1038/nature12127. [DOI] [PubMed] [Google Scholar]
- 56.Lambert JF. Adsorption and polymerization of amino acids on mineral surfaces: A review. Orig Life Evol Biosph. 2008;38(3):211–242. doi: 10.1007/s11084-008-9128-3. [DOI] [PubMed] [Google Scholar]
- 57.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 58.Bonomi M, et al. PLUMED: A portable plugin for free energy calculations with molecular dynamics. Comput Phys Commun. 2009;180(10):1961–1970. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.