

Significance
Pressure unfolding of proteins is a fundamental aspect of their thermodynamic response, the origins of which remain controversial. Here, we use high-pressure solution NMR to investigate the pressure response of a model protein, T4 lysozyme, under various conditions. Our data resolve longstanding controversies regarding the pressure response of this protein and the hydration of the internal hydrophobic cavity. It is shown that local packing (cavities) and the availability of conformational space have important and nonlocal impacts on the protein pressure response. Overall, the findings presented here reveal a previously unappreciated complexity in the pressure response of protein structure.
Keywords: protein stability, protein folding and cooperativity, protein hydration, high-pressure NMR, reverse micelle encapsulation
Abstract
It is well known that high hydrostatic pressures can induce the unfolding of proteins. The physical underpinnings of this phenomenon have been investigated extensively but remain controversial. Changes in solvation energetics have been commonly proposed as a driving force for pressure-induced unfolding. Recently, the elimination of void volumes in the native folded state has been argued to be the principal determinant. Here we use the cavity-containing L99A mutant of T4 lysozyme to examine the pressure-induced destabilization of this multidomain protein by using solution NMR spectroscopy. The cavity-containing C-terminal domain completely unfolds at moderate pressures, whereas the N-terminal domain remains largely structured to pressures as high as 2.5 kbar. The sensitivity to pressure is suppressed by the binding of benzene to the hydrophobic cavity. These results contrast to the pseudo-WT protein, which has a residual cavity volume very similar to that of the L99A–benzene complex but shows extensive subglobal reorganizations with pressure. Encapsulation of the L99A mutant in the aqueous nanoscale core of a reverse micelle is used to examine the hydration of the hydrophobic cavity. The confined space effect of encapsulation suppresses the pressure-induced unfolding transition and allows observation of the filling of the cavity with water at elevated pressures. This indicates that hydration of the hydrophobic cavity is more energetically unfavorable than global unfolding. Overall, these observations point to a range of cooperativity and energetics within the T4 lysozyme molecule and illuminate the fact that small changes in physical parameters can significantly alter the pressure sensitivity of proteins.
The destabilization of proteins by pressure is a fundamental and highly informative probe of their structural free energy landscape but remains inadequately understood (1). The underlying determinants of pressure-induced unfolding have recently been a subject of several detailed investigations (2–10). Fundamentally, pressure-induced unfolding of proteins results from the population of nonnative conformations having a lower total system volume than the native structure seen at ambient pressure. Various mechanisms for pressure-induced unfolding have been proposed including changes in water structure that weaken the hydrophobic effect at high pressure (11, 12), increases in solvent density at the protein surface that contribute to a reduction in the total volume of the protein–water system (13, 14), and the elimination of cavities in the protein interior through exposure to solvent (3). With the development of high-pressure sample cells compatible with modern solution NMR probes (15), detailed measurements of proteins unfolding under pressure with atomic resolution have now become possible (5, 16–18). Recent studies of staphylococcal nuclease (SNase) compellingly argue that the filling of void volumes present in the native state is the primary determinant of pressure-induced unfolding (4–6). A critical aspect of a “destruction of voids” mechanism for pressure-induced unfolding of proteins is whether the voids or cavities are occupied with water in the folded state. Early investigations of buried hydrophobic pockets indicated that even large cavities are typically not hydrated, whereas hydrophilic cavities generally are occupied by water (19, 20). Many of the key studies impacting this question used the L99A single-point mutant of the model enzyme T4 lysozyme (20).
The L99A mutation creates an internal cavity with an estimated volume of ∼150–160 Å3, large enough to accommodate three or four water molecules (21) (Fig. 1). Crystallographic investigation found no electron density within this pocket at ambient pressure (22, 23). In contrast, solution NMR and molecular-dynamics simulations suggest that the region of the protein around the hydrophobic pocket is highly dynamic, possibly to the extent that the pocket may be transiently accessible to solvent (22, 24–27). Crystallographic studies conducted at high pressure conversely suggested that the region around the pocket is rigid and exhibits increasing rigidity with increased pressure (23). Electron density also increased within the cavity as the hydrostatic pressure was increased (22), consistent with a pressure-induced filling of the hydrophobic cavity with water molecules. In contrast, fluorescence and small-angle X-ray scattering studies in bulk solution demonstrated that the protein is unfolded at these elevated pressures (2), suggesting that the crystal packing effects stabilize the protein. The hydrophobic cavity also provides a general, moderate-affinity binding site for small, relatively nonpolar ligands (28).
Fig. 1.
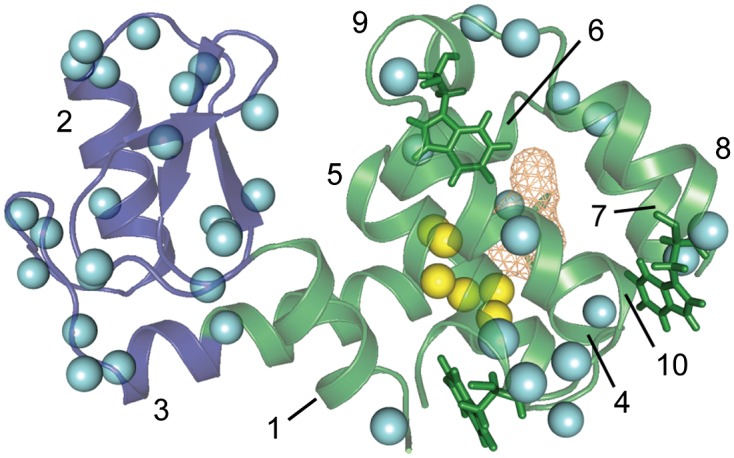
Hydration of T4 lysozyme L99A at ambient pressure (∼1 bar). A backbone ribbon representation of L99A [Protein Data Bank (PDB) ID code 1L90 (63)] is shown with the N-terminal domain (residues 13–65) illustrated in blue, and the C-terminal domain (residues 1–12 and 66–164) is colored green. The hydrophobic pocket created by the L99A mutation is shown as orange mesh, and the three tryptophan side chains are shown as stick representations. The helices are numbered as a reference for discussion in the text. Cyan spheres are shown at the positions of amide hydrogens where an NOE to the water resonance was detected. Yellow spheres indicate the positions of amide hydrogens within NOE distance (5 Å) of the interior of the hydrophobic pocket, but outside NOE distance to the protein surface. These are the sites where detection of NOEs to the water resonance would indicate hydration of the pocket. No NOE cross-peaks from these sites to the water resonance were observed, suggesting that the pocket is not hydrated at ambient pressure.
T4 lysozyme is one of the smallest known proteins to contain more than one cooperative folding unit. The folding of WT T4 lysozyme has been examined in detail by using hydrogen–deuterium exchange approaches and has been shown to contain two domains that fold cooperatively and with distinct free energy profiles (29–32). The N-terminal domain is ∼6 kcal/mol less stable than the C-terminal domain. The cavity created by the L99A mutation is in the center of the C-terminal domain. The thermal stability of the L99A mutant is reduced compared with the WT protein by 16 °C (5 kcal/mol) (33), an effect that is partially abrogated by binding hydrophobic ligands to the cavity (28, 34).
The L99A mutant of T4 lysozyme provides a unique system to examine the hydration of internal pockets and the details of pressure-induced unfolding. In principle, protein–water interactions can be characterized by solution NMR methods (35), but severe artifacts often render the approach quite limited (36). Recently, it has been shown that various advantageous properties of proteins and water encapsulated within reverse micelles largely overcome these artifacts (37, 38). Here, we use this approach to directly measure the hydration of the internal cavity. High-pressure NMR is used to examine the pressure-induced response of the protein in bulk solution and under confinement by the reverse micelle. We demonstrate that the hydrophobic pocket appears to be essentially dehydrated at ambient pressure (∼1 bar) and that the pressure response of the protein is an unfolding of the C-terminal domain only, representing an inversion of the relative stability of the domains as a result of the cavity-creating mutation. This result is in contrast to the unfolding of the cysteine-free WT (WT*) protein, which shows only the earliest stages of pressure-induced subglobal unfolding. Furthermore, the L99A mutant with benzene occupying the cavity shows no evidence of pressure unfolding. Nanoscale confinement of the protein also suppresses the L99A pressure-induced unfolding transition (Pu) as a result of the restriction of conformational space imposed by the reverse micelle. In lieu of the pressure unfolding transition, the volume reduction imposed by increasing pressure is compensated for in the reverse micelle by progressively increasing incorporation of water into the cavity interior, essentially recapitulating the observations from high-pressure crystallography in a solution measurement. These findings have important implications with respect to the nature of pressure-induced unfolding, the roles of cavities in protein structural stability, and the effects of confinement, a critical parameter when considering the intracellular milieu, in which proteins must fold and carry out their functions.
Results
Reverse Micelle Encapsulation of T4 L99A and Absence of Hydration of the Hydrophobic Cavity at Ambient Pressure.
T4 lysozyme L99A was encapsulated in reverse micelles with high structural fidelity based on analysis of 15N heteronuclear single quantum coherence (HSQC) spectra of free and encapsulated protein (Fig. S1). To assess the hydration of the internal cavity of encapsulated L99A, nuclear Overhauser effects (NOEs) between protein hydrogens and water were resolved using a three-dimensional 15N NOESY-HSCQ spectrum recorded at ambient pressure. Encapsulation suppresses hydrogen exchange artifacts that generally prevent the use of this simple approach in free aqueous solution (37). There are many amide hydrogens within NOE distance (∼5 Å) of the interior of the hydrophobic pocket, but only a handful of these are sufficiently buried to avoid the possibility of NOE contacts to water at the surface of the protein (Fig. 1). None of these sites showed cross-relaxation to the water resonance, indicating that the pocket is unlikely to be hydrated in the reverse micelle at ambient pressure. These data do not preclude the possibility of very transient water access to the cavity interior, but they do establish the absence of long-lived (greater than ∼100 ps) water residing in the cavity.
Site-Resolved Analysis of the Pressure-Induced Unfolding.
Nitrogen-15 HSQC spectra were recorded as a function of hydrostatic pressure to investigate the pressure-induced unfolding of L99A in bulk aqueous solution (Fig. S2). A significant fraction of the protein molecule unfolds with increasing pressure, as is evident from the collapse of the amide N-H chemical shift correlation spectrum. The pressure response of the WT* mutant and the L99A mutant with benzene bound in the hydrophobic pocket were also examined (Figs. S3 and S4, respectively). The WT* protein showed only a few sites that locally unfold over the pressure range examined here, whereas the binding of benzene to the hydrophobic pocket of the L99A mutant completely suppressed pressure-induced unfolding. Amide cross-peaks of the folded state of the L99A mutant (all resolved sites) and WT* (23 residues) that decreased in intensity by at least 25% over the pressure range were used to determine the thermodynamics of unfolding across the protein structure as described in Materials and Methods and illustrated in Fig. S5 and compiled in Tables S1 and S2. The midpoint of the Pu for each amide resonance in the protein is illustrated in Fig. 2. It should be emphasized that the responses observed here are indicative of subglobal and local unfolding events and largely correspond to the cooperative unfolding of secondary structural units and domains. Localized pressure-induced perturbations of the protein structure, resulting in chemical shift changes, were quite similar under all conditions examined, including reverse micelle encapsulation (Figs. S2–S4 and S6). For simplicity, we refer later to all conformational changes as unfolding events based on a simplified two-state modeling of the pressure response.
Fig. 2.
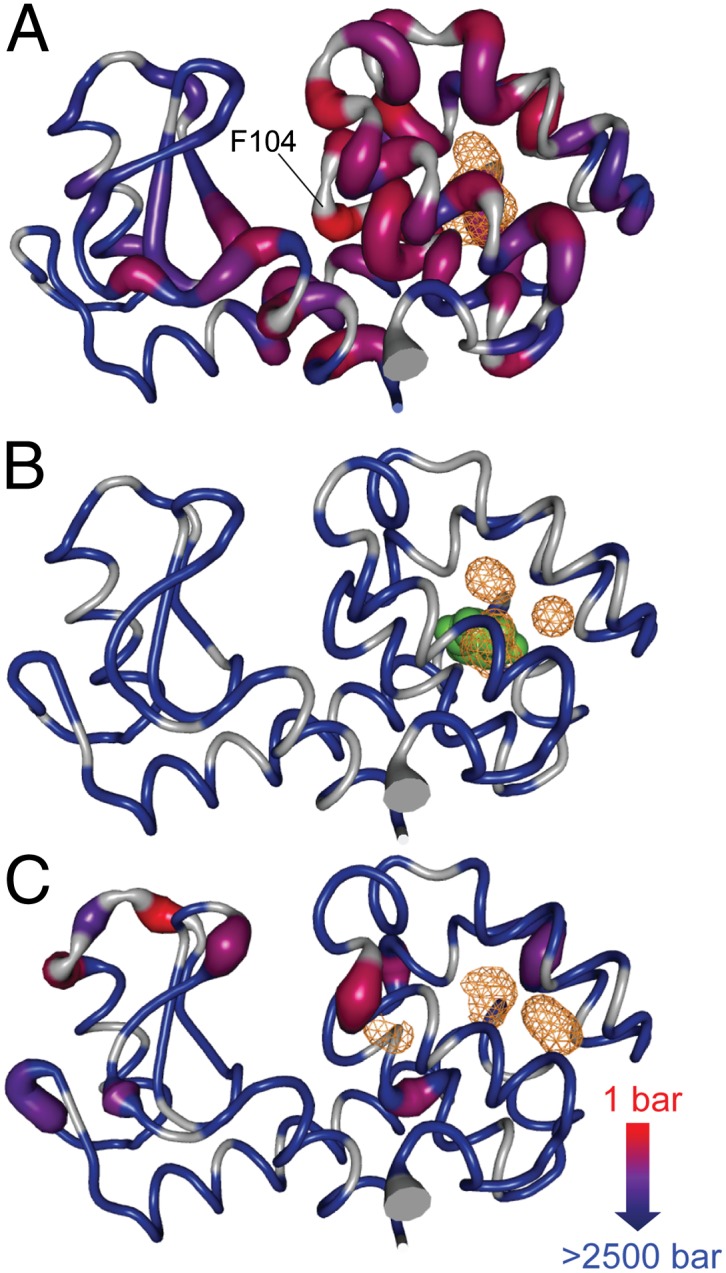
The intensities of cross-peaks in the 15N HSQC spectra shown in Figs. S2 and S3 were fitted to a two-state pressure unfolding model to extract thermodynamic parameters of the unfolding process. The Pu is mapped to the backbone structures of (A) T4 lysozyme L99A [PDB ID code 1L90 (63)], (B) L99A with benzene bound [PDB ID code 1L84 (34)], and (C) WT* [PDB ID code 1L63 (64)] with sites linearly color coded as indicated at bottom right. To further illustrate these data, the thickness of the backbone cartoon is scaled with the color code such that the thickest sites are those that unfold at the lowest pressures. Sites at which data could not be quantitatively analyzed are colored gray and are shown at the minimal thickness.
For the L99A mutant, sites with the lowest Pu are those corresponding to the points of primary contact between the two domains and involve helix 1, the C-terminal end of helix 5, and the middle of helix 3. The ends of the helices surrounding the hydrophobic pocket exhibit end-fraying at these lower pressures and result in essentially complete unfolding of the C-terminal domain within the measured pressure range. Helices 4 and 7 seem to retain some residual contact at pressures beyond those required to unfold the remainder of the C-terminal domain. In distinct contrast to the behavior of the C-terminal domain, the N-terminal domain remains largely structured up to 2.5 kbar. These results are in general agreement with the internal cooperative substructure of T4 lysozyme determined by native-state hydrogen exchange methods (30). The binding of benzene to the hydrophobic pocket of the L99A mutant stabilizes the protein to pressure such that not a single residue (locally) unfolds at less than 2.5 kbar. In contrast, the WT* protein shows pressure destabilization at several sites in the C-terminal domain in the vicinity of the cavity. The WT* mutant also exhibits unfolding of a pair of loops in the N-terminal domain that is not observed in the L99A mutant with or without benzene bound.
The Effect of Confinement on the Pressure Response of L99A.
The application of high hydrostatic pressure to a free solution of the L99A cavity mutant of T4 lysozyme results in unfolding of the protein (2). In principle, confinement of the protein to a nanoscale cavity will favor the more compact folded state of the protein (39–41). Forced folding of a protein of marginal stability within the confines of a reverse micelle water core has been previously demonstrated (42). L99A was most stably encapsulated in reverse micelles at a molar ratio of water to surfactant of 18 (Fig. S1). The radius of the water pool of protein-containing reverse micelles derived from the rotational correlation time of the encapsulated protein by using the transverse relaxation-optimized spectroscopy for rotational correlation times (TRACT) approach (43–45) is estimated to be ∼25 Å. Confinement of L99A within a reverse micelle stabilizes the protein to pressures as high as 2 kbar (Fig. S6). The maximal intensity is reduced for many cross-peaks. This effect arises primarily from line broadening caused by the slowed tumbling of the reverse micelle particle as a result of the increase in the viscosity of pentane with increasing pressure. The viscosity of pentane at 1 bar is one fourth that of water. As the reverse micelle particle is approximately four times larger than the protein in free solution, the encapsulated protein tumbles with approximately the same correlation time as aqueous protein at ambient pressure (46). The viscosity of pentane at 2 kbar, however, is approximately the same as that of water, so the tumbling time of the reverse micelle particle increases by a factor of four over the pressure range examined here (47). Although substantial line-broadening results, the spectrum at 2 kbar demonstrates that the pressure unfolding transition is suppressed in the reverse micelle.
Suppression by the reverse micelle of pressure-induced unfolding allows examination of the hydration of the apolar cavity at high pressures. Nitrogen-15 NOESY-HSQC spectra were collected at 1 kbar and 2 kbar to detect the entrance of water into the cavity. One new NOE between the amide hydrogen of I100 and the water resonance was observed at 1 kbar, which increased in intensity at 2 kbar. An additional protein–water cross peak appeared at 2 kbar and was assigned to the amide hydrogen of residue N101. These two residues are at the center of the most deeply buried helix in the C-terminal domain of the protein (Fig. 3). Only six hydrogens of the 164 resolved amide NH correlations are spatially located in the protein such that NOE cross peaks between them and any water molecules in the hydrophobic pocket would not be contaminated by similar interactions with surface water. Residues 100 and 101 have amide hydrogens that are among the closest to the interior surface of the pocket while also being deeply buried from the outer surface of the protein. Unfortunately, the slow reorientation of the reverse micelle at high-pressure results in transverse relaxation times that are too short to permit acquisition of high-quality rotating frame NOESY spectra. This precludes measurement of the time scale of the protein–water interactions in the pocket (35, 37). Nevertheless, the occupancy of the cavity by water at increased pressure is unequivocally demonstrated by the detection of NOE cross-peaks between the water resonance and the amide hydrogens of I100 and N101.
Fig. 3.

Hydration of T4 lysozyme L99A at elevated pressure. (Upper) Slices of 15N-resolved NOESY-HSQC spectra of reverse micelle-encapsulated L99A at the 1H resonance of water (4.875 ppm). Cross-peaks centered at this resonance are labeled according to the amide hydrogen resonance of the residue from which they arise. Unlabeled cross-peaks are those centered at planes above and below the water resonance. Planes are shown at 1 bar (red), 1 kbar (purple), and 2 kbar (blue). At 1 kbar, an NOE is seen from the amide hydrogen of residue 101 to the water resonance. This cross-peak increases in intensity at 2 kbar and is joined by the appearance of an NOE cross-peak between the amide hydrogen of residue 100 and the water resonance. (Lower) The structure of L99A is shown with the ribbon and pocket illustrated as in Fig. 1. Sites where a cross-peak to the water resonance is observed at 1 bar are again shown in cyan. Approximately half of the protein–water cross-peaks at these sites are broadened into the noise as pressure is increased as a result of slower macromolecular tumbling caused by increased viscosity of the alkane solvent. Residues 100 and 101 are highlighted in magenta. These are the only sites where new protein–water cross-peaks are observed as pressure is increased, providing conclusive evidence of water entry into the hydrophobic pocket under solution conditions.
Discussion
The Pressure-Induced Unfolding of a Multidomain Protein.
T4 lysozyme L99A presents an opportunity to examine the pressure-induced unfolding of a multidomain protein at atomic resolution without the use of chemical denaturants. Unfolding pressures for typical proteins are generally substantially higher (4–8 kbar) (48) than the highest pressures used here, necessitating the use of low levels of chemical denaturants to destabilize the proteins of interest, thereby lowering their unfolding pressure to a range accessible via NMR (i.e., less than 2.5 kbar in this context). Because the mechanism of destabilization of the various perturbations (e.g., chemical denaturants, pH, temperature, pressure) likely varies, it is advantageous to use a single denaturing perturbation (although see ref. 49).
Two explanations for pressure-induced unfolding are most prevalent in the literature. One is that pressure alters the water–water hydrogen bonding energetics such that the unfolded state is favored at high pressure (11, 12, 50). Pressure-driven changes in the energetics of solvation of hydrophobic surfaces and of charged side chains have been argued to be significant (11, 12, 14). A more mechanical view is that pressure-induced unfolding is essentially a result of the relief of packing defects in the native conformations of proteins (3, 5, 51). Recent studies of cavity mutants of SNase, in which a correlation between the site-resolved volume changes upon unfolding and void volumes within the native state was observed, reinforce this interpretation (4, 5, 52). The SNase results provide compelling evidence that the reflection of Le Chatelier’s principle in the elimination of void spaces within the folded state can dominate pressure-induced unfolding. Nevertheless, it remains possible that the destabilization produced by chemical denaturants may tip the balance between changes in hydration chemistry and the contribution of void volumes to the pressure destabilization of proteins.
As previously noted, fluorescence spectroscopy was used to characterize the pressure sensitivity of several cavity mutants of T4 lysozyme, including L99A, which were found to globally unfold at unusually low pressures. A correlation between calculated unfolding volumes and cavity volumes in the native state was also observed (2). However, all three tryptophan residues are located in the C-terminal domain (Fig. 1). The side chains of two of these (126 and 158) are largely exposed to solvent in the native state, so their spectral properties have limited utility. The remaining tryptophan (W138) is entirely buried in the native state and is therefore likely to be the primary reporter of the pressure unfolding transition. Thus, tryptophan fluorescence is likely insensitive to the conformational behavior of the N-terminal domain. The high-pressure NMR data presented here demonstrate that the N-terminal domain remains largely intact at pressures much greater than the unfolding pressure of the C-terminal domain. Folding studies of WT T4 lysozyme have shown that the C-terminal domain is typically more stable than the N-terminal domain by ∼6 kcal/mol (30); thus, the result of the cavity creating mutation is a large destabilization of the C-terminal domain with respect to pressure. The creation of a pressure-destabilizing cavity in one domain of a small protein with minimal effects on the other WT domain supports the notion of a critical role for void volume in determining pressure-induced unfolding.
Closer inspection of the pressure sensitivity of L99A reveals a cascade of responses. The midpoint pressures of the various localized structural transitions indicate that interdomain contacts are the least stable to pressure (Fig. 2). When these tertiary contacts have been lost, the C-terminal domain unfolds by progressive fraying of the helices with increasing pressure. The residues along the interdomain interface correlate with previously identified regions of internal motion on the millisecond to microsecond time scale at ambient pressure where the interdomain motion of T4 lysozyme WT* was characterized as an interconversion between the “closed” conformation, exemplified by the crystal structure, and a more “open” conformation of the protein in which a flexing of helix 3 increases the distance between the domains (53). Of particular note is the N-terminal helix 1 that cooperatively folds with the C-terminal domain in the WT protein and provides a large number of interdomain contacts (Figs. 1 and 2A). The remaining interdomain contacts reside in the center of helix 3 and show a bifurcated response with pressure that corresponds closely with the division of this helix between the two domains. The portion of the helix in the C-terminal domain largely unfolds at relatively low pressures, whereas the N-terminal domain portion remains completely structured at the highest pressures examined. Indeed, essentially the entire N-terminal domain remains structured throughout the pressure perturbation.
The majority of the C-terminal domain unfolds at moderate pressures. In concert with the loss of tertiary contacts between the domains, helices 5, 6, 9, and 10 become disordered from the ends at relatively low pressures. Helices 4 and 7 show only a slight loss of structure. Interestingly, substantial backbone conformational exchange has been observed at 1 bar in helices 5, 6, 7, and 9 of L99A (24) but is not present in the WT* protein. Inclusion of a hydrophobic ligand in the cavity interior quenches the motion, suggesting that conformational exchange controls access of the cavity to solvent and ligands. The portions of helices 5, 6, and 9 where conformational exchange was detected correlate closely with those sites that unfold at relatively low pressures, further indicating that the pressure-sensitive portions of the C-terminal domain of the L99A mutant are coupled to exposure of the cavity to solvent. In this regard, a minor conformation of the L99A mutant is seen at ambient pressure (54) and involves a rearrangement that allows the aromatic side chain of phenylalanine 114 to partially fill the hydrophobic cavity (55). Chemical shift analysis indicates that the pressure response described here does not correspond to an increase in the population of this minor state (Fig. S2). Simulations have also suggested coupling of F114 dynamics with gating of solvent access to the hydrophobic cavity at high pressure (22). Interestingly, F104 (indicated in Fig. 2A) has the lowest unfolding pressure midpoint and the largest ΔVu0 (volume change upon unfolding) in the L99A protein (Fig. S7), confirming that the pressure sensitivity and conformational exchange at ambient pressure are distinct.
Comparison of L99A, WT*, and the L99A–benzene complex reveals further insights. The WT* mutant contains smaller hydrophobic spaces in the C-terminal domain than the L99A protein—33 Å3 vs. ∼150 Å3, respectively—so the comparative pressure stability of WT* is consistent with the idea that increased void volume destabilizes the L99A protein. However, the majority of residues in the WT* protein that unfold at moderate pressures involve interdomain contacts (helices 5 and 10), just as seen in the L99A protein. The remaining sites, which unfold at moderate pressures, are found in the N-terminal domain of the WT* protein. This surprising result clearly indicates the presence of long-range coupling. More striking is the stabilization of the L99A mutant to pressure perturbation by the binding of benzene to the hydrophobic cavity. The structure of L99A with benzene bound contains residual cavity volume of 32 Å3, nearly identical to that of WT* (34), yet no pressure-induced unfolding was detected.
The free energy of benzene binding to the L99A hydrophobic cavity is −5 kcal/mol (28) and improves the thermal stability by ∼1.9 kcal/mol (34). Based on the average free energy of pressure unfolding for unliganded L99A from the data presented here (∼2.4 kcal/mol), the complete suppression of pressure-induced denaturation indicates that the benzene stabilizes the protein more with respect to pressure than with temperature. Furthermore, the close similarity between the void volume present in the native state of the benzene-bound L99A and the WT* argues that the observed stabilization from benzene binding is not simply a ΔVu-driven effect. Clearly, the presence of a small hydrophobic molecule stabilizes the cavity to pressure denaturation beyond a simple localized filling of void volume, as the pressure stability of the benzene-bound complex is higher than that of WT* not only in the C-terminal domain, but in the N-terminal domain as well. We propose that the fluidity of a free ligand vs. a side chain provides additional pressure stabilization by virtue of its freedom to dynamically fill void volume, thereby optimizing hydrophobic interactions, as the structure of the protein adjusts in response to pressure. This suggests long-range effects on protein stability from packing interactions beyond the local interactions implicated in recent studies of the pressure response of SNase (4–6). As previously stated, the L99A protein is known to undergo considerable conformational exchange at 1 bar. These motions are quenched by the binding of the hydrophobic ligand xenon to the cavity at 4 bar (24). This contrast suggests a possible relevance for motional modes in the millisecond-microsecond time frame in the pressure response of T4 lysozyme. Overall, the observed pressure response of T4 lysozyme has revealed a range of subglobal cooperativities.
Nanoscale Confinement Suppresses the Pressure-Unfolding of the L99A Mutant and Promotes Solvation of the Cavity Under High Pressure.
The encapsulation of L99A in reverse micelles provides clarity to the seemingly conflicting results of the high-pressure crystallographic and solution studies of this protein. The pressure-unfolded state has nearly twice the radius of the native state (∼32 Å vs. ∼17 Å) (2), and the absence of pressure-induced unfolding in the crystalline state was attributed to crystal packing forces. The ∼25-Å radius of protein-containing reverse micelle core is insufficient to contain the fully extended pressure-unfolded L99A protein. This confinement completely suppresses the pressure unfolding transition (Fig. S6).
The treatment of confinement by Zhou and Dill (39) suggests that encapsulation will stabilize the native state by ∼15 kcal/mol. This is considerably greater than the local stabilities of the protein to pressure denaturation, which averaged ∼2.4 kcal/mol. It should be noted that suppression of unfolding in the reverse micelle does not appear to be related to insufficient solvation of the unfolded state. Here, each protein-containing reverse micelle contains ∼4,000 water molecules, enough for approximately three or four layers of water surrounding the protein in its native state. The solvent accessible surface area of the L99A mutant is predicted to increase by a factor of 2.3 upon complete unfolding (56). The pressures used here unfold only the C-terminal domain, and thus ample water needed to solvate this state is available in the reverse micelle core.
Pressure-induced hydration of the hydrophobic cavity was also observed (Fig. 3), again in agreement with the crystallographic results (22). These observations provide conclusive evidence that the pressure unfolding of the L99A mutant is sensitive to spatial restriction and that high-pressure cavity hydration is energetically more unfavorable than unfolding i.e., is seen only if Pu is suppressed. The energetic penalty for insertion of water into a bulk hydrophobic solvent is ∼4.6 kcal/mol (57). Assuming as many as approximately three water molecules may hydrate the cavity (22), a maximum of ∼14 kcal/mol is needed, which is less than the stabilization of the native state as a result of confinement. This estimate is similar in magnitude to the estimated potential energy of waters in the cavity from high-pressure simulations (22). It is interesting to note that the pressure at which water apparently enters the hydrophobic pocket in the reverse micelle coincides closely with that observed by crystallography. It is tempting to interpret this as indicative that denaturation of the protein with pressure in aqueous solution occurs as a result of water being forced into the cavity and destabilizing the C-terminal domain, a mechanism that has been previously suggested (11). The present data suggest, however, that hydration of the cavity in a native-like state followed by denaturation is unlikely. Rather, the exposure of the cavity to solvent appears to occur in concert with denaturation as pressure is raised in bulk aqueous solution. Previous investigations of the high-pressure hydration of ubiquitin also failed to detect entrance of water to the hydrophobic core (58).
As a final note, the nature of pressure adaptations in proteins has attracted considerable attention, and various evolutionary optimizations of protein structure have been suggested (7–10). Although there are important distinctions between crowding and confinement (7–10, 40), the confinement used here is comparable to the spaces in the cytosol (44, 59). The stabilization of T4 lysozyme by confinement suggests that extensive adaptation may not be necessary for protein stability in piezophilic organisms.
Materials and Methods
Sample Preparation.
Appropriately isotopically labeled cysteine-free T4 lysozyme (i.e., WT*) and the single-point L99A mutant of WT* were purified as described previously (21, 60). Aqueous samples were composed of 500 µM T4 lysozyme WT* or L99A in 50 mM sodium chloride, 50 mM sodium acetate, pH 5, with 8% (vol/vol) D2O. For benzene-bound L99A measurements, 20 mM benzene was dissolved in the buffer, and saturation of benzene binding was confirmed from chemical shift perturbations. Optimal reverse micelle conditions were determined (details provided in SI Materials and Methods) to be 80 µM T4 lysozyme L99A in 75 mM decanoyl-1-rac-glycerol/lauryldimethylammonium-N-oxide/dodecyltrimethylammonium bromide (52.5 mM/15 mM/7.5 mM) (45) at a water:surfactant molar ratio of 18 in d-pentane (98% d; Cambridge Isotopes). Reverse micelle surfactants were purchased from Sigma except lauryldimethylammonium N-oxide, which was purchased from Affymetrix.
NMR Spectroscopy.
Standard heteronuclear NMR experiments (61) were carried out at 600 MHz (1H; L99A and WT*) or 500 MHz (L99A–benzene) by using Bruker Avance III NMR spectrometers equipped with triple-resonance cryoprobes. High-pressure NMR spectra were acquired by using a 2.5 kbar rated 5 mm o.d./3 mm i.d. NMR cell and computer-controlled pressure generator (Daedalus Innovations). Spectra were collected at 25 °C and processed by using Felix (Accelrys) or ALNMR (62). Structural fidelity of reverse micelle-encapsulated T4 L99A and at elevated pressures was confirmed by comparison of 15N HSQC spectra (Fig. S1) and by standard triple-resonance spectroscopy (61). For examination of pocket hydration for encapsulated T4 L99A, 15N NOESY-HSQC spectra were collected at 1 bar, 1 kbar, and 2 kbar using 50 ms for NOE mixing. Further details are provided in SI Materials and Methods.
Thermodynamic Analysis.
Nitrogen-15 HSQC cross-peak intensities were used to obtain the fraction in the native state at each amide site as a function of applied hydrostatic pressure. The intensities at 1 bar were taken as representative of 100% natively folded at that site and intensities at the noise level as representative of 0%. The apparent equilibrium constant (Ku) was determined at each pressure, and the corresponding free energy was calculated as ΔGu(P) = −RT ln Ku. These data for each residue were fitted to a linear function [ΔGu(P) = ΔGu0 − PΔVu], where ΔGu(P) is the unfolding free energy as a function of pressure, ΔGu0 is the free energy of unfolding at zero pressure, P is the pressure, and ΔVu is the volume change upon pressure-induced unfolding (5). Further details and tabulated data (Tables S1 and S2) are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
The authors thank Drs. Kathy Valentine and Sabrina Bédard for technical assistance and advice; Professor Frederick Dahlquist for generously providing the T4 lysozyme L99A and WT* vectors; and Dr. Igor Dodevski for helpful discussions. This work was supported by National Science Foundation Grant MCB-115803 and National Institutes of Health Postdoctoral Fellowship GM087099 (to N.V.N.).
Footnotes
Conflict of interest statement: A.J.W. declares a competing financial interest as a member of Daedalus Innovations, LLC, a manufacturer of reverse micelle and high-pressure NMR apparatus.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410655111/-/DCSupplemental.
References
- 1.Zipp A, Kauzmann W. Pressure denaturation of metmyoglobin. Biochemistry. 1973;12(21):4217–4228. doi: 10.1021/bi00745a028. [DOI] [PubMed] [Google Scholar]
- 2.Ando N, et al. Structural and thermodynamic characterization of T4 lysozyme mutants and the contribution of internal cavities to pressure denaturation. Biochemistry. 2008;47(42):11097–11109. doi: 10.1021/bi801287m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Royer CA. Revisiting volume changes in pressure-induced protein unfolding. Biochim Biophys Acta. Protein Struct Mol Enzymol. 2002;1595:201–209. doi: 10.1016/s0167-4838(01)00344-2. [DOI] [PubMed] [Google Scholar]
- 4.Roche J, et al. Remodeling of the folding free energy landscape of staphylococcal nuclease by cavity-creating mutations. Biochemistry. 2012;51(47):9535–9546. doi: 10.1021/bi301071z. [DOI] [PubMed] [Google Scholar]
- 5.Roche J, et al. Cavities determine the pressure unfolding of proteins. Proc Natl Acad Sci USA. 2012;109(18):6945–6950. doi: 10.1073/pnas.1200915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rouget J-B, et al. Size and sequence and the volume change of protein folding. J Am Chem Soc. 2011;133(15):6020–6027. doi: 10.1021/ja200228w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hay S, et al. Are the catalytic properties of enzymes from piezophilic organisms pressure adapted? ChemBioChem. 2009;10(14):2348–2353. doi: 10.1002/cbic.200900367. [DOI] [PubMed] [Google Scholar]
- 8.Sineva EV, Davydov DR. Constrained water access to the active site of cytochrome P450 from the piezophilic bacterium Photobacterium profundum. High Press Res. 2010;30(4):466–474. doi: 10.1080/08957959.2010.535208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davydov DR. Merging thermodynamics and evolution: How the studies of high-pressure adaptation may help to understand enzymatic mechanisms. J Thermodyn Catal. 2012;3:1000e110/1–1000e110/3. [Google Scholar]
- 10.Kato C, et al. Protein adaptation to high‐pressure environments. In: Siddiqui KS, Thomas T, editors. Protein Adaptation in Extremophiles. Hauppauge, NY: Nova Science Publishers; 2008. pp. 167–191. [Google Scholar]
- 11.Hummer G, Garde S, García AE, Paulaitis ME, Pratt LR. The pressure dependence of hydrophobic interactions is consistent with the observed pressure denaturation of proteins. Proc Natl Acad Sci USA. 1998;95(4):1552–1555. doi: 10.1073/pnas.95.4.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rick SW. Free energy, entropy and heat capacity of the hydrophobic interaction as a function of pressure. J Phys Chem B. 2000;104:6884–6888. [Google Scholar]
- 13.Chalikian TV, Macgregor RB., Jr Origins of pressure-induced protein transitions. J Mol Biol. 2009;394(5):834–842. doi: 10.1016/j.jmb.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Danielewicz-Ferchmin I, Banachowicz EM, Ferchmin AR. Role of electromechanical and mechanoelectric effects in protein hydration under hydrostatic pressure. Phys Chem Chem Phys. 2011;13(39):17722–17728. doi: 10.1039/c1cp21819k. [DOI] [PubMed] [Google Scholar]
- 15.Urbauer JL, Ehrhardt MR, Bieber RJ, Flynn PF, Wand AJ. High-resolution triple-resonance NMR spectroscopy of a novel calmodulin peptide complex at kilobar pressures. J Am Chem Soc. 1996;118:11329–11330. [Google Scholar]
- 16.Collins MD, Kim CU, Gruner SM. High-pressure protein crystallography and NMR to explore protein conformations. Annu Rev Biophys. 2011;40:81–98. doi: 10.1146/annurev-biophys-042910-155304. [DOI] [PubMed] [Google Scholar]
- 17.Fourme R, Girard E, Akasaka K. High-pressure macromolecular crystallography and NMR: Status, achievements and prospects. Curr Opin Struct Biol. 2012;22(5):636–642. doi: 10.1016/j.sbi.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Vajpal N, Nislus L, Wiktor M, Grzesiek S. High-pressure NMR reveals close similarity between cold and alcohol protein denaturation in ubiquitin. Proc Natl Acad Sci USA. 2013;110(5):E368–76. doi: 10.1073/pnas.1212222110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard SJ, Gross KH, Argos P. Intramolecular cavities in globular proteins. Protein Eng. 1994;7(5):613–626. doi: 10.1093/protein/7.5.613. [DOI] [PubMed] [Google Scholar]
- 20.Matthews BW, Liu L. A review about nothing: Are apolar cavities in proteins really empty? Protein Sci. 2009;18(3):494–502. doi: 10.1002/pro.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eriksson AE, et al. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255(5041):178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 22.Collins MD, Hummer G, Quillin ML, Matthews BW, Gruner SM. Cooperative water filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation. Proc Natl Acad Sci USA. 2005;102(46):16668–16671. doi: 10.1073/pnas.0508224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins MD, Quillin ML, Hummer G, Matthews BW, Gruner SM. Structural rigidity of a large cavity-containing protein revealed by high-pressure crystallography. J Mol Biol. 2007;367(3):752–763. doi: 10.1016/j.jmb.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulder FAA, Hon B, Muhandiram DR, Dahlquist FW, Kay LE. Flexibility and ligand exchange in a buried cavity mutant of T4 lysozyme studied by multinuclear NMR. Biochemistry. 2000;39(41):12614–12622. doi: 10.1021/bi001351t. [DOI] [PubMed] [Google Scholar]
- 25.Mulder FAA, Hon B, Mittermaier A, Dahlquist FW, Kay LE. Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2002;124(7):1443–1451. doi: 10.1021/ja0119806. [DOI] [PubMed] [Google Scholar]
- 26.Mulder FAA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: Application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2001;123(5):967–975. doi: 10.1021/ja003447g. [DOI] [PubMed] [Google Scholar]
- 27.Skrynnikov NR, Mulder FA, Hon B, Dahlquist FW, Kay LE. Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: application to methionine residues in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2001;123(19):4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- 28.Morton A, Baase WA, Matthews BW. Energetic origins of specificity of ligand binding in an interior nonpolar cavity of T4 lysozyme. Biochemistry. 1995;34(27):8564–8575. doi: 10.1021/bi00027a006. [DOI] [PubMed] [Google Scholar]
- 29.Llinás M, Marqusee S. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 1998;7(1):96–104. doi: 10.1002/pro.5560070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Llinás M, Gillespie B, Dahlquist FW, Marqusee S. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat Struct Biol. 1999;6(11):1072–1078. doi: 10.1038/14956. [DOI] [PubMed] [Google Scholar]
- 31.Cellitti J, et al. Exploring subdomain cooperativity in T4 lysozyme I: Structural and energetic studies of a circular permutant and protein fragment. Protein Sci. 2007;16(5):842–851. doi: 10.1110/ps.062628607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cellitti J, Bernstein R, Marqusee S. Exploring subdomain cooperativity in T4 lysozyme II: Uncovering the C-terminal subdomain as a hidden intermediate in the kinetic folding pathway. Protein Sci. 2007;16(5):852–862. doi: 10.1110/ps.062632807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baase WA, Liu L, Tronrud DE, Matthews BW. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19(4):631–641. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355(6358):371–373. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 35.Otting G, Liepinsh E, Wüthrich K. Protein hydration in aqueous solution. Science. 1991;254(5034):974–980. doi: 10.1126/science.1948083. [DOI] [PubMed] [Google Scholar]
- 36.Halle B. Protein hydration dynamics in solution: A critical survey. Philos Trans R Soc London Ser B. 2004;359:1207–1224. doi: 10.1098/rstb.2004.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nucci NV, Pometun MS, Wand AJ. Site-resolved measurement of water-protein interactions by solution NMR. Nat Struct Mol Biol. 2011;18(2):245–249. doi: 10.1038/nsmb.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nucci NV, Pometun MS, Wand AJ. Mapping the hydration dynamics of ubiquitin. J Am Chem Soc. 2011;133(32):12326–12329. doi: 10.1021/ja202033k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H-X, Dill KA. Stabilization of proteins in confined spaces. Biochemistry. 2001;40(38):11289–11293. doi: 10.1021/bi0155504. [DOI] [PubMed] [Google Scholar]
- 40.Zhou H-X, Rivas G, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou HX. Protein folding in confined and crowded environments. Arch Biochem Biophys. 2008;469(1):76–82. doi: 10.1016/j.abb.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peterson RW, Anbalagan K, Tommos C, Wand AJ. Forced folding and structural analysis of metastable proteins. J Am Chem Soc. 2004;126(31):9498–9499. doi: 10.1021/ja047900q. [DOI] [PubMed] [Google Scholar]
- 43.Lee D, Hilty C, Wider G, Wüthrich K. Effective rotational correlation times of proteins from NMR relaxation interference. J Magn Reson. 2006;178(1):72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 44.Nucci NV, et al. Optimization of NMR spectroscopy of encapsulated proteins dissolved in low viscosity fluids. J Biomol NMR. 2011;50(4):421–430. doi: 10.1007/s10858-011-9528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dodevski I, et al. Optimized reverse micelle surfactant system for high-resolution NMR spectroscopy of encapsulated proteins and nucleic acids dissolved in low viscosity fluids. J Am Chem Soc. 2014;136(9):3465–3474. doi: 10.1021/ja410716w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nucci NV, Valentine KG, Wand AJ. High-resolution NMR spectroscopy of encapsulated proteins dissolved in low-viscosity fluids. J Magn Reson. 2014;241:137–147. doi: 10.1016/j.jmr.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemmon EW, McLinden MO, Friend DG. In: NIST Chemistry WebBook, NIST Standard Reference Database Number 69. Linstrom PJ, Mallard WG, editors. Gaithersburg, MD: National Institute of Standards and Technology; 2014. [Google Scholar]
- 48.Akasaka K. Probing conformational fluctuation of proteins by pressure perturbation. Chem Rev. 2006;106(5):1814–1835. doi: 10.1021/cr040440z. [DOI] [PubMed] [Google Scholar]
- 49.Fuentes EJ, Wand AJ. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry. 1998;37(28):9877–9883. doi: 10.1021/bi980894o. [DOI] [PubMed] [Google Scholar]
- 50.Bianco V, Iskrov S, Franzese G. Understanding the role of hydrogen bonds in water dynamics and protein stability. J Biol Phys. 2012;38(1):27–48. doi: 10.1007/s10867-011-9235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frye KJ, Royer CA. Probing the contribution of internal cavities to the volume change of protein unfolding under pressure. Protein Sci. 1998;7(10):2217–2222. doi: 10.1002/pro.5560071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kitahara R, et al. Structural plasticity of staphylococcal nuclease probed by perturbation with pressure and pH. Proteins. 2011;79(4):1293–1305. doi: 10.1002/prot.22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goto NK, Skrynnikov NR, Dahlquist FW, Kay LE. What is the average conformation of bacteriophage T4 lysozyme in solution? A domain orientation study using dipolar couplings measured by solution NMR. J Mol Biol. 2001;308(4):745–764. doi: 10.1006/jmbi.2001.4614. [DOI] [PubMed] [Google Scholar]
- 54.Mulder FAA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Studying excited states of proteins by NMR spectroscopy. Nat Struct Biol. 2001;8(11):932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- 55.Bouvignies G, et al. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature. 2011;477(7362):111–114. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Estrada J, Bernadó P, Blackledge M, Sancho J. ProtSA: A Web application for calculating sequence specific protein solvent accessibilities in the unfolded ensemble. BMC Bioinformatics. 2009;10:104. doi: 10.1186/1471-2105-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polak J, Lu BCY. Mutual solubilities of hydrocarbons and water at 0.deg. and 25.deg. Can J Chem. 1973;51:4018–4023. [Google Scholar]
- 58.Fu Y, et al. Coupled motion in proteins revealed by pressure perturbation. J Am Chem Soc. 2012;134(20):8543–8550. doi: 10.1021/ja3004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Medalia O, et al. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science. 2002;298(5596):1209–1213. doi: 10.1126/science.1076184. [DOI] [PubMed] [Google Scholar]
- 60.McIntosh LP, Wand AJ, Lowry DF, Redfield AG, Dahlquist FW. Assignment of the backbone 1H and 15N NMR resonances of bacteriophage T4 lysozyme. Biochemistry. 1990;29(27):6341–6362. doi: 10.1021/bi00479a003. [DOI] [PubMed] [Google Scholar]
- 61.Rule GS, Hitchens TK. In: Fundamentals of Protein NMR Spectroscopy. Kaptein R, editor. Dordrecht, The Netherlands: Springer; 2006. [Google Scholar]
- 62.Gledhill JM, Jr, Wand AJ. Al NMR: a novel NMR data processing program optimized for sparse sampling. J Biomol NMR. 2012;52(1):79–89. doi: 10.1007/s10858-011-9584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eriksson AE, Baase WA, Matthews BW. Similar hydrophobic replacements of Leu99 and Phe153 within the core of T4 lysozyme have different structural and thermodynamic consequences. J Mol Biol. 1993;229(3):747–769. doi: 10.1006/jmbi.1993.1077. [DOI] [PubMed] [Google Scholar]
- 64.Nicholson H, Anderson DE, Dao-pin S, Matthews BW. Analysis of the interaction between charged side chains and the α-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry. 1991;30(41):9816–9828. doi: 10.1021/bi00105a002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.