

Significance
Commensal microbiota are known to be required for the elicitation of host Th17 responses, which may mediate autoimmune diseases. Here, we demonstrate that the IL-23 pathway dynamically regulates the abundance of certain commensals and maintains barrier function. Barrier disruption results in systemic dissemination of microbial products, which invokes the IL-23 pathway, with both beneficial and potentially deleterious consequences. Through induction of IL-22, IL-23 contributes to barrier repair, and through induction of the Th17 response, it aims to neutralize escaped commensal microbes. Thus, barrier disruption results in a pro-Th17 environment in which not only antimicrobial but also potentially antihost Th17 cells can develop.
Keywords: microbiome, mucosal immunity, interleukin-17, endotoxemia
Abstract
Mammalian hosts are colonized with commensal microbes in various mucosal and epithelial tissues, including the intestinal tract. In mice, the presence of segmented filamentous bacteria (SFB) promotes Th17 differentiation and the development of autoimmune disease. Here, we demonstrate that the IL-23 pathway dynamically regulates the abundance of SFB as well as mucosal barrier function in the adult animal. Genetic or pharmacological inactivation of the pathway selectively perturbs the abundance of a small group of commensals, including SFB, and results in an impaired mucosal barrier. Defective barrier function leads to systemic dissemination of microbial products, provoking induction of the IL-23 pathway with dual consequences: IL-23 drives IL-22 production to reinforce mucosal barrier function and elicit antimicrobial activities, and it also drives the differentiation of Th17 cells in an attempt to combat escaped microbes in the lamina propria and in distal tissues. Thus, barrier defects generate a systemic environment that facilitates Th17 development.
The human gastrointestinal tract harbors an estimated 500–1,000 distinct bacterial species, and the total numbers of intestinal bacteria are in the trillions (1–3). Although the presence of microbiota is required for immune system development, the host depends on an intact barrier to contain the microbiota in their assigned niches, and on an effective immune system to destroy bacteria that may have escaped and invaded host tissues. It is thought that diseases such as inflammatory bowel disease and atopic dermatitis result, in part, from barrier defects, leading to bacterial invasion, breakdown of tolerance, and inflammation. Interestingly, germ-free mice or antibiotic-treated mice are resistant to intestinal as well as extraintestinal autoimmune diseases and defective in the generation of Th17 cells, a lineage of CD4+ T cells contributing to autoimmune pathologies (4–6), suggesting that immune cell education takes place in colonized tissues and may have consequences elsewhere in the organism.
Genetic modification of the host was reported to result in altered composition of the intestinal microbial community (7, 8). However, it remains largely unknown to what extent commensal homeostasis can be modulated by host factors in the adult WT animal. Furthermore, previous literature strongly implicates IL-22 in the maintenance of gut homeostasis (9, 10). IL-22 production by T cells and innate lymphoid cells is potently induced by IL-23, an antigen-presenting cell–derived cytokine that is induced upon sensing of pathogen-associated molecular patterns or damage-associated molecular patterns (11–13). Thus, we hypothesized that IL-23 induction in response to segmented filamentous bacteria (SFB) colonization and/or systemic dissemination of microbial components following barrier disruption has two main consequences. First, IL-23 triggers IL-22 production to elicit antimicrobial peptide production, which reestablishes microbial containment. Second, IL-23 drives Th17 cell differentiation, which is critical to neutralize escaped commensal microbes in the lamina propria, and perhaps even in distal tissues. Here, we present data that are consistent with this model and suggest that microbial induction of the IL-23 pathway, while protecting the host through IL-22 activity, also generates a systemic environment supportive of Th17 cell development.
Results
Homeostatic IL-23 Receptor/IL-22 Signaling Controls Intestinal SFB Colonization.
To assess if homeostatic IL-23 expression is required to control the level of SFB colonization in the gut, we quantitated the abundance of bacteria species in Interleukin-23 subunit p19 deficient (IL-23p19−/−) or IL-23 receptor deficient (IL-23R−/−) mice vs. WT littermates by quantitative PCR (qPCR). SFB abundance was increased by fivefold in the KO mice. In contrast, the total amount of bacteria, Clostridium coccoides, or Clostridium leptum was not changed (Fig. 1 A and B and Fig. S1A). Colonization of SFB in the WT ileum was confirmed by SEM, and comparison with Il-23r−/− further revealed SFB dysregulation in the Il-23r−/− ileum (Fig. S1B). Activation of IL-23R signaling induces several downstream effectors mediating immune responses, including IL-17 and IL-22. Using mice deficient in IL-22, IL-17F, or IL-17 receptor C (IL-17Rc), we further demonstrated that the SFB suppressing effect of IL-23 was mediated by IL-22 signaling but not by IL-17A/F signaling (Fig. 1C and Fig. S1 C and D).
Fig. 1.

Endogenous IL-23R/IL-22 signaling in ILC3 and T cells regulates intestinal SFB homeostasis. (A–D) qPCR analysis of intestinal bacteria quantity in indicated gene-deficient mice and their respective littermate controls. Signals were normalized to the abundance of respective bacteria species in control mice. EUB, total bacteria. (E) qPCR analysis of intestinal bacteria abundance in Rag2−/− mice depleted for Thy1+ cells, natural killer (NK) cells, or both Thy1+ and NK cells. Mice were killed 2 d after the last treatment, and signals were normalized as in A. (F) qPCR comparing intestinal SFB abundance in Rag2−/− or Il-23r−/−Rag2−/− mice 14 d after reconstitution with the indicated gene-deficient CD4+ T cells. Error bars represent mean ± SEM with three to five mice in each group. *P < 0.05; **P < 0.01; ***P < 0.001. ns, not significant.
IL-23R is expressed on both innate and adaptive immune cell types, including T cells and group 3 innate lymphoid cells (ILC3s). To evaluate the contribution of these cell types in IL-23–mediated SFB control, we first examined the innate immune compartment by comparing intestinal SFB abundance between recombination activating enzyme 2 deficient (Rag2−/−), Il-23r−/−Rag2−/−, and Interleukin-2 receptor gamma chain deficient (IL-2Rγ−/−)Rag2−/− mice. Genetic ablation of IL-23R or IL-2Rγ led to a marked increase of SFB quantity in comparison with rag2−/− mice, suggesting that IL-23R+ and IL-2Rγ+ ILC3s are essential in controlling SFB colonization (Fig. 1D). In further support of this hypothesis, depletion of Thy1+ ILCs led to SFB expansion, whereas depletion of natural killer cells by anti-asialo GM1 treatment (14) did not perturb SFB homeostasis (Fig. 1E). Because Rag2-deficient mice are more dependent on IL-23R–mediated mechanisms for SFB control than Rag2-sufficient mice (30-fold vs. fivefold), and T cell-derived IL-22 is essential for antimicrobial host defense (15), we next tested whether reintroduction of T cells could restore normal SFB control in Il-23r−/−Rag2−/− mice. Transfer of WT T cells reestablished normal SFB homeostasis, whereas Il-23r−/− or Il-22−/− T cells failed to suppress elevated SFB levels fully (Fig. 1F), indicating that the IL-23/IL-22 pathway in T cells contributes to SFB regulation. We noted that IL-23R or IL-22–deficient T cells were able to exert some control over SFB. Although the mechanism for this residual activity is unknown, it implies that T cells have IL-23/IL-22–independent antimicrobial effector mechanisms at their disposal.
IL-22 Suppresses Local and Systemic Th17 Responses.
Defective control of SFB colonization in the Il-23r−/−Rag2−/− mice prompted us to investigate how this dysbiotic microenvironment may affect local T-cell responses. We first tested whether SFB specifically affects intestinal Th17 generation, similar to the WT mice, in an adoptive transfer system. When we transferred purified CD4+ cells into recombination activating gene 1 deficient (rag1−/−) recipients, we found that SFB colonization potentiates IL-17–producing but not IFN-γ–producing CD4+ T cells upon adoptive transfer (Fig. S2A). Next, we adoptively transferred WT and Il-22−/− CD4+ T cells into Rag2−/− and Il-23r−/−Rag2−/− recipients and examined T-cell activation 2 wk after reconstitution. Intriguingly, colonic lamina propria T cells defective in producing IL-22 showed elevated RAR-related orphan receptor-γ (ROR-γ) and IL-17 expression in Il-23r−/−Rag2−/− hosts compared with Rag2−/− recipients. In contrast, a similar percentage of WT T cells expressed ROR-γ and IL-17, regardless of which host they were transferred into (Fig. 2 A and B and Fig. S2 B and C). Thus, dysregulation of IL-17 production by Il-22−/− T cells in the Il-23r−/−Rag2−/− hosts correlated with their inability to control SFB (Fig. 1F). Similar to the reported function of SFB in potentiating IL-17 generation but not IFN-γ production (11), we did not observe dysregulated IFN-γ production in conditions tested (Fig. 2B). Furthermore, recipients did not develop colitis in our adoptive transfer system, likely because the donor graft was not depleted of T-regulatory cells (Fig. S2D).
Fig. 2.
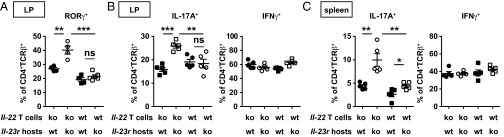
IL-22 negatively regulates local and systemic Th17 responses. Transcription factor and cytokine expression in WT or Il-22−/− CD4+ T cells adoptively transferred into Rag2−/− or Il-23r−/−Rag2−/− mice were assessed 14 d after reconstitution. Gated CD4+TCR-β+ lymphocytes derived from colonic lamina propria (LP) (A and B) or spleen (C) were analyzed. Frequency of gated cells expressing Ror-γ (A), IL-17A, or IFN-γ (B and C) is shown in the dot plots. Error bars represent mean ± SEM with at least four mice in each group. *P < 0.05; **P < 0.01; ***P < 0.001.
To determine whether the increased Th17 differentiation extended beyond the lamina propria, we analyzed T-cell responses in mesenteric lymph nodes and the spleen (Fig. 2C and Fig. S3 A and B). To our surprise, Il-22−/− T cells also overproduced IL-17, but not IFN-γ, compared with WT T cells in peripheral tissues. T cells coexpressing IL-17 and IFN-γ were also elevated in Il-23r−/−Rag2−/− hosts receiving Il-22−/− T cells compared with hosts receiving WT T cells (Fig. S3B). A closer look at our adoptive transfer system revealed that a substantial amount of IL-22 is produced by IL-17–producing cells located in both the lamina propria and spleen (Fig. S3C). Furthermore, roughly one-half of the IL-22–producing cells in the lamina propria are TCR-β− cells. Upon further gating, the majority of TCR-β− cells are negative for lineage markers and have high expression of Thy1 antigen, consistent with the notion that they are indeed ILCs (Fig. S3D). Collectively, these data suggest that IL-22–mediated control of microflora is associated with suppression of not only local but also systemic Th17 responses.
Defective Containment of Commensal Microflora Led to IL-17–Dependent Splenomegaly.
To investigate the mechanism underlying systemic Th17 responses in Il-23r−/−Rag2−/− mice reconstituted with Il-22−/− T cells further, we tested whether the dissemination of microbial products to the peripheral organs observed in ILC-deficient mice (16) was recapitulated in Il-23r−/−Rag2−/− mice, which are essentially devoid of IL-22. Interestingly, we detected elevated amounts of LPS in Il-23r−/−Rag2−/− liver and spleen (Fig. S4A). In our Rag2−/− model, this endotoxemia did not result in a change in spleen weight as is typically seen in infections in lymphocyte-replete hosts (Fig. S4B). Reconstitution of Il-23r−/−Rag2−/− mice with WT T cells led to a mild increase in spleen size, whereas reconstitution with Il-22−/− T cells caused a much more dramatic splenic enlargement (Fig. 3 A and B). In either case, the enlargement was driven by neutrophil influx (Fig. 3C), which was completely suppressed when mice were treated with antibiotics (Fig. 3 B and C). The spleen weight change and neutrophil influx correlated with expression levels of Ror-γ and IL-17 in T cells (Fig. S4C). To corroborate further that the splenomegaly was an IL-17–dependent effect, we treated mice with IL-17 blocking antibodies, which normalized splenic weight gain but had no impact on peripheral LPS levels (Fig. 3 D and E). IL-17 blockade largely normalized the histological appearance of spleens, which is characterized by increased cellularity and loss of white pulp in Il-23r−/−Rag2−/− mice that are recipients of Il-22−/− T cells (Fig. S4D). To test whether IL-17 production depends on IL-23 in this model, we transferred Il-23r−/− T cells into Il-23r−/−Rag2−/− mice. These IL-23–unresponsive T cells produced much less IL-17 and failed to promote neutrophil infiltration and splenic weight gain compared with IL-22–deficient T cells, which can still respond to IL-23 and respond by producing IL-17 (Fig. 3 F and G and Fig. S4E). Together, our data demonstrate that disruption of the IL-23/IL-22 axis leads to dissemination of microbial products into distal tissues, which, in turn, provides an environment in which Th17 cells can readily develop.
Fig. 3.
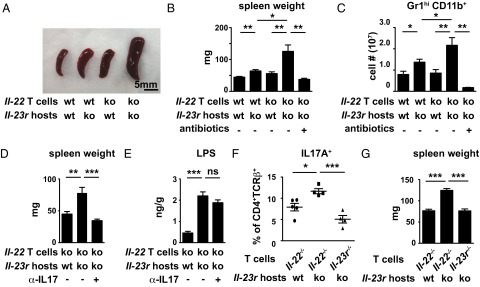
Defective containment of commensal microflora leads to IL-17–dependent splenomegaly. Spleen sizes (A), spleen weights (B), and splenic Gr1hiCD11b+ cell numbers (C) 2 wk after adoptive transfer of WT or Il-22−/− CD4+ T cells into Rag2−/− or Il-23r−/−Rag2−/− hosts are shown. Mice were maintained with regular water or orally fed with a mixture of antibiotics in the drinking water as indicated. (D and E) Il-22−/− CD4+ T cells adoptively transferred into Rag2−/− or Il-23r−/−Rag2−/− hosts treated with isotype control or a combination of anti–IL-17A and anti–IL-17F antibodies as indicated. Spleen weights (D) and LPS concentration in liver homogenates (E) were measured 4 wk after reconstitution. The percentage of IL-17A+ splenic CD4+TCR-β+ lymphocytes (F) and spleen weights (G) 2 wk after adoptive transfer of Il-23R−/− or Il-22−/− CD4+ T cells into Il-23r−/−Rag2−/− hosts is shown. Error bars represent mean ± SEM of four to five mice in each group. *P < 0.05; **P < 0.01; ***P < 0.001. (Scale bar: 5 mm.)
Pharmacological Modulation of IL-23/IL-22 Signaling Controls Intestinal SFB Abundance and Th17 Development.
We next wanted to interrogate whether our findings are relevant in normal adult WT mice with no innate barrier deficiency. In line with the genetic models, blocking IL-23 signaling with p40 neutralizing antibody led to increased SFB abundance in WT mice but not in Il-23p19−/− or Il-23r−/− mice, implying that IL-23, but not IL-12, is the primary p40-containing cytokine regulating SFB (Fig. S5A). Consistent with this notion, gavage of Jackson Laboratory mice free of SFB colonization with feces containing SFB resulted in increased SFB quantity and elevated IL-23p19 gene induction in the gut (Fig. S5 B and C). Conversely, dysbiosis of SFB in Il-23r−/− mice can be rescued by administration of recombinant IL-22–Fc (Fig. S5D). Furthermore, IL-22 blockade by neutralizing antibody resulted in increased SFB quantity (Fig. S5E). To determine a time course of SFB suppression, we monitored SFB abundance for 2 wk after a single dose of IL-22–Fc. We found that the quantity of SFB in the colonic feces started to decrease 1 d after IL-22–Fc treatment and reached the trough level within 48 h. SFB colonization then gradually increased and reestablished to its homeostatic level 2 wk after IL-22–Fc administration (Fig. 4A). The decrease and subsequent recolonization of SFB correlated with the expression kinetics of Reg3α, Reg3β, and Reg3γ (Fig. 4B), consistent with a previous report implicating Reg3γ in controlling SFB abundance (17). Conversely, expression of Reg3β and Reg3γ was significantly reduced when IL-22 signaling was blocked (Fig. S5F). Although a mild increase of SFB abundance in Il-22−/− mice did not lead to a significant increase in Th17 generation (Fig. S6A), IL-22–Fc administration that resulted in a more than 20-fold reduction of SFB markedly suppressed the frequencies of T cells expressing ROR-γ and IL-17, but not IFN-γ (Fig. 4 C and D and Fig. S6 B and C). Interestingly, reduction of the commensal bacteria burden by means of a mixture of antibiotics or with vancomycin abrogated IL-22–mediated suppression of IL-17 expression (Fig. 4E), indicating that IL-22 limits Th17 development through modulation of the microflora.
Fig. 4.

IL-22–mediated suppression of Th17 maintenance is dependent on commensal microflora. Kinetics of microflora regulation (A) and ileum gene expression (B) upon single dose IL-22–Fc treatment by qPCR. Signals were normalized to the respective abundance in untreated mice. Dot plots revealing the frequency of Ror-γ (C) and IL-17A and IFN-γ (D and E) expression in CD4+TCR-β+ lymphocytes isolated from the WT ileal LP treated with 30 μg of control antibody or IL-22–Fc for a week are shown. Mice were maintained with regular water (C–E) or water supplemented with a mixture of antibiotics (Abx) or single vancomycin (van; E) as indicated. Percentages of cells expressing the indicated proteins are shown. Error bars represent mean ± SEM with at least four mice in each group. *P < 0.05; **P < 0.01; ***P < 0.001.
Specificity of the IL-23/IL-22–Mediated Control of Commensal Microbiota.
To explore the level of specificity by which the IL-23/IL-22 axis controls the composition of the microflora beyond SFB, we performed 16S rRNA gene sequencing. We compared intestinal microbial compositions in Rag2−/− vs. Il-23r−/−Rag2−/− mice that were cohoused for 6 wk and the effect of IL-22–Fc treatment in WT mice. Consistent with our earlier data (Fig. 1), we did not observe a significant difference in microbial compositions at the phylum (Fig. S7) or family (Fig. 5A) level in either comparison. A closer look at the differential abundance of operational taxonomic units (OTUs) revealed that a total of 1,442 OTUs were present; 21 of them were significantly increased in Il-23r−/−Rag2−/− mice relative to Rag2−/− mice, and 31 of them were significantly decreased in mice treated with IL-22–Fc vs. control mice. Interestingly, 48% of the OTUs that expanded in Il-23r−/−rag2−/− mice and 61% of the OTUs that showed a reduction mediated by IL-22–Fc belong to the Rikenellaceae family (Tables S1 and S2). Comparison of the two datasets further confirmed SFB modulation by IL-22 signaling (Fig. 5B) and revealed similar IL-22–dependent modulation of four other taxa (OTUs 385422, 214403, 177425, and 209377), all of which belong to the Rikenellaceae family (Tables S1 and S2). Interestingly, mice obtained from the Jackson Laboratory, which are known not to be colonized with SFB (11), were also devoid of these Rikenellaceae taxa (Fig. 5B).
Fig. 5.

16S rRNA gene sequencing to identify IL-23/IL-22–regulated microbiota. 16S rRNA gene sequencing of intestinal fecal contents collected from indicated gene-deficient mice or WT mice treated with 50 μg of IL-22–Fc or control antibody is shown. (A) Bar graphs revealing compositions of intestinal commensal microbiota compared between Rag2−/− and Il-23r−/−Rag2−/− mice (Left) or mice treated with IL-22–Fc and IgG (Right) at the family level. (B) Sequence counts of SFB (Upper) or OTU 385422 (Lower) in Rag2−/− vs. Il-23r−/−Rag2−/− mice and WT mice treated with IL-22–Fc, which were obtained from Taconic (Tac) or the Jackson Laboratory (Jax). Error bars represent mean ± SEM with four to six mice per group. A.U., arbitrary units.
Discussion
The current body of literature suggests that altered composition of the gut microflora occurs in various genetically modified mouse strains, including IL-10−/−, IL-22−/−, and TLR5−/− mice (9, 18, 19). Our study prompts inclusion of IL-23p19−/− and IL-23R−/− in this list, because these mice have impaired IL-22 expression and harbor elevated amounts of SFB. This finding is consistent with recent reports showing that IL-22 administration can lower elevated abundance of SFB in Ltβr−/− or Ahr−/− mice (10, 20). We further demonstrated that modulation of intestinal flora can be elicited in the immune-competent adult animal with normal barrier function. Blockade of IL-23/IL-22 signaling by anti-p40 or anti–IL-22 antibody led to SFB expansion, whereas IL-22–Fc administration suppresses SFB colonization. Our data suggests that microbial homeostasis is the result of a dynamic calibration process in which host cells continuously sample gut microbiota and elaborate appropriate control mechanisms.
Furthermore, the regulation of microbiota through the IL-23/IL-22 pathway appears to be highly specific. Our 16S rRNA gene sequencing analysis revealed no changes at the phylum level, and only identified a handful of other taxa that were also specifically regulated by IL-23/IL-22, among which members of the Rikenellaceae family were prominent. One caveat is that the resolution of our sequencing effort did not permit analysis below the family level, and thus it is possible that subtle difference were missed for technical reasons. Nevertheless, we can conclude that the IL-23/IL-22 axis exerts a highly selective effect on a small subset of commensals. How this specificity is achieved and whether a change in the abundance of Rikenellaceae contributes to the functional consequences on Th17 development are issues that require further clarification.
One question was whether manipulation of the microflora elicits changes in the makeup of the host immune response. Unfortunately, this question is experimentally challenging to address, because the regulatory effects of the IL-23/IL-22 pathway on the microbiota cannot easily be segregated from the direct activities of these cytokines on host cells. We therefore initially resorted to an artificial system and performed T-cell reconstitution experiments to analyze the connection between microbiota and T-cell fate. ROR-γ expression and IL-17 production by WT CD4+ T cells upon transfer into either Rag2−/− or Il-23r−/−Rag2−/− recipients were comparable, and SFB levels were normalized in Il-23r−/−Rag2−/− recipients. However, when IL-22−/− CD4+ T cells were transferred into Il-23r−/−Rag2−/− recipients, they not only failed to regulate SFB abundance in the gut but exhibited a remarkable propensity to develop into Th17 cells in proximal and even distal tissues in an IL-23–dependent manner. Il-23r−/−Rag2−/− recipients had elevated levels of endotoxin in their peripheral tissues, similar to the low-grade bacteremia state reported in ILC-depleted animals (16). To evaluate the significance of this regulatory pathway further, we extended our studies to WT mice. Treatment of WT mice with IL-22–Fc, similar to antibiotic treatment, suppressed SFB colonization and resulted in Th17 suppression in a microbiota-dependent manner. We noted that IL-22−/− mice do not display Th17 misregulation in the gut, suggesting a threshold of SFB colonization has to be bypassed to elicit Th17 dysregulation. Alternatively, another immune compartment that can regulate SFB abundance (21) may compensate for the loss of IL-22 in SFB regulation and Th17 generation.
Our finding is consistent with a recent study reporting the association of SFB dysregulation and an enhanced intestinal Th17 response in Ahr−/− mice, which correlated with reduced IL-22 production (20). Our data further suggest that IL-22, by promoting intestinal barrier integrity, is suppressing not only local but systemic aberrant Th17 responses. Consistent with the finding that Il-22−/− mice do not develop spontaneous colitis (9), our Il23r−/−Rag2−/− mice reconstituted with Il-22−/− T cells, although unable to constrain SFB colonization and intestinal Th17 generation, did not display colonic inflammation. The observation that mice with AhR deficiency harbor an elevated amount of SFB and develop exacerbated colitis (20) therefore suggests that AhR may regulate immune homeostasis through additional pathways. Furthermore, SFB dysregulation alone is not sufficient to trigger colonic inflammation in the absence of IL-22.
One limitation of our study is that we cannot strictly distinguish whether the effects on Th17 cells are due to IL-23/IL-22–mediated control of SFB levels or regulation of barrier function, because they appear to be highly correlated in all of our experiments. However, they may merely be two sides of the same coin. Commensals are required for in vivo Th17 development (22, 23) but will not elicit inflammation unless the mucosal barriers are breached. SFB is a very potent agonist of the Th17 phenotype, perhaps because unlike other commensals, it has the ability to penetrate the mucus layer and make direct contact with epithelial cells (24). However, a barrier breach will also allow for dissemination of other organisms, or components thereof, and additional studies will be required to determine the precise nature of the components mediating systemic Th17 development.
Th17 cells are involved in the pathogenesis of most models of autoimmune disease (25). Our data prompt a model in which microflora components can act as an adjuvant for T cells to assume the pathogenic Th17 phenotype. We speculate that presentation of an autoantigen in the context of a breached barrier, be it through injury or colonization with specific microbes, such as SFB, may lead to expansion and terminal differentiation of autoantigen-specific, autoimmune Th17 cells. Our data thus provide a plausible explanation for the requirement for commensals in the context of autoimmune models.
Materials and Methods
Mice.
Mice on a C57BL/6 background were bred and maintained under specific pathogen-free conditions. All mice except rag1−/− mice were rederived in a Genentech facility and were free of Helicobacter. The rag1−/− mice were purchased from the Jackson Laboratory and confirmed to be free of SFB colonization. SFB-colonized rag1−/− mice were established by housing at the Children’s Hospital of Pittsburgh. Further details are provided in SI Materials and Methods.
Bacterial Abundance Quantitation by 16S Ribosomal RNA Gene Sequencing.
Bacteria genomic DNA was isolated from colonic intestinal stool using a QIAamp DNA Stool Mini Kit (Qiagen). The 16S rRNA gene sequencing and data analysis were performed by Second Genome. Quantitation of bacteria abundance was performed with an ABI real-time PCR system detecting SYBR (Applied Biosystems). Primers and PCR condition used were as described (26, 27) and are listed in Table S3. Further details are provided in SI Materials and Methods.
Animal Dosing and Antibiotic Treatment.
Mice were dosed i.p. with 30 or 50 μg of IL-22–Fc as indicated (Genentech, Inc.), 150 μg of anti–IL-22 (8E11.9; Genentech, Inc.), 100 μg of anti-p40 (C17.8, BD Biosciences), 1 mg of anti-Thy1 (TIB107, Genentech, Inc.), 50 μL of anti-asialo GM1 (WAKO Chemicals), and a combination of 200 μg of anti–IL-17A (Genentech, Inc.) and 200 μg of anti–IL-17F (6D10.5.8; Genentech, Inc.) to neutralize endogenous IL-17 signaling, or isotype control antibodies. Mice were dosed three times per week for 2 wk unless otherwise indicated. To reduce the abundance of intestinal bacteria, 0.5 g/L vancomycin (MP Biomedicals) or a mixture of 1 g/L each of ampicillin, neomycin, metronidazole, and gentamicin (Sigma) plus 0.5 g/L vancomycin was added to the drinking water each week for 4 wk before CD4+ T-cell adoptive transfer or IL-22–Fc dosing, and antibiotic treatment continued throughout the study.
Mice Cohousing.
Rag2−/− and Il23r−/−rag2−/− mice were cohoused starting from birth, with their female parents present until weaning. Cohoused animals were euthanized 6 wk after initiation of cohousing, and colonic feces were collected and subjected to 16S rRNA sequencing analysis.
CD4+ T-Cell Adoptive Transfer.
A single-cell suspension was prepared from spleens and peripheral lymph nodes, and CD4+ T cells were purified by magnetic cell sorting with a mouse CD4+ T-cell isolation kit (Miltenyi Biotec). Isolated CD4+ T cells were more than 95% pure, as confirmed by flow cytometry. A total of 107 unfractionated CD4+ T cells were adoptively transferred i.v. into the recipients.
Lamina Propria Leukocyte Isolation, ex Vivo Stimulation, and Flow Cytometry.
Colons or 10-cm segments of small intestine close to cecum were separated from mesentery, and Peyer’s patches were carefully excised. Lamina propria cells were harvested by digesting tissues enzymatically. For ex vivo stimulation, lamina propria leukocytes or single-cell suspensions from mesenteric lymph node and spleen were restimulated with phorbol 12-myristate 13-acetate (50 ng/mL) and ionomycin (750 ng/mL) in the presence of Brefeldin A (5 μg/mL) for 4 h. For flow cytometry, all cell preparations were blocked with anti-mouse CD16/32 (Fc block) and stained with appropriate antibodies. Further details are provided in SI Materials and Methods.
Measurement of Tissue Endotoxin Contents.
Organs were removed aseptically and homogenized with sterile PBS. LPS was measured in homogenates with the Limulus Amebocyte Lysate Chromogenic Endotoxin Quantitation Kit (Thermo Scientific).
Gene Expression Analyses.
mRNA was isolated from distal small intestine using an RNeasy Mini Kit (Qiagen) per the manufacturer’s protocol. Gene expression was assessed by real-time qPCR with a TaqMan Detection System (Applied Biosystems) using probes listed in Table S4. Further details are provided in SI Materials and Methods.
Histological Sections.
Mice spleens were removed and fixed with 10% (vol/vol) buffered formalin and embedded in paraffin, and 4- to 5-μm sections were stained with H&E.
Statistical Analysis.
Statistical analysis was performed with Prism software (GraphPad) using an unpaired Student t test (*P < 0.05; **P < 0.01; ***P < 0.001). Error bars denote ±SEM.
Supplementary Material
Acknowledgments
We thank S. Rutz and R. Noubade for technical advice, M. Thayer and L. Nguyen for animal husbandry, K. Schroeder for antibody preparation, and the Second Genome team for microbiome analysis.
Footnotes
Conflict of interest statement: V.F.-S.S., J.C., N.M.K., H.S.D., M.R., L.R., L.D., W.O., and N.G. are present or former employees of Genentech, Inc., a member of the Roche group.
This article is a PNAS Direct Submission. C.T.W. is a Guest Editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323852111/-/DCSupplemental.
References
- 1.Eckburg PB, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochoa-Repáraz J, et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183(10):6041–6050. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- 5.Wu HJ, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garrett WS, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131(1):33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zenewicz LA, et al. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol. 2013;190(10):5306–5312. doi: 10.4049/jimmunol.1300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Upadhyay V, et al. Lymphotoxin regulates commensal responses to enable diet-induced obesity. Nat Immunol. 2012;13(10):947–953. doi: 10.1038/ni.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8(6):630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 13.Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol Rev. 2008;226:112–131. doi: 10.1111/j.1600-065X.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasai M, et al. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature. 1981;291(5813):334–335. doi: 10.1038/291334a0. [DOI] [PubMed] [Google Scholar]
- 15.Basu R, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37(6):1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonnenberg GF, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336(6086):1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaishnava S, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vijay-Kumar M, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117(12):3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devkota S, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487(7405):104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu J, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39(2):386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki K, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci USA. 2004;101(7):1981–1986. doi: 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atarashi K, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 23.Ivanov II, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4(4):337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schnupf P, Gaboriau-Routhiau V, Cerf-Bensussan N. Host interactions with Segmented Filamentous Bacteria: An unusual trade-off that drives the post-natal maturation of the gut immune system. Semin Immunol. 2013;25(5):342–351. doi: 10.1016/j.smim.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 26.Barman M, et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76(3):907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atarashi K, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331(6015):337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.