

This work shows that the oral iron chelator deferiprone can be retina-protective in mice. In ceruloplasmin/hephaestin (Cp/Heph)-deficient mice, which have age-dependent retinal iron accumulation and degeneration, the drug protects against retinal degeneration.
Abstract
Purpose.
Iron-induced oxidative stress may exacerbate age-related macular degeneration (AMD). Ceruloplasmin/Hephaestin double-knockout (DKO) mice with age-dependent retinal iron accumulation and some features of AMD were used to test retinal protection by the oral iron chelator deferiprone (DFP).
Methods.
Cultured retinal pigment epithelial (ARPE-19) cells and mice were treated with DFP. Transferrin receptor mRNA (Tfrc), an indicator of iron levels, was quantified by qPCR. In mice, retinal oxidative stress was assessed by mass spectrometry, and degeneration by histology and electroretinography.
Results.
DFP at 60 μM decreased labile iron in ARPE-19 cells, increasing Tfrc and protecting 70% of cells against a lethal dose of H2O2. DFP 1 mg/mL in drinking water increased retinal Tfrc mRNA 2.7-fold after 11 days and also increased transferrin receptor protein. In DKOs, DFP over 8 months decreased retinal iron levels to 72% of untreated mice, diminished retinal oxidative stress to 70% of the untreated level, and markedly ameliorated retinal degeneration. DFP was not retina toxic in wild-type (WT) or DKO mice, as assessed by histology and electroretinography.
Conclusions.
Oral DFP was not toxic to the mouse retina. It diminished retinal iron levels and oxidative stress and protected DKO mice against iron overload–induced retinal degeneration. Further testing of DFP for retinal disease involving oxidative stress is warranted.
Iron is crucial for optimal cellular metabolism, but is also a potent generator of oxidative stress if present in excess, especially in the form of labile ferrous iron. Inability of the body to actively excrete excess iron leads to age-dependent iron accumulation in certain tissues, including the macula.1 Excess tissue iron generates reactive oxygen species (ROS) via the Fenton reaction, leading to oxidative damage. Free radicals and oxidative stress have been implicated in a growing number of conditions, from normal aging to cancer, diabetes, and neurodegenerative diseases, making iron overload or metabolic mishandling of iron an important target for therapeutic intervention.2–6
Since iron catalyzes the production of the hydroxyl radical, the most damaging of the free radicals, it is likely to exacerbate oxidative damage in a tissue that is already prone to oxidative insult. Retinal pigment epithelial (RPE) cells and photoreceptors are especially vulnerable to oxidative damage due to high oxygen tension, ROS production by large numbers of mitochondria, and abundant, easily oxidized polyunsaturated fatty acids in photoreceptor membranes.7 Indeed, several neurodegenerative disorders with iron dysregulation feature retinal degeneration.8 These include the rare hereditary disorders aceruloplasminemia, Friedreich's ataxia, and pantothenate kinase-associated neurodegeneration. Further, traumatic siderosis causes rapid retinal degeneration.9 Similarly, retinal degeneration in several mouse models is associated with retinal iron dysregulation.10–12
Age-related macular degeneration (AMD) is the most common cause of irreversible vision loss in the elderly worldwide. Although the pathogenesis of AMD is incompletely understood, growing evidence suggests that, in addition to inflammation, complement activation, and other hereditary and environmental influences,13–19 oxidative stress20–24 and iron may play important roles. We have demonstrated higher iron levels in AMD retinas than in age-matched controls, suggesting that iron-mediated oxidative stress may contribute to retinal degeneration in AMD.9 Supporting this hypothesis, patients lacking the ferroxidase ceruloplasmin (Cp) as a result of the autosomal recessive condition aceruloplasminemia, have retinal iron accumulation and early-onset macular degeneration.25 Similarly, Cp and hephaestin (Heph) double knockout (DKO) mice have age-dependent retinal iron accumulation, and, presumably as a result of the iron accumulation, have increased retinal oxidative stress, and retinal degeneration. This retinal degeneration shares some features of AMD, including photoreceptor and RPE death, RPE hypertrophy and autofluorescence, sub-RPE deposits including activated complement factor 3 (C3), and subretinal neovascularization. DKO mice also exhibit sparse macrophage infiltration between the RPE and outer segments, suggesting a chronic inflammatory component in their pathologic retinas.26,27 There are also differences in the pathologic features of the DKO versus AMD retinas: The width of the sub-RPE deposits in DKOs is smaller than in AMD retinas, the number of hypertrophic RPE cells is greater in DKOs than in AMD, and the subretinal neovascularization in DKOs more often originates from the retinal vasculature than from the choroid.
To determine whether iron dysregulation is the cause of retinal degeneration in DKOs and to develop a therapeutic model, we tested whether deferiprone can protect DKO retinas against iron accumulation and degeneration.7 Chelation therapy has, until recently, been used mainly for the treatment of acute iron toxicity and chronic transfusional iron overload in thalassemia and other conditions.2 Recently, iron-chelating drugs have been tested in additional categories of patients with normal body iron load, such as those with neurodegenerative,28 renal, and infectious diseases.29–31 Three widely used iron chelating drugs are deferoxamine, deferiprone (DFP), and deferasirox. Deferoxamine has been used for decades as the main iron chelating agent to treat transfusion-related hemosiderosis. It is administrated via slow subcutaneous infusion over 8 to 12 hours or intravenously in some patients. Deferoxamine's potential as a therapeutic agent is limited by the route of administration, as well as severe side effects at higher doses that include pigmentary retinopathy,32 bone dysplasia, and auditory toxicity.33 DFP is a low-molecular-weight iron chelator that can readily penetrate cells and is approved for use in Europe and Asia. The drug can decrease liver and cardiac iron levels in patients with transfusional iron overload. DFP can cross the blood–brain barrier34 and decrease brain iron levels in patients with Friedreich's ataxia,35 which is associated with improved motor function in some patients. DFP is orally absorbed and binds iron in multiple subcellular and extracellular locations.36,37 Approximately 1% to 2% of patients with thalassemia given oral DFP develop reversible agranulocytosis,38 necessitating constant blood cell count monitoring. Deferasirox, another oral iron chelator, received FDA (U.S. Federal Drug Administration) approval in 2005 for treatment of transfusional iron overload, but there is no evidence to date that it can decrease brain or retinal iron levels.
Iron chelators may serve as protective agents against human retinal disorders in which iron accumulation or oxidative stress play an important role. Systemic chelation with deferoxamine protects the rodent retina from degeneration caused by light-induced damage39 and ischemia reperfusion injury40 and the rabbit retina from electroretinogram (ERG) changes induced by subretinal blood (Youssef TA, et al. IOVS 2002;43:ARVO E-Abstract 3000). Yet, deferoxamine's clinical potential for retina protection is limited by route of administration and retinal toxicity, as discussed earlier. DFP seems to be an attractive candidate as it is orally absorbed, has been shown to cross the blood–brain barrier, and has shown efficacy and low toxicity in human clinical trials for diabetic nephropathy and primary glomerulonephritis.41 We tested whether oral DFP can chelate retinal iron in wild-type (WT) mice without adverse effects on retinal function. Further, we tested whether it can protect cultured retinal cells from oxidative stress and iron overloaded Cp/Heph double knockout (DKO) mouse retinas from degeneration.
Materials and Methods
Cell Culture
The spontaneously immortalized human RPE cell line, ARPE-19 (ATCC, Manassas, VA) was cultured until confluent in 24-well plates (Falcon; BD Biosciences, Bedford, MA) in 1:1 DMEM/F12, 20 μM l-glutamine (Invitrogen, Carlsbad, CA) supplemented with heat inactivated 10% FBS (Hyclone, Logan, UT) and 1% penicillin-streptomycin. The cells were grown in 5% CO2 at 37°C. To evaluate the protective effect of DFP, confluent ARPE-19 cells were washed three times with MEM (Invitrogen, Carlsbad, CA) then pretreated for 30 minutes with various concentrations of DFP in MEM, followed by a 15 hour co-treatment with different concentrations of DFP and 200 μM H2O2 (Sigma-Aldrich, St. Louis, MO). Cell death was assayed after 15 hours (LDH Release Assay Kit; Roche, Basel, Switzerland). Cytotoxicity was expressed as a ratio between maximum LDH release in hydrogen peroxide–treated cells (200 μM) and minimum LDH release in cells incubated in MEM, and the percent viability was calculated. To visualize cell death, a cell viability assay was performed as suggested by the manufacturer (Live/Dead Assay; Invitrogen).
Animals
Female, 6-month-old C57BL/6 mice were obtained from The Jackson Laboratory. Female and male C57BL/6 WT mice, C57BL/6 mice with a targeted mutation in the Cp gene (Cp−/−),42 and mice with a naturally occurring mutation in the Heph gene (Hephsla/sla or Hephsla/Y)43 were generated, as previously published.26 Cp−/−Hephsla/sla (or Cp−/−Hephsla/Y) mice are referred to herein as DKO and Hephsla/sla or Hephsla/Y as Heph KO. The mice were housed in the same facilities in equivalent conditions and were fed ad libitum with a mouse standard growth diet containing 213 ppm of iron (ASAP; Animal Specialities and Provisions, Quakertown, PA). All procedures were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and complied with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Eyes were enucleated immediately after death and were fixed overnight in either 2% paraformaldehyde (PFA) and 2% glutaraldehyde for morphologic analysis or in 4% PFA for immunofluorescence.
DFP Treatment
The mice were treated per os with 1 mg/mL DFP (ApoPharma, Inc., Toronto, Ontario, Canada) in drinking water for various amounts of time, as indicated in the Results section.
Quantitative Real-Time PCR
Gene expression in the neurosensory retina and RPE/choroid samples obtained from DFP-treated and untreated WT and DKO mice was analyzed by quantitative (q)RT-PCR. The samples were acquired by removing the anterior segment and ciliary body and detaching the neurosensory retina from the underlying RPE/choroid tissue. RNA was isolated (RNeasy Mini Kit; Qiagen, Inc., Valencia, CA) according to the manufacturer's protocol. The RNA was quantified with a spectrophotometer and stored at −80°C. cDNA was synthesized (TaqMan Reverse Transcription Reagents; Applied Biosystems, Inc. [ABI], Carlsbad, CA), according to the manufacturer's protocol. Gene expression assays were obtained (Taqman; ABI) and used for qPCR analysis. Probes used were transferrin receptor (Tfrc, Mm00441941_m1), vascular endothelial growth factor A (Vegfa, Mm00437304_m1), erythropoietin (Epo, Mm00433126_m1), CD68 antigen (Cd68, Mm03047343_m1*), and complement component 3 (C3, Mm01232779_m1). Eukaryotic 18S rRNA (Hs99999901_s1) was used as an endogenous control. Real-time RT-PCR (Taqman; ABI) was performed on a sequence detection system (Prism model 7500; ABI) using the ΔΔCT method, which provides normalized expression values. The amount of target mRNA was compared among the groups of interest. All reactions were performed in biological (three mice or tissue culture wells) and technical (three qPCR replicates per biological sample) triplicates.
Immunofluorescence
The globes fixed in 4% PFA were rinsed in PBS, and the eye cups were generated by removing the anterior segment. The eye cups were cryoprotected in 30% sucrose overnight and embedded in optimal cutting temperature compound (OCT; Tissue-Tek, Sakura Finetek, Torrance, CA). Immunofluorescence was performed on 10-μm-thick sections as previously published.44 The primary antibody was rabbit anti-TfR at 1:80 dilution (Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibody was detected using fluorophore labeled secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Control sections were treated identically but with omission of primary antibody (data not shown). The sections were analyzed by fluorescence microscopy with identical exposure parameters (model TE300 microscope; Nikon, Tokyo, Japan, with ImagePro software; Media Cybernetics, Bethesda, MD).
Electroretinogram Recordings
ERG recordings followed procedures described previously.45,46 In brief, mice were dark-adapted overnight and then anesthetized with a cocktail containing (in mg/kg body weight): 25 ketamine, 10 xylazine, and 1000 urethane. In each mouse, the pupils were dilated with 1% tropicamide saline solution (Mydriacil; Alconox, New York, NY) and the mouse was placed on a stage maintained at 37°C. Two miniature cups made of UV-transparent plastic with embedded platinum wires serving as recording electrodes were placed in electrical contact with the corneas. A platinum wire loop placed in the mouth served as the reference and ground electrode. ERGs were then recorded (Espion Electrophysiology System; Diagnosys LLC, Lowell, MA). The apparatus was modified by the manufacturer for experiments with mice by substituting LEDs with emission maximum at 365 nm for standard blue ones. A stage with the mouse was positioned in such a way that the mouse's head was located inside the stimulator (ColorDome; Diagnosys LLC), thus ensuring full-field uniform illumination. Methods for light stimulation and calibration of light stimuli are described elsewhere.45 The a- and b-wave amplitudes are reported for saturating light stimuli.
Quantitative Iron Detection
The eyes from age-matched DFP-treated and untreated WT mice, as well as age-matched DFP-treated and untreated DKO mice were fixed in 4% PFA for several days. Eye cups were made by removing the anterior segment. The ciliary body was removed with a curved scalpel blade, and the neurosensory retina was detached from the underlying RPE/choroid tissue. Samples of the neurosensory retina and RPE/choroid (with sclera) were placed in separate tubes and allowed to dry at room temperature. Total nonheme iron was quantified using the bathophenanthroline-based spectrophotometric protocol described by Torrance and Bothwell.47 Briefly, preweighed tissues were snap frozen and kept at −80°C. Tissue digestion was performed overnight at 65°C in acid digest solution (0.1% trichloroacetic acid and 0.03 M HCl). After digestion, the tissue-acid mixture was vigorously vortexed, cooled to room temperature, and then centrifuged for 25 minutes at 3500 rpm in a centrifuge at room temperature (model 5415D; Eppendorf, Fremont, CA). The supernatant (20 μL) was added to 1 mL of chromogen reagent (2.25 M sodium acetate pretreated with Chelex 100 [Bio-Rad, Hercules, CA], 0.01% bathophenanthroline, and 0.1% thioglycolic acid). The absorbances were read at 535 nm. Iron level was assessed by comparing absorbances of tissue-chromogen samples with serial dilutions of iron standard (Sigma-Aldrich, Inc.).
Morphologic Analysis
After fixation in 2% PFA and 2% glutaraldehyde, eye cups were made. The tissues were then dehydrated in increasing concentrations of ethanol, infiltrated overnight, and embedded the next day in plastic (JB4 Solution A; Polysciences, Inc., Warrington, PA). For standard histology 3-μm-thick plastic sections were toluidine blue–stained by incubation of the sections in 1% toluidine blue O and 1% sodium tetraborate decahydrate (Sigma-Aldrich) for 5 seconds. Stained sections were observed and photographed with the fluorescence microscope (model TE300; Nikon).
Assessment of Oxidative Stress
Mass spectrometry was used for biochemical analysis of isoprostane F2α-VI levels, a specific marker of oxidative stress. Immediately after euthanatization, the retinas were isolated and snap frozen on dry ice. The retinas were homogenized, and total lipids were extracted as previously published.48
Statistical Analysis
The mean ± SE were calculated for each comparison pair. The means between each pair were compared by t-test. P < 0.05 was considered statistically significant (GraphPad software; San Diego CA).
Results
DFP Chelated Labile Iron and Protected against H2O2-Induced Cell Death in Cultured Retinal Cells
To determine whether DFP can protect retinal cells against iron-catalyzed oxidative stress, we used confluent human retinal pigment epithelial cells (ARPE-19 cell line). First, we assessed DFP's ability to chelate labile iron within these cells. Since Tfrc mRNA stability is regulated by labile iron levels, with decreased labile iron resulting in increased Tfrc mRNA,49 we quantified Tfrc mRNA levels after exposing the cells to various concentrations of DFP. At DFP concentrations of 60 μM and higher, Tfrc mRNA levels were increased (Fig. 1A). Next, the cells were treated with 200 μM H2O2, which caused death of 100% of the cells, as measured by the LDH release assay and the fluorescent cell viability assay (Figs. 1B, 1C). This cytotoxicity was markedly diminished when DFP was applied 30 minutes before and during H2O2 exposure. The lowest concentration that chelated enough labile iron to cause a measurable increase in Tfrc mRNA (60 μM) was also the lowest concentration able to protect more than 50% of the cells. DFP itself was not cytotoxic up to the highest concentration tested, 10 mM (data not shown).
Figure 1.
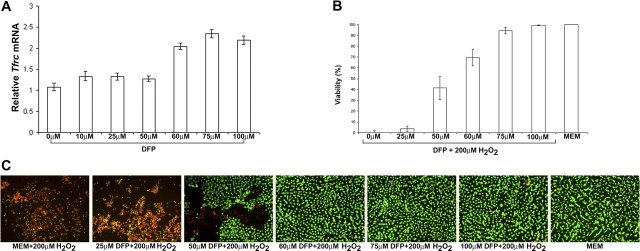
DFP treatment decreased labile iron and protected ARPE19 cells from H2O2-induced cell death. (A) Relative Tfrc mRNA levels as determined by qPCR in ARPE-19 cells after 15 hours of treatment with different concentrations of DFP, as indicated. (B, C) ARPE-19 cells were treated for 15 hours with MEM (100% viability control), 200 μM H2O2 alone, or 200 μM H2O2 with different concentrations of DFP, as indicated. (B) Percentage of cell viability as determined by LDH assay. DFP protected the ARPE-19 cells against H2O2-induced cell death. (C) Fluorescence photomicrographs of ARPE-19 cells labeled in a fluorescent cell viability assay. Red: dead cells as detected by ethidium bromide homodimer fluorescence. Green: live cells as detected by calcein fluorescence. Error bars ± SEM.
Oral DFP in WT Mice Decreased Labile and Total Retinal Iron
Wild-type mice, 6 months of age, that were given DFP in drinking water for 24 hours and killed the following morning had decreased retinal labile iron levels, as indicated by increased Tfrc mRNA levels in the neural retina (Fig. 2A). RPE/choroid Tfrc mRNA levels did not change (Fig. 2B). Longer DFP treatment, for 11 days, resulted in a larger increase in Tfrc mRNA (almost threefold) in the neural retina (Fig. 2C). The RPE/choroid showed a similar trend, but the increase was not statistically significant (Fig. 2D). To test whether transferrin receptor protein (TfR) levels change with the treatment and to localize TfR, retinas from mice treated for 11 days with DFP were compared with untreated controls using immunofluorescence with an anti-TfR antibody. The DFP-treated mice had increased TfR immunoreactivity in all retinal cell layers (Figs. 2E1, 2E2). In 9-month-old WT mice after 3 months of treatment, DFP decreased total nonheme iron levels in the neural retina (Fig. 2F), but not in the RPE/choroid (Fig. 2G).
Figure 2.
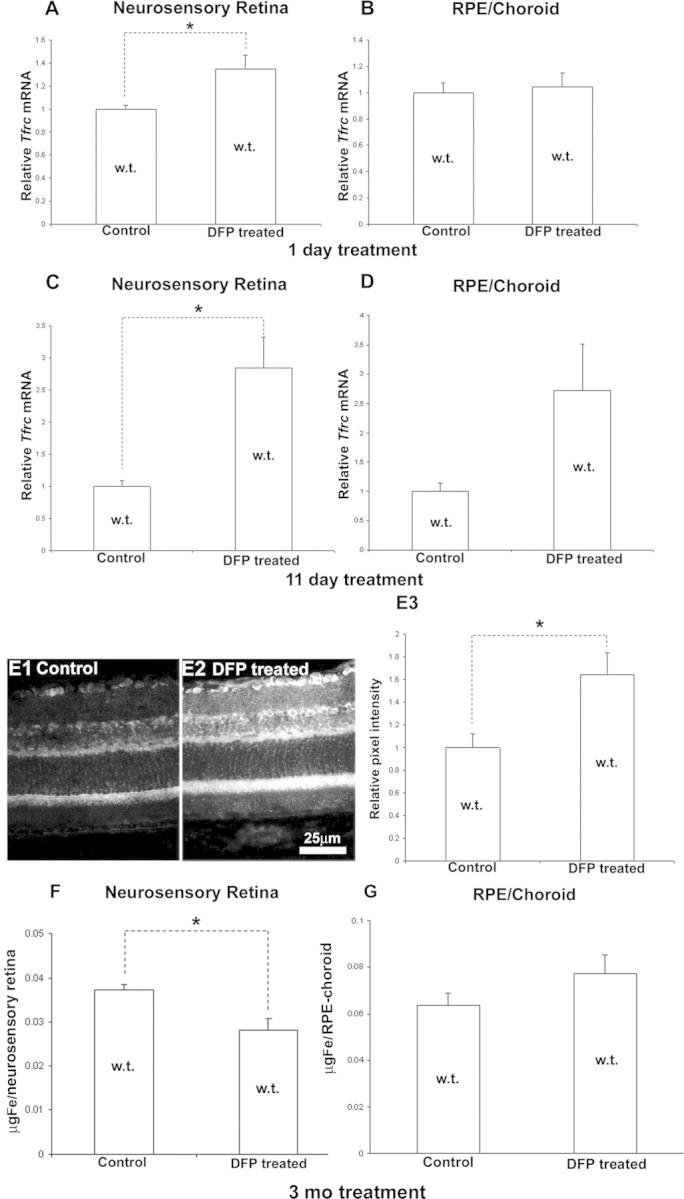
DFP treatment in WT mice chelated labile iron, increasing retinal Tfrc mRNA and protein levels. Wild-type mice had significantly increased Tfrc mRNA levels, as measured by qPCR in the neural retina after DFP treatment for 1 day (A; n = 3 mice per group, 6 months old) and 11 days (C; n = 4 mice per group, 6 months old). The difference in Tfrc expression is not statistically significant in the RPE/choroid in both groups (B, D). Representative fluorescence retinal photomicrographs from 11-day untreated control (E1) and DFP-treated (E2) WT mice. The DFP-treated mice showed increased anti-TfR immunoreactivity throughout the retina. Scale bar, 25 μm. Immunoreactivity was quantified by measuring the mean pixel intensity within the RPE and neural retinas (n = 3 mice per group, 6 months old) and is shown as the mean (E3). Three months of treatment with DFP significantly reduced total iron in the neural retina (F), but not in the RPE/choroid (G). *Significant difference (P < 0.05). Error bars ± SEM.
Oral DFP Exhibited No Retinal Toxicity as Assessed by ERG and Histology
After treatment with DFP, no retinal toxicity was detected by ERG. Wild-type mice treated for 11 days or 3 months with DFP manifested no changes in ERG relative to age- and genotype-matched untreated controls (Table 1). The absence of retinal toxicity in 9-month-old WT mice was also confirmed by the normal retinal morphology after 3 months of treatment (Fig. 3). After 6 months of treatment with DFP, 8-month-old DKO mice had an increase in all ERG wave amplitudes (though not statistically significant at this age; see the Discussion section) relative to untreated age-matched DKO controls (Table 2).
Table 1.
Scotopic and Photopic ERG Wave Amplitudes from WT Mice
| A. Amplitudes with or without 11 Days' Treatment with DFP | |||
|---|---|---|---|
| Group | Scotopic ERG (μV) |
Photopic ERG (μV) |
|
| a-Wave | b-Wave | b-Wave | |
| Control | 372 ± 23.7 | 394 ± 30.56 | 188 ± 15.4 |
| DFP treated | 361 ± 31.3 | 398 ± 36.9 | 183 ± 7.8 |
| P | 0.789 | 0.933 | 0.781 |
| B. Amplitudes with or without 3 Months' Treatment with DFP | |||
|---|---|---|---|
| Group | Scotopic ERG (μV) |
Photopic ERG (μV) |
|
| a-Wave | b-Wave | b-Wave | |
| Control | 216 ± 31.8 | 286 ± 52.5 | 154 ± 33.1 |
| DFP treated | 209 ± 37 | 261 ± 23.9 | 145 ± 16.5 |
| P | 0.906 | 0.660 | 0.802 |
n = 4/group (A) and 5/group (B).
Data are the mean amplitudes ± SEM in 6-month-old mice with access to drinking water, with or without DFP added. The a- and b-wave amplitudes are shown for saturating light stimuli.
Figure 3.
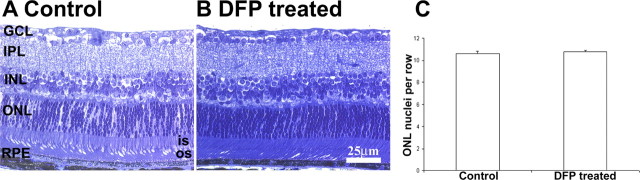
Normal retinal morphology after DFP treatment. Representative bright-field photomicrographs from WT DFP-treated (B) and untreated (A) 9-month-old mice (n = 3 each). After 3 months of treatment, the retinas showed no changes in morphology or in the number of ONL nuclei compared with the controls (C, P > 0.05). OS, photoreceptor outer segment; IS, photoreceptor inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar, 25 μm.
Table 2.
Scotopic and Photopic ERG Wave Amplitudes from DKO Mice
| Group | Scotopic ERG (μV) |
Photopic ERG (μV) |
|
|---|---|---|---|
| a-Wave | b-Wave | b-Wave | |
| WT | 243.5 ± 13.1 | 255 ± 2.3 | 156 ± 2.4 |
| DKO untreated | 54 ± 38.7 | 139 ± 46.5 | 73 ± 35.3 |
| DKO DFP treated | 98 ± 42.3 | 189 ± 11.6 | 107 ± 33 |
| P* | 0.484 | 0.357 | 0.540 |
Mice (n = 3 per group) had access to drinking water, with or without DFP, starting at 2 months of age for 6 months and were killed at 8 months of age. The a- and b-wave amplitudes are shown for saturating light stimuli. Data are the mean ± SEM amplitudes at the 6-month treatment time point.
Calculated for DKO-untreated and DKO DFP-treated mice.
DFP Treatment Decreased Total Iron Pools within the Neural Retina and RPE/Choroid of DKO Mice
Similar to its effects on WT mice, in 9-month-old DKOs, DFP significantly reduced the total iron pool within the neural retina and the RPE/choroid after 6 months of treatment (Figs. 4A, 4B). DFP also decreased retinal labile iron, as 6 months of treatment upregulated Tfrc mRNA in the neural retinas of treated DKO mice relative to untreated age- and genotype-matched controls (Fig. 4C).
Figure 4.

DFP treatment decreased total nonheme and labile iron levels in DKO mice. Shown is total nonheme iron levels in 9-month-old DKO mice treated with DFP for 6 months relative to age-matched, untreated controls. Treated mice had significantly reduced total iron levels in the neural retina (A) and RPE/choroid (B). After 6 months of treatment with DFP, labile iron was significantly reduced in 8-month-old treated DKO mice as assessed by TfR mRNA upregulation (C). *Significant difference (P < 0.05). n = 3 mice per group. Error bars ± SEM.
DFP Protected against Iron–Induced Retinal Degeneration and Reduced Lipofuscin-Like Material Accumulation within the RPE of DKO Mice
The few untreated DKO mice that survived to age 12 to 13 months had iron-laden, massively hypertrophic RPE cells throughout more than 90% of the total retina (n = 3). There was also focal loss of photoreceptor inner and outer segments and thinning of the outer nuclear layer (Figs. 5B, 5E). When treated with DFP for 8 months, age-matched or older DKO mice had marked protection against retinal degeneration (n = 4), with less than 10% of the total retina showing RPE hypertrophy or loss of photoreceptors (Figs. 5C, 5F). The hypertrophic RPE cells in DKO mice were autofluorescent, presumably due to buildup of a form of lipofuscin, an aggregate of lipids and proteins found in aged cells.27 Spectral analysis of autofluorescent, hypertrophic RPE cells from DKO mice and human AMD samples exhibited several peak emission intensities at similar wavelengths,50 suggesting that some of the lipofuscin components are similar in the two types of RPE cells. While untreated DKO mice had bright autofluorescence in more than 90% of the RPE cells (Fig. 5H), age matched DFP-treated DKOs had autofluorescence in fewer than 10% of the RPE cells (Fig. 5I). Age-matched and older WT mice had no RPE autofluorescence, according to these imaging parameters (Fig. 5A).
Figure 5.
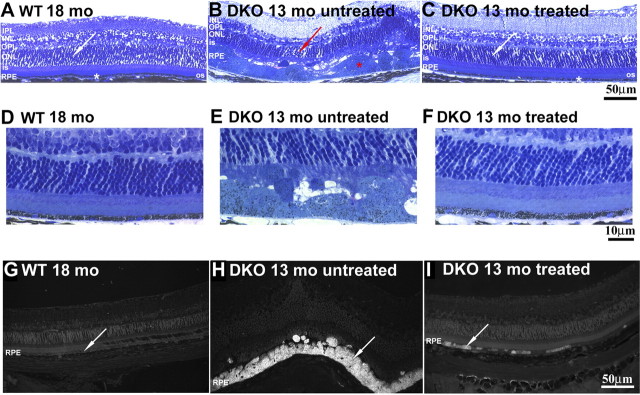
Eight months of DFP treatment protected against iron-induced retinal degeneration and reduced the accumulation of lipofuscin-like material. Bright-field photomicrographs of plastic sections showed that 13-month-old untreated DKO mice (B, n = 3) had massively hypertrophic RPE cells (red asterisk) and focal thinning of the ONL (red arrow). In contrast, 13- to 14-month-old DKO mice treated for 8 months with DFP had normal morphology in most of the retina (C, white asterisk and white arrow; n = 4). (D–F) Bright-field photomicrographs of plastic sections showing the same retinal changes under higher magnification. Fluorescence photomicrographs (Cy2 filter set) of plastic sections show a decrease in the number of autofluorescent RPE cells in DFP-treated DKO mice (I, arrow; n = 4), while the age-matched, untreated DKOs have autofluorescent RPE cells (H, arrow; n = 3) throughout most of the retina. All comparisons were made relative to 18-month WT mice that showed normal retinal architecture and absence of RPE autofluorescence (A, G, arrow, n = 3). RPE, retinal pigment epithelium; OS, photoreceptor outer segment; IS, photoreceptor inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar: (A–C, G–I) 50 μm; (D–F) 10 μm.
DFP Treatment Decreased Retinal Markers of Oxidative Stress and Expression of Genes Involved in Inflammation and Complement Activation
Isoprostane F2α-VI is a prostaglandin-like compound formed by the free radical-catalyzed peroxidation of arachidonic acid. It serves as a quantitative, specific marker of oxidative stress in the retina48 and the brain.51 DFP treatment for 5 months significantly reduced isoprostane F2α-VI levels in the neural retinas of DKO mice (age 9 months) relative to untreated age-matched control DKOs (Fig. 6A). Erythropoietin (Epo), a cytokine regulating hematopoiesis, can be considered a marker of oxidative stress because Epo mRNA levels can be upregulated by oxidative and nitrosative stress.52 Consistent with a decrease in oxidative stress, DFP treatment for 6 months significantly lowered retinal Epo mRNA levels (Fig. 6B), whereas mRNA levels of another HIF regulated proangiogenic gene, Vegfa, remained unchanged in both neurosensory retina (Fig. 6C) and RPE/choroid (Fig. 6D). DFP treatment significantly reduced neurosensory retina mRNA levels of the monocyte/macrophage specific glycoprotein Cd68 (Fig. 6E) and was associated with a trend toward decreased C3 mRNA levels in the RPE/choroid (Fig. 6F).
Figure 6.

DFP treatment decreases retinal markers of oxidative stress. Graph showing retinal isoprostane F2α-VI levels in the retinas of 9-month-old DKO mice treated with DFP for 5 months relative to untreated, age-matched DKO and WT mice (A, n = 3 mice per group). DFP treatment significantly reduced Epo mRNA levels, as measured by qPCR, in the retinas of DKO mice treated for 6 months with DFP relative to untreated 9-month-old DKOs (B, n = 3 mice per group). DFP did not change Vegfa mRNA levels in either neurosensory retina (C) or RPE/choroid (D), but it significantly reduced Cd68 mRNA levels in neurosensory retinas of treated DKO relative to untreated DKO controls (E) and showed a trend toward C3 mRNA level reduction in the RPE/choroid (F). *Significant difference (P < 0.05). Error bars ± SEM.
Discussion
In this study, we tested whether chelation with a low-molecular-weight, cell-permeant, central nervous system–permeant iron chelator, such as DFP, is a safe and effective therapy for iron-exacerbated retinal degeneration. Our data showed that in mice, oral DFP was not retina toxic, caused a decrease in retinal labile iron after short-term administration, and decreased retinal iron stores after long-term administration. Further, it diminished oxidative stress in DKO mouse retinas and provided marked protection against retinal degeneration.
Since transferrin receptor (Tfrc) mRNA is stabilized by a decrease in labile iron, we tested chelation of labile retinal iron in WT mice through quantification of Tfrc mRNA. After only 1 day of oral DFP treatment, qPCR demonstrated a significant increase in retinal Tfrc mRNA levels (Fig. 2). Eleven days of treatment was sufficient to upregulate TfR protein enough to detect increased anti-TfR immunofluorescence throughout retinas of treated mice compared with untreated controls. Unlike chelation of free, labile iron, longer DFP treatment was required to produce a measurable reduction in total retinal iron levels. Three months of treatment in WT mice or 6 months of treatment in DKOs significantly reduced total neural retina iron stores.
Unlike deferoxamine, which is a large, positively charged molecule, DFP is only 139 Da in mass and is neutral in the circulation, whether free or bound to iron, and readily penetrates cells.53 Retinal toxicity manifesting as a pigmentary retinopathy with altered retinal function documented by ERG has been reported in some patients treated with deferoxamine.32,54 The mechanism of deferoxamine's retinal toxicity has not been defined. We detected no DFP-induced decreases in ERG amplitudes in WT or DKO mice at the doses used. On the contrary, the mean amplitudes were higher for all three wave types in the treated DKOs compared with the untreated DKOs. This difference was not statistically significant, most likely because 8-month DKOs have a variable amount of focal retinal degeneration resulting in large ERG standard errors. At 13 months, untreated DKOs have widespread retinal degeneration, but we were unable to obtain ERGs on those, as most of them die from ataxia associated with brain iron accumulation by 6 to 9 months, before they develop widespread retinal degeneration.
The small number of untreated DKO mice that survived until 12 to 13 months had RPE hypertrophy in more than 90% of the retina. These RPE cells were autofluorescent, suggesting lipofuscin accumulation.44 When treated with oral DFP, DKO mice were protected from harmful effects of excess iron. Even at 14 months of age, DFP-treated DKO mice had less than 10% of the total retina affected by RPE hypertrophy, increased photoreceptor survival, and markedly reduced areas of RPE autofluorescence (Fig. 5). The mechanism of protection is most likely chelation of redox active iron within the cytosol or within organelles such as lysosomes, diminishing oxidative stress. Supporting this, isoprostane levels, a marker of lipid peroxidation, were diminished in the retinas of DFP-treated DKO mice relative to untreated DKOs (Fig. 6). The fact that DFP provided marked protection against the retinal degeneration in DKOs supports the hypothesis that the age-dependent iron accumulation within retinas of these mice causes their retinal degeneration.
DKO mice were treated with DFP beginning at age 5 months or younger. At 5 months, the retina has measurable increases in iron, but this is before the onset of retinal degeneration. Thus, this study shows that DFP prevents retinal degeneration after iron accumulation. Future studies will test whether retinal degeneration can be reversed by DFP administration beginning at an older age.
In DKO retinas, two HIF-regulated proangiogenic genes are upregulated relative to WT: vascular endothelial growth factor (Vegf) and erythropoietin (Epo). One theoretical concern regarding the therapeutic use of an iron chelator in AMD patients is that it might stabilize HIF through inactivation of the iron-dependent prolyl hydroxylase that tags HIF for degradation, resulting in Vegf and Epo upregulation, which, in turn, could cause harmful neovascularization. Yet, in DKOs, DFP treatment decreases Epo mRNA levels (Fig. 6), whereas Vegf mRNA levels remain unchanged. WT mice treated with oral DFP do not have changes in retinal Vegf and Epo mRNA levels (not shown). Thus, oral DFP does not appear to upregulate retinal Vegf. The DFP-induced reduction in Epo in DKOs may result from diminished retinal oxidative stress (which has been shown to upregulate Epo)55 or from improved retinal oxygenation. The former explanation is more likely, as anemic 3-month-old HephKO mice, which do not have retinal iron overload, do not have retinal Epo mRNA upregulation compared with age-matched WT mice (not shown).
While testing for retinal protection, we observed other systemic effects of DFP.
Most DKO mice manifest progressive ataxia and die by 6 to 9 months, because of brain iron accumulation within specific brain regions, especially the substantia nigra and striatum, with loss of dopaminergic neurons (Harris ZL et al., manuscript in preparation). DFP treatment decreased nonheme iron in DKO brains (not shown). This was associated with prevention of ataxia in these mice and a markedly increased lifespan (not shown).
Our study suggests that DFP may serve as a protective agent against retinal diseases in which iron-induced oxidative stress has been implicated, including AMD. The dose used in this study, 1 mg/mL in drinking water, results in an approximate dose in mice of 150 mg/kg/d. This dose is higher than that typically used in thalassemic patients (75–100 mg/kg/d), but provides proof-of-principle that DFP can be therapeutic in the retina. Experiments are in progress to assess ocular pharmacokinetics in mice and larger animals. In humans, a single 50-mg/kg dose can result in blood levels as high as 300 μM, which is more than five times the dose needed to provide significant protection to ARPE-19 cells exposed to a lethal dose of hydrogen peroxide.56
Although DFP is approved for use for iron overload disorders in Europe and Asia, approximately 1% of thalassemic patients taking oral DFP develop reversible agranulocytosis, requiring frequent blood cell count monitoring.53,57 DFP binds iron with higher affinity than it does copper or zinc, but may diminish levels of these latter metals. Thus levels of iron, copper, and zinc should be monitored in any future clinical trial of DFP for AMD, especially given the evidence that zinc supplementation is protective against AMD58 and that the elderly may have reduced zinc intake. Unlike intracellular zinc chelation, which could be detrimental, it is possible that copper chelation would be helpful, as copper can produce reactive oxygen species. Extracellular zinc chelation could also be helpful, as zinc deposits in Bruch's membrane and may promote drusen formation.59 Since DFP is currently approved only for treatment of patients with iron overload, any consideration of its potential use in patients with localized iron dysregulation, as opposed to iron overload, must incorporate careful assessment of dose, as novel adverse effects and/or adverse effects at lower doses could be anticipated.
While further safety and pharmacokinetic studies are needed before initiating a clinical trial for retinal disease, this study suggests that DFP holds promise for prevention or treatment of retinopathies involving iron overload or oxidative stress, including AMD, diabetic retinopathy, and aceruloplasminemia.
Acknowledgments
The authors thank Xinyu Zhao for technical support.
Footnotes
Supported by The American Health Assistance Foundation, the Pennsylvania Lions Eye Research Foundation, ApoPharma, the F. M. Kirby Foundation, and the Paul and Evanina Bell Mackall Foundation Trust. The University of Pennsylvania and ApoPharma share intellectual property rights to the use of deferiprone for treatment of eye disease.
Disclosure: M. Hadziahmetovic, None; Y. Song, None; N. Wolkow, None; J. Iacovelli, None; S. Grieco, None; J. Lee, None; A. Lyubarsky, None; D. Pratico, None; J. Connelly, ApoPharma (E), M. Spino, ApoPharma (E), P; Z.L Harris, None; J.L. Dunaief, ApoPharma (F), P
References
- 1. Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch Ophthalmol. 2003;121:1099–1105 [DOI] [PubMed] [Google Scholar]
- 2. Kontoghiorghes GJ, Efstathiou A, Kleanthous M, Michaelides Y, Kolnagou A. Risk/benefit assessment, advantages over other drugs and targeting methods in the use of deferiprone as a pharmaceutical antioxidant in iron loading and non iron loading conditions. Hemoglobin. 2009;33:386–397 [DOI] [PubMed] [Google Scholar]
- 3. Snyder AM, Connor JR. Iron, the substantia nigra and related neurological disorders. Biochim Biophys Acta. 2009;1790:606–614 [DOI] [PubMed] [Google Scholar]
- 4. Sullivan JL. Iron in arterial plaque: modifiable risk factor for atherosclerosis. Biochim Biophys Acta. 2009;1790:718–723 [DOI] [PubMed] [Google Scholar]
- 5. Zacharski LR, Chow BK, Howes PS, et al. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. J Natl Cancer Inst. 2008;100:996–1002 [DOI] [PubMed] [Google Scholar]
- 6. Duce JA, Tsatsanis A, Cater MA, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell. 2010;142:857–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614 [DOI] [PubMed] [Google Scholar]
- 8. Loh A, Hadziahmetovic M, Dunaief JL. Iron homeostasis and eye disease. Biochim Biophys Acta. 2009;1790:637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong RW, Richa DC, Hahn P, Green WR, Dunaief JL. Iron toxicity as a potential factor in AMD. Retina. 2007;27:997–1003 [DOI] [PubMed] [Google Scholar]
- 10. Yefimova MG, Jeanny JC, Keller N, et al. Impaired retinal iron homeostasis associated with defective phagocytosis in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 2002;43:537–545 [PubMed] [Google Scholar]
- 11. Deleon E, Lederman M, Berenstein E, Meir T, Chevion M, Chowers I. Alteration in iron metabolism during retinal degeneration in rd10 mouse. Invest Ophthalmol Vis Sci. 2009;50:1360–1365 [DOI] [PubMed] [Google Scholar]
- 12. Gnana-Prakasam JP, Thangaraju M, Liu K, et al. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: role of cystine/glutamate exchanger. Biochem J. 2009;424:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li CM, Chung BH, Presley JB, et al. Lipoprotein-like particles and cholesteryl esters in human Bruch's membrane: initial characterization. Invest Ophthalmol Vis Sci. 2005;46:2576–2586 [DOI] [PubMed] [Google Scholar]
- 14. Takeda A, Baffi JZ, Kleinman ME, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460:225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cousins SW, Espinosa-Heidmann DG, Csaky KG. Monocyte activation in patients with age-related macular degeneration: a biomarker of risk for choroidal neovascularization? Arch Ophthalmol. 2004;122:1013–1018 [DOI] [PubMed] [Google Scholar]
- 16. Spraul CW, Lang GE, Grossniklaus HE. Morphometric analysis of the choroid, Bruch's membrane, and retinal pigment epithelium in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:2724–2735 [PubMed] [Google Scholar]
- 17. Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGeer EG, Klegeris A, McGeer PL. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26(suppl 1):94–97 [DOI] [PubMed] [Google Scholar]
- 19. Wu Y, Yanase E, Feng X, Siegel MM, Sparrow JR. Structural characterization of bisretinoid A2E photocleavage products and implications for age-related macular degeneration. Proc Natl Acad Sci U S A. 2010;107:7275–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai J, Nelson KC, Wu M, Sternberg P, Jr, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221 [DOI] [PubMed] [Google Scholar]
- 22. Cano M, Thimmalappula R, Fujihara M, et al. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res. 2010;50:652–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Age-Related Eye Disease Study Research Group. The Age-Related Eye Disease Study: a clinical trial of zinc and antioxidants. Age-Related Eye Disease Study Report No. 2. J Nutr. 2000;130:1516S–1519S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen JK, Dong A, Hackett SF, Bell WR, Green WR, Campochiaro PA. Oxidative damage in age-related macular degeneration. Histol Histopathol. 2007;22:1301–1308 [DOI] [PubMed] [Google Scholar]
- 25. Dunaief JL, Richa C, Franks EP, et al. Macular degeneration in a patient with aceruloplasminemia, a disease associated with retinal iron overload. Ophthalmology. 2005;112:1062–1065 [DOI] [PubMed] [Google Scholar]
- 26. Hahn P, Qian Y, Dentchev T, et al. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci U S A. 2004;101:13850–13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hadziahmetovic M, Dentchev T, Song Y, et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boddaert N, Le Quan Sang KH, Rotig A, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–408 [DOI] [PubMed] [Google Scholar]
- 29. Kontoghiorghes GJ, Kolnagou A, Peng CT, Shah SV, Aessopos A. Safety issues of iron chelation therapy in patients with normal range iron stores including thalassaemia, neurodegenerative, renal and infectious diseases. Expert Opin Drug Saf. 2010;9:201–206 [DOI] [PubMed] [Google Scholar]
- 30. Liu G, Men P, Perry G, Smith MA. Nanoparticle and iron chelators as a potential novel Alzheimer therapy. Methods Mol Biol. 2010;610:123–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boelaert JR, Weinberg GA, Weinberg ED. Altered iron metabolism in HIV infection: mechanisms, possible consequences, and proposals for management. Infect Agents Dis. 1996;5:36–46 [PubMed] [Google Scholar]
- 32. Hidajat RR, McLay JL, Goode DH, Spearing RL. EOG as a monitor of desferrioxamine retinal toxicity. Doc Ophthalmol. 2004;109:273–278 [DOI] [PubMed] [Google Scholar]
- 33. Karimi M, Asadi-Pooya AA, Khademi B, Asadi-Pooya K, Yarmohammadi H. Evaluation of the incidence of sensorineural hearing loss in beta-thalassemia major patients under regular chelation therapy with desferrioxamine. Acta Haematol. 2002;108:79–83 [DOI] [PubMed] [Google Scholar]
- 34. Fredenburg AM, Sethi RK, Allen DD, Yokel RA. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1′ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology. 1996;108:191–199 [DOI] [PubMed] [Google Scholar]
- 35. Kakhlon O, Manning H, Breuer W, et al. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112:5219–5227 [DOI] [PubMed] [Google Scholar]
- 36. Kontoghiorghes GJ, Neocleous K, Kolnagou A. Benefits and risks of deferiprone in iron overload in thalassaemia and other conditions: comparison of epidemiological and therapeutic aspects with deferoxamine. Drug Saf. 2003;26:553–584 [DOI] [PubMed] [Google Scholar]
- 37. Sohn YS, Breuer W, Munnich A, Cabantchik ZI. Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood. 2008;111:1690–1699 [DOI] [PubMed] [Google Scholar]
- 38. Galanello R, Campus S. Deferiprone chelation therapy for thalassemia major. Acta Haematol. 2009;122:155–164 [DOI] [PubMed] [Google Scholar]
- 39. Michon JJ, Li ZL, Shioura N, Anderson RJ, Tso MO. A comparative study of methods of photoreceptor morphometry. Invest Ophthalmol Vis Sci. 1991;32:280–284 [PubMed] [Google Scholar]
- 40. Zhu Y, Zhang L, Gidday JM. Deferroxamine preconditioning promotes long-lasting retinal ischemic tolerance. J Ocul Pharmacol Ther. 2008;24:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shah SV, Rajapurkar MM. The role of labile iron in kidney disease and treatment with chelation. Hemoglobin. 2009;33:378–385 [DOI] [PubMed] [Google Scholar]
- 42. Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999;96:10812–10817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199 [DOI] [PubMed] [Google Scholar]
- 44. Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442 [DOI] [PubMed] [Google Scholar]
- 45. Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN., Jr UV- and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lyubarsky AL, Lem J, Chen J, Falsini B, Iannaccone A, Pugh EN., Jr Functionally rodless mice: transgenic models for the investigation of cone function in retinal disease and therapy. Vision Res. 2002;42:401–415 [DOI] [PubMed] [Google Scholar]
- 47. Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci. 1968;33:9–11 [PubMed] [Google Scholar]
- 48. Dentchev T, Yao Y, Pratico D, Dunaief J. Isoprostane F2alpha-VI, a new marker of oxidative stress, increases following light damage to the mouse retina. Mol Vis. 2007;13:190–195 [PMC free article] [PubMed] [Google Scholar]
- 49. Rouault TA, Tang CK, Kaptain S, et al. Cloning of the cDNA encoding an RNA regulatory protein: the human iron-responsive element-binding protein. Proc Natl Acad Sci U S A. 1990;87:7958–7962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hadziahmetovic M, Song Y, Ponnuru P, et al. Age-dependent retinal iron accumulation and degeneration in Hepcidin knockout mice. Invest Ophthalmol Vis Sci. Published online September 10, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pratico D. The neurobiology of isoprostanes and Alzheimer's disease. Biochim Biophys Acta. 2010;1801:930–933 [DOI] [PubMed] [Google Scholar]
- 52. Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647 [DOI] [PubMed] [Google Scholar]
- 53. Cappellini MD, Pattoneri P. Oral iron chelators. Annu Rev Med. 2009;60:25–38 [DOI] [PubMed] [Google Scholar]
- 54. Baath JS, Lam WC, Kirby M, Chun A. Deferoxamine-related ocular toxicity: incidence and outcome in a pediatric population. Retina. 2008;28:894–899 [DOI] [PubMed] [Google Scholar]
- 55. Pialoux V, Mounier R, Brown AD, Steinback CD, Rawling JM, Poulin MJ. Relationship between oxidative stress and HIF-1 alpha mRNA during sustained hypoxia in humans. Free Radic Biol Med. 2009;46:321–326 [DOI] [PubMed] [Google Scholar]
- 56. al-Refaie FN, Sheppard LN, Nortey P, Wonke B, Hoffbrand AV. Pharmacokinetics of the oral iron chelator deferiprone (L1) in patients with iron overload. Br J Haematol. 1995;89:403– 408 [DOI] [PubMed] [Google Scholar]
- 57. Cappellini MD, Piga A. Current status in iron chelation in hemoglobinopathies. Curr Mol Med. 2008;8:663–674 [DOI] [PubMed] [Google Scholar]
- 58. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS Report no. 8. Arch Ophthalmol. 2001;119:1417–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lengyel I, Flinn JM, Peto T, et al. High concentration of zinc in sub-retinal pigment epithelial deposits. Exp Eye Res. 2007;84:772–780 [DOI] [PubMed] [Google Scholar]