

Abstract
Purpose
Aurora kinase A (AURKA) overexpression is associated with poor prognosis in neuroblastoma and has been described to upregulate VEGF in gastric cancer cells. However, the exact role of AURKA in the regulation of neuroblastoma tumorigenesis remains unknown. We hypothesize that AURKA-mediated stabilization of N-Myc may affect VEGF expression and angiogenesis in neuroblastoma. Therefore, we sought to determine whether inhibition of AURKA modulates neuroblastoma angiogenesis.
Methods
Cell viability and anchorage-independent growth were determined after silencing AURKA or after treatment with MLN8237, AURKA inhibitor. Immunofluorescence was used to determine N-Myc localization. Human umbilical vein endothelial cells (HUVECs) were used to assess angiogenesis in vitro. Real time-PCR and ELISA were performed to determine VEGF transcription and secretion, respectively.
Results
Knockdown of AURKA significantly reduced cell proliferation and inhibited anchorage-independent growth. It also decreased N-Myc protein levels and nuclear localization. AURKA inhibition also decreased HUVECs tubule formation along with VEGF transcription and secretion. Similarly, MLN8237 treatment decreased neuroblastoma tumorigenicity in vitro.
Conclusions
Our findings demonstrate that AURKA plays a critical role in neuroblastoma angiogenesis. AURKA regulates nuclear translocation of N-Myc in neuroblastoma cells, thus potentially affecting cell proliferation, anchorage-independent cell growth, and angiogenesis. Targeting AURKA might provide a novel therapeutic strategy in treating aggressive neuroblastomas.
Keywords: Neuroblastoma, AURKA, Angiogenesis, N-Myc
Introduction
Clinical heterogeneity is a hallmark of neuroblastoma with certain tumors undergoing spontaneous regression and others progressing to advanced disease. To help guide treatment, patients are stratified into high- and low-risk groups based on MYCN status, age at diagnosis, and DNA ploidy (1). Despite advances in treatment modalities, high-risk group of patients remain difficult to cure with dismal long-term survival of 40% (1, 2). In light of this, we and others continue to discern intracellular signaling in neuroblastoma that are associated aggressive tumor phenotypes with the goal of developing highly specialized treatment against specific biologic targets.
Aurora kinase A (AURKA), also known as STK15/BTAK, is part of a family of serine/threonine kinases that are integral to the regulation of mitosis and cytokinesis. Amplification of AURKA has been reported in breast (3), and colon (4) cancers, as well as in neuroblastoma cell lines (5). Dysregulation of this oncogene is associated with chromosomal instability, aneuploidy, cell cycle delay and centrosomal abnormalities (6). In neuroblastoma, AURKA overexpression is associated with high-risk group of tumors, MYCN amplification, disease-relapse and decreased progression free survival (7). Furthermore, AURKA has been shown to stabilize MYCN protein levels in neuroblastoma (8). Previously, we have demonstrated that N-Myc regulates PI3K-mediated vascular endothelial growth factor (VEGF) and angiogenesis in neuroblastoma (9). Besides the established role that AURKA plays in promoting carcinogenesis, AURKA overexpression is associated with increased VEGF transcription (10). AURKA contributes to poor prognosis in neuroblastoma via its own overexpression and by directly interacting with N-Myc to stabilize its protein levels.
Several AURKA inhibitors are currently being used in clinical settings. Specifically, MLN8237 is a second generation, orally bioavailable, selective AURKA inhibitor that has been shown to induce cytotoxicity and cell cycle arrest in multiple myeloma (11), enhance chemosensitivity in esophageal cancer, medulloblastoma, and neuroblastoma (12, 13). Preclinical studies using MLN8237 showed significant in vitro growth inhibition and a positive impact on in vivo event-free survival in several pediatric cancers, including neuroblastoma (14), thus prompting phase I clinical trials (15). Based on these, MLN8237 shows a promise for clinical use; however, it remains critical to elucidate the signaling pathways involved in AURKA-mediated tumorigenesis in neuroblastoma.
In this study, silencing AURKA, with shRNA or MLN8237, inhibited cell proliferation and anchorage-independence. For the first time in neuroblastoma, to our knowledge, we also show that targeting AURKA decreases in vitro angiogenesis. Here, we demonstrate that knockdown of AURKA results in decreased nuclear translocation and expression of N-Myc and decreased VEGF secretion, suggesting that AURKA may be upstream of critical oncogene and indirectly regulating angiogenesis in neuroblastoma. Our results further solidify the role that AURKA has in promoting malignant neuroblastoma and the rationale behind creating and using biologic inhibitors, such as MLN8237, as part of the treatment for children with this disease.
Materials and Methods
Materials
Antibodies against AURKA, N-Myc and cell lysis buffer were obtained from Cell Signaling Technology (Beverly, MA). Antibody against β-actin and fetal bovine serum (FBS) were from Sigma (St. Louis, MO). NuPAGE Novex 4–12% Bis–Tris Gel and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). Horseradish Peroxidase (HRP)-conjugated secondary antibodies against mouse and rabbit IgG were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Chemiluminescence (ECL) HRP substrate were purchased from Millipore (Immobilon Western) and Perkin Elmer (Western Lightning). MLN8237 was from Selleckchem (Houston, TX). Human VEGF antibody and VEGF neutralizing antibody were from R&D Systems, Inc. (Minneapolis, MN).
Cell culture, plasmids and transfection
Human neuroblastoma cell lines were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI 1640 medium with L-glutamine (CellGro Mediatech, Inc. Herndon, VA) supplemented with 10% FBS. Cells were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Human umbilical vein endothelial cells (HUVECs, obtained from Dr. M. Freeman, Vanderbilt University Medical Center) were cultured in EMM-2 supplemented with growth factors (EGM-2 SingleQuot kit, Lonza, Walkersville, MD) at 37°C and humidified 5% CO2. shRNA against AURKA (shAURKA) and non-targeting control (shCON) were purchased from Sigma-Aldrich. For transfection, cells were plated in 6-well plates and transfected with shRNA using Lipofectamine 2000 as per manufacturer's protocol.
Cell viability and soft agar colony formation assays
Cells were seeded onto 96-well plates at a density of 1 × 104 cells per well in RPMI culture media with 10% FBS, and cell number was assessed using Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD) for cell viability. For soft agar assay, cells were trypsinized and resuspended in RPMI 1640 media containing 0.4% agarose and 10% FBS. Cells were overlaid onto a bottom layer of solidified 0.8% agarose in RPMI 1640 media containing 5% FBS, at cell concentrations of 3×103 cells per well of a 6-well plate, and incubated for 3 weeks. Colonies were stained with 0.05% crystal violet, photographed, and quantified.
Immunoblotting and immunofluorescence
Whole-cell lysates were prepared using cell lysis buffer with 1 mM PMSF and incubated on ice for 30 to 60 min. Total protein (50 μg/lane) was resolved on NuPAGE Novex 4–12% Bis–Tris gels and electrophoretically transferred to polyvinylidene difluoride membranes. Nonspecific binding sites were blocked with 5% milk in TBST (120 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 0.05% Tween 20) for 1 h at room temperature (RT) or overnight at 4 °C. Target proteins were detected by using rabbit or mouse anti-human antibodies (1:500 to 1000 dilution) for 3 h at RT or overnight at 4 °C. The membranes were washed three times and incubated with secondary antibodies (1:5000 dilution) conjugated with HRP. Immune complexes were visualized using the enhanced ECL system. Equal loading and transfer were confirmed by blotting the same membrane with β-actin antibody. Data are representative of three independent experiments.
For immunofluorescence, transfected BE(2)-C cells were plated on glass coverslips and incubated for 48 h. The treated cells were washed once with phosphate buffered saline (PBS) and fixed in formalin for 20 min at RT. Fixed cells were rehydrated for 30 min at RT. Samples were then blocked in 1% BSA/PBS buffer for 30 min, and incubated for 60 min in 1:100 dilution of anti-N-Myc antibody. Cells were washed three times with PBS and incubated 30 min in PBS containing 1% BSA and 1:500 secondary antibody Alexa Flour 488 goat anti-rabbit, then stained with DAPI for 5 min. Coverslips were mounted onto the slide glasses. The mounted cells were viewed with a fluorescence microscope using a 40X objective lens.
Endothelial cell tube formation assay
HUVECs grown to ∼70% confluence were trypsinized, counted, and seeded with 48,000 cells per well in 24-well plates coated with 300 μl of Matrigel (BD Biosciences, Bedford, MA). These cells were periodically observed by microscope as they differentiated into capillary-like tubule structures. After 6 h, cells were stained with Hematoxylin & Eosin and photographs were taken via microscope. The average number of tubules was calculated from examination of three separate microscopic fields (200X) and representative photographs obtained.
VEGF ELISA
The supernatant of cultured cells was collected at various time points, and subsequently frozen at -70°C. For assay, samples were thawed and centrifuged, and then VEGF levels were measured using a human VEGF ELISA kit according to manufacturer's instructions (R&D Systems, Inc.).
Reverse transcription and Real Time-PCR
Total RNA was isolated using RNAqueous™ kit (Ambion, Austin, TX, USA) according to the manufacturer's instructions. Isolated RNA was used to synthesize cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Austin, TX, USA). Primers designed to amplify human VEGF fragment (NM_003376) were: forward primer 5′-AGGAGGAGGGCAGAATCATCAC-3′; reverse primer 5′-ATGTCCACCAGGGTCTCGATTG-3′. Glyceraldehyde 3-phosphate dehydrogenase-specific oligonucleotide primers used were previously published (16). VEGF mRNA levels were measured by quantitative real-time PCR using SsoFast™ EvaGreen Supermix with CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA, USA)
Statistical analysis
Scoring index, relative luciferase activity were expressed as means ± SEM; statistical analyses were performed using one-way analysis of variance for comparisons between the treatment groups. A P value of <0.05 was considered significant. Image J was used for densitometric analysis of immunoblot band intensities.
Results
Constitutive mRNA and protein levels of AURKA
We first examined constitutive mRNA and protein levels of AURKA in our 13 human neuroblastoma cell lines. Several of our cell lines demonstrated high AURKA mRNA levels (Fig. 1A), with the highest expression found in SK-N-DZ, LA-N-1 and MC-IXC. Western blotting also revealed significantly high protein expression of AURKA in several of our cell lines (Fig. 1B), which corroborated mRNA levels. We then chose to work with BE(2)-C cell for this study since it is known to have increased malignant potential and tumorigenic properties (17). BE(2)-C cells were transfected with plasmids containing shRNA against AURKA and stable subclonal populations were cultured. After transfection, there was reduced expression of AURKA as compared to cells transfected with control vector, shCON (Fig. 1C).
Figure 1. Constitutive mRNA and protein levels AURKA in neuroblastoma cell lines and AURKA silencing in BE(2)-C cells.

(A) Endogenous AURKA mRNA transcription activity in 13 cell lines. GAPDH was used as an internal control. (B) AURKA protein expression was examined by immunoblotting. The numbers indicate the relative expression of AURKA to β-actin measured with densitometry. (C) Immunoblotting confirmed the decrease in the protein levels after targeted silencing (shAURKA). β-actin was used as a loading control for (B) and (C).
Inhibition of AURKA decreased neuroblastoma cell proliferation
We then focused on identifying the phenotypic effects of RNA-mediated silencing of AURKA in BE(2)-C cells. Cell viability assay (CCK-8) was used to assess cell proliferation at 48, 72, and 96 h after transfection with shRNA against AURKA (shAURKA) or non-targeted control (shCON). Cell proliferation was significantly decreased at 72 and 96 h in BE(2)-C/shAURKA cells when compared to BE(2)-C/shCON (Fig. 2A). We then used MLN8237, AURKA inhibitor, at varying dosages to treat BE(2)-C cells at 72 h. After treatment with MLN8237, there was markedly decreased cell proliferation at 72 h at concentrations of 100 and 500 nM (Fig. 2B).
Figure 2. AURKA inhibition decreased cell proliferation.

(A) Cell proliferation, assessed using CCK-8 kit, was decreased in BE(2)-C cells transfected with shAURKA in comparison to controls (shCON). (B) MLN8237, a specific AURKA inhibitor, also decreased BE(2)-C cell proliferation at 72 h. (mean ± SEM; *=p<0.05 vs. control or control vector)
Inhibition of AURKA suppressed anchorage-independent colony growth
We next wanted to examine the effects of AURKA inhibition on soft agar colony formation. BE(2)-C cells were transfected with a control vector or shRNA against AURKA. These cells were then plated in soft agar and colony-forming property was assessed over the course of 3 weeks.
Silencing AURKA resulted in a significant decrease in the number of colonies formed as compared to control (Fig. 3A). We then plated BE(2)-C cells in soft agar in triplicates and treated with MLN8237, and assessed for colony formation after 3 weeks. We observed inhibition of colony formation after MLN8237 treatment in a dose-dependent manner (Fig. 3B). These results suggest that AURKA may be involved in promoting malignant properties such as anchorage-independence and that the use of AURKA inhibitor may have a significant impact on preventing neuroblastoma progression.
Figure 3. Inhibition of soft agar colony formation after shAURKA or MLN8237 treatment.

(A) Soft agar colony formation was decreased in BE(2)-C/shAURKA cells as compared to controls (shCON). (B) MLN8237 treatment also decreased colony formation of BE(2)-C cells in dose-dependent manner when compared to vehicle alone (10% FBS RPMI media). (mean ± SEM; *=p<0.05 vs. control)
Inhibition of AURKA reduced angiogenesis in vitro by modulating VEGF
Neo-vascularization is a critical component to tumor growth and survival. In neuroblastoma, increased vascularity and expression of VEGF is associated with aggressive behavior (18). We investigated whether silencing AURKA would have an effect on tumor angiogenesis. Supernatant of stably transfected cell lines, BE(2)-C/shCON and BE(2)-C/shAURKA, was collected and used to treat HUVECs grown on a Matrigel. We observed that HUVEC tubule formation was significantly decreased in cells treated with supernatant from BE(2)-C/shAURKA cells as compared to control (Fig. 4A). We then used MLN8237 (100 nM) to treat BE(2)-C cells. Supernatant from MLN8237-treated BE(2)-C cells showed decrease trend in HUVEC tubule formation (Fig. 4B). To identify more specifically whether AURKA was modulating angiogenesis via altered secretion of VEGF, an ELISA was performed to assess levels of VEGF from BE(2)-C cells after knockdown of AURKA. The amount of VEGF secreted from cells where AURKA was silenced was significantly less than in control cells (Fig. 4C). Finally, to determine the effect on VEGF transcription after AURKA silencing, Real Time-PCR was used to asses VEGF mRNA levels using BE(2)-C/shCON and BE(2)-C/shAURKA cells. There was a marked (∼ 6-fold) decrease in the transcription level of VEGF in silenced AURKA cells as compared to controls (Fig. 4D). Taken together, these findings suggest that AURKA is involved in angiogenesis in neuroblastoma by upregulation of transcriptional activity of VEGF.
Figure 4. Inhibition of AURKA reduced angiogenesis in vitro and decreased VEGF mRNA levels and secretion.

(A) HUVECs cultured in cell culture supernatant from BE(2)-C/shAURKA cells resulted in decreased tubule formation when compared to cells grown in supernatant from BE(2)-C/shCON cells. (B) HUVECs cultured in supernatant from BE(2)-C cells treated with MLN8237 resulted in decreased tubule formation when compared to cells grown in supernatant from BE(2)-C cells treated with vehicle alone. (C) BE(2)-C/shCON and BE(2)-C/shAURKA cells were seeded in a 6-well plate, then serum-starved for 48 h. Cell culture supernatant was then collected and VEGF ELISA was performed. (D) AURKA silencing decreased mRNA levels of VEGF by ∼6 fold as assessed by real Time-PCR. GAPDH was used as an internal control. (mean ± SEM; *=p<0.05 vs. control).
Silencing AURKA decreased N-Myc expression and inhibited nuclear translocation
As a transcription factor, N-Myc is able to activate multiple genes that promote cellular pathways critical to malignant tumor development. Although it is known that AURKA directly interacts with N-Myc to stabilize its protein levels by inhibiting its degradation (8), we sought to determine whether AURKA had any effect on its cellular activity. Using the same cell lines that we had established with shRNA against AURKA, we plated cells and then performed immunofluorescence staining to detect N-Myc. After AURKA silencing, there was decreased protein expression (Fig. 5A) and nuclear localization of N-Myc (Fig. 5B). Given that MYCN amplification strongly correlates with poor prognosis, identifying interactions with other oncogenes that enhance tumorigenic properties of neuroblastoma becomes critically important. Our data demonstrating that silencing AURKA can modulate cellular location of N-Myc is a key finding since nuclear translocation is critical for N-Myc transcription activity (19) and inhibiting this ability could interfere with neuroblastoma development and progression.
Figure 5. Silencing AURKA decreased N-Myc expression and inhibited nuclear translocation.
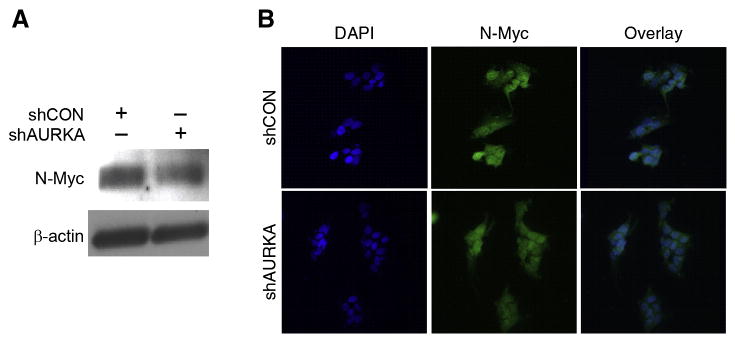
(A) N-Myc expression was decreased in BE(2)-C/shAURKA in comparison to control cells, BE(2)-C/shCON, as assessed by immunoblotting. β-actin was used as a loading control. (B) Immunofluorescence analysis demonstrated decreased nuclear translocation of N- Myc (red) in BE(2)-C/shAURKA cells when compared to controls. DAPI (blue) was used as a nuclear stain.
Discussion
To explore the effects of AURKA on neuroblastoma progression and angiogenesis, we used shRNA to silence AURKA and MLN8237, a specific inhibitor of AURKA, in BE(2)-C cells. Inhibition of AURKA resulted in decreased neuroblastoma cell proliferation, anchorage-independent colony formation and angiogenesis in vitro. We also demonstrated that AURKA silencing decreased the protein expression of N-Myc, and inhibited nuclear translocation of N-Myc along with downregulation of VEGF. It has been well established that AURKA works as an oncogene in many cancers, with a particular focus on its effect on cell proliferation and aberrancies in mitosis or cell cycle checkpoints. In the present study, we identify two novel potential mechanisms, in which AURKA enhances the tumorigenicity of neuroblastoma cells, by regulating the nuclear translocation of N-Myc and its significant effect on VEGF-mediated angiogenesis.
Crosstalk between cellular signaling pathways can sometimes highlight key interactions between previously unassociated targets. AURKA interacts with several substrates in various oncogenic pathways that are critical to tumor progression. We demonstrated that silencing AURKA leads to decreased cell viability and soft agar colony formation. A potential mechanism for this phenomenon could be related to the fact AURKA regulates the expression of key factors involved in neuroblastoma cell proliferation and adhesion. As described by Zhang and colleagues, AURKA has been reported regulate the expression of MMP, pFAK and FAK, key regulators of cell adhesion and anchorage-independence (20). Furthermore, our lab has recently shown that FAK is a critical regulator of cell viability and soft agar colony formation in BE(2)-C cells (21). Although it is beyond the scope of this paper, we speculate that AURKA regulates these critical downstream targets to induce malignant phenotype in neuroblastoma cells, thus offering another avenue of investigation to understand how AURKA contributes to tumor progression.
Tumor neovascularization is required to help initiate and sustain the growth of tumors. Increased VEGF expression is known to be associated with unfavorable Shimada classification and aggressive behavior in neuroblastoma. In adult gastric cancer, AURKA overexpression resulted in increased VEGF mRNA expression (10). After demonstrating that silencing AURKA in BE(2)-C cells resulted in decreased VEGF secretion and in vitro angiogenesis, we wanted to identify whether the transcription activity was also affected. As demonstrated, silencing AURKA resulted in a decrease in mRNA levels of VEGF, providing an explanation for the inhibition of angiogenesis seen in the Matrigel assay. Several modes of VEGF transcriptional regulation have been cited in the literature including AKT-mediated induction in breast (22) and bladder (23) cancers. Interestingly, in our study, in addition to the effects seen on in vitro angiogenesis, silencing AURKA also resulted in decreased expression of pAKT (data not shown). Regulation of Myc proteins occurs through several pathways, including phosphorylation via PI3K/AKT signaling (24). Our data supports the notion that AURKA may be upstream of both N-Myc and AKT, subsequently modulating downstream tumor characteristics such as angiogenesis. Additional studies are required to clarify the relationship between AURKA and MYCN in the regulation of VEGF-induced angiogenesis.
The future of neuroblastoma treatment largely rests upon identifying accessible biologic targets that can be specifically inhibited by drug therapy. The development of AURKA inhibitors has generated a large amount of attention because of its interaction and influence on multiple downstream targets (25). As demonstrated by our studies, targeting AURKA with MLN8237 mimicked the decrease in cell proliferation, colony formation and angiogenesis seen with shRNA-mediated knockdown. MLN8237 also has promising potential to be used in treatment of multiple kinds of cancers (12). The results from our experiments describe a novel role for AURKA in the regulation of N-Myc and tumor vascularization in neuroblastoma.
Acknowledgments
This work was supported by grants R01 DK61470 from the National Institutes of Health and Rally Foundation for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.van Noesel MM, Versteeg R. Pediatric neuroblastomas: genetic and epigenetic ‘danse macabre’. Gene. 2004;325:1–15. doi: 10.1016/j.gene.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 3.Kallioniemi A, Kallioniemi OP, Piper J, Tanner M, Stokke T, Chen L, et al. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(6):2156–60. doi: 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlegel J, Stumm G, Scherthan H, Bocker T, Zirngibl H, Ruschoff J, et al. Comparative genomic in situ hybridization of colon carcinomas with replication error. Cancer research. 1995;55(24):6002–5. [PubMed] [Google Scholar]
- 5.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nature genetics. 1998;20(2):189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 6.Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK. Biology of Aurora A kinase: Implications in cancer manifestation and therapy. Medicinal research reviews. 2010 doi: 10.1002/med.20203. [DOI] [PubMed] [Google Scholar]
- 7.Shang X, Burlingame SM, Okcu MF, Ge N, Russell HV, Egler RA, et al. Aurora A is a negative prognostic factor and a new therapeutic target in human neuroblastoma. Molecular cancer therapeutics. 2009;8(8):2461–9. doi: 10.1158/1535-7163.MCT-08-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer cell. 2009;15(1):67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Kang J, Rychahou PG, Ishola TA, Mourot JM, Evers BM, Chung DH. N-myc is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene. 2008;27(28):3999–4007. doi: 10.1038/onc.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28(6):866–75. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115(25):5202–13. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sehdev V, Peng D, Soutto M, Washington MK, Revetta F, Ecsedy J, et al. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Molecular cancer therapeutics. 2012;11(3):763–74. doi: 10.1158/1535-7163.MCT-11-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muscal JA, Scorsone KA, Zhang L, Ecsedy JA, Berg SL. Additive effects of vorinostat and MLN8237 in pediatric leukemia, medulloblastoma, and neuroblastoma cell lines. Investigational new drugs. 2013;31(1):39–45. doi: 10.1007/s10637-012-9831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatric blood & cancer. 2010;55(1):26–34. doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children's Oncology Group Phase I Consortium study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(21):6058–64. doi: 10.1158/1078-0432.CCR-11-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chatzistamou I, Schally AV, Sun B, Armatis P, Szepeshazi K. Inhibition of growth of OV-1063 human epithelial ovarian cancers and c- jun and c- fos oncogene expression by bombesin antagonists. British journal of cancer. 2000;83(7):906–13. doi: 10.1054/bjoc.2000.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, et al. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia. 2004;6(6):838–45. doi: 10.1593/neo.04310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langer I, Vertongen P, Perret J, Fontaine J, Atassi G, Robberecht P. Expression of vascular endothelial growth factor (VEGF) and VEGF receptors in human neuroblastomas. Medical and pediatric oncology. 2000;34(6):386–93. doi: 10.1002/(sici)1096-911x(200006)34:6<386::aid-mpo2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Luscher B, Vervoorts J. Regulation of gene transcription by the oncoprotein MYC. Gene. 2012;494(2):145–60. doi: 10.1016/j.gene.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Chen X, Liu B, Zhou L. Effects of stable knockdown of Aurora kinase A on proliferation, migration, chromosomal instability, and expression of focal adhesion kinase and matrix metalloproteinase-2 in HEp-2 cells. Molecular and cellular biochemistry. 2011;357(1-2):95–106. doi: 10.1007/s11010-011-0879-1. [DOI] [PubMed] [Google Scholar]
- 21.Lee S, Qiao J, Paul P, O'Connor KL, Evers MB, Chung DH. FAK is a critical regulator of neuroblastoma liver metastasis. Oncotarget. 2012;3(12):1576–87. doi: 10.18632/oncotarget.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu C, Qi X, Chen Y, Sun B, Dai Y, Gu Y. PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in IGF-1-induced VEGF-C upregulation in breast cancer. Journal of cancer research and clinical oncology. 2011;137(11):1587–94. doi: 10.1007/s00432-011-1049-2. [DOI] [PubMed] [Google Scholar]
- 23.Pore N, Liu S, Shu HK, Li B, Haas-Kogan D, Stokoe D, et al. Sp1 is involved in Akt-mediated induction of VEGF expression through an HIF-1-independent mechanism. Molecular biology of the cell. 2004;15(11):4841–53. doi: 10.1091/mbc.E04-05-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29(9):1249–59. doi: 10.1038/onc.2009.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors--rising stars in cancer therapeutics? Molecular cancer therapeutics. 2010;9(2):268–78. doi: 10.1158/1535-7163.MCT-09-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]