

Abstract
Herein, we report chemistry that enables excitation energy transfer (EET) to be accurately measured via action spectroscopy on gaseous ions in an ion trap. It is demonstrated that EET between tryptophan or tyrosine and a disulfide bond leads to excited state, homolytic fragmentation of the disulfide bond. This phenomenon exhibits a tight distance dependence, which is consistent with Dexter exchange transfer. The extent of fragmentation of the disulfide bond can be used to determine the distance between the chromophore and disulfide bond. The chemistry is well suited for the examination of protein structure in the gas phase because native amino acids can serve as the donor/acceptor moieties. Furthermore, both tyrosine and tryptophan exhibit unique action spectra, meaning that the identity of the donating chromophore can be easily determined in addition to the distance between donor/acceptor. Application of the method to the Trpcage miniprotein reveals distance constraints that are consistent with a native-like fold for the +2 charge state in the gas phase. This structure is stabilized by several salt bridges, which have also been observed to be important previously in proteins that retain native-like structures in the gas phase. The ability of this method to measure specific distance constraints, potentially at numerous positions if combined with site-directed mutagenesis, significantly enhances our ability to examine protein structure in the gas phase.
Introduction
Meaningful examination of protein structure in the gas phase is complicated by several fundamental challenges. Perhaps most obviously, the gas phase is a foreign environment where proteins are not typically found. Indeed, intact transfer of proteins into the gas phase proved to be a challenge for some time, and the eventual solutions to this problem were recognized with Nobel prizes.1,2 Subsequent improvements in speed and sensitivity have now made mass spectrometry (MS) the method of choice for protein identification and sequencing.3 For these same reasons, considerable effort is currently aimed at developing methods that can extract information about protein structure in the gas phase.4 However, our understanding of how proteins are transferred into the gas phase and how this process impacts structure is both imperfect and rapidly evolving. Important factors to be considered include the instrument/source conditions and relevant time scales over which protein structure can be preserved. Although it is clear that significant features of solution phase protein structure can be retained in the gas phase in some cases,5,6 there are also examples where solution phase structure appears to be lost.7,8
Ion mobility is the most commonly implemented experimental technique for examining protein structure in the gas phase.9 Although ion mobility provides direct measurement of the overall collision cross-section of a protein, no specific information about substructural elements is revealed. It can be difficult to confidently discriminate numerous structural possibilities with this type of global structural information. Recent experiments relying on radical based chemistry have helped to provide more specific information about the distances between particular residues. For example, radicals can easily be generated at specific sites with atomic precision in proteins. Subsequently, radical directed dissociation (which typically takes place at different sites than where the initial radical was created) can easily be monitored by mass spectrometry. In this fashion, distance constraints between the initial and final radical positions can be estimated.5,7 Similarly, MS/MS experiments and ion/molecule reactions can be used to probe the probability of recombination of two radicals created at two separate, specific sites.10 Although these experiments provide detailed substructural information, the difficulty with these methods is that radical migration within a protein is not easily modeled or a function of any simple chemical or physical property.
An alternative way of probing specific distances (and thus structural features) in proteins relies on excitation energy transfer (EET).11 EET encompasses a number of mechanisms including Dexter energy transfer and more popularly, Förster resonance energy transfer (FRET). FRET is notable for the transfer of energy at distances that can exceed 40 Å, making it a valuable tool for examining protein conformation, enzymatic activity, metabolic pathways, and many other aspects of biology where distance probes are useful.12−15 Dexter energy transfer or Dexter exchange transfer is also a distance-sensitive form of EET that requires overlapping orbitals between chromophores and results from the synchronous exchange of two electrons.16 Because of the requirement that molecular orbitals must overlap, Dexter energy transfer occurs at much shorter distances than those that are typically associated with FRET. Additionally, the distance dependence for Dexter energy transfer is exponential and is rarely observed to occur at distances over 15 Å.11
The implementation of traditional EET based methods in gas phase experiments is difficult. Both the number of ions that can be interrogated in gas phase experiments and optical access to them make direct observation of emission difficult to achieve. Nevertheless, a few groups have succeeded with the traditional approach.17−26 Similar challenges exist for gas phase spectroscopy experiments, which led to the development of action spectroscopy.27 In these experiments fragmentation, which is easily and sensitively measured by a mass spectrometer, is monitored rather than absorption or emission. In theory, a similar scheme should work for EET, where photon absorption would occur at one chromophore, followed by energy transfer and fragmentation at the other. There is a subtle (but very important) difference between action EET and action spectroscopy, namely, for action EET to work, energy transfer must lead to excitation of a dissociative excited state28,29 in the acceptor.30 The reason for this requirement is simple. If dissociation occurred following internal conversion of the energy into vibrational excitation, then intramolecular vibrational energy redistribution would lead to identical products whether energy conversion occurred in the donor or the acceptor. Therefore, no characteristic peaks would be generated that would uniquely indicate energy transfer. On the other hand, if excitation of the donor leads to specific fragmentation of the acceptor prior to internal conversion, then energy transfer is straightforwardly identified.
In solution, previous studies have found that disulfide reduction by UV light is enhanced by the presence of tryptophan.31 Additional studies have found that disulfides participate in photochemical reactions in conjunction with tryptophan.32 Accordingly, there is some evidence that disulfides may participate with tryptophan in EET. However, to the best of our knowledge, no such investigation has been performed on the gas phase chemistry of this system.
In this work, we reveal an EET pair suitable for action spectroscopy. It is demonstrated that excitation of tryptophan/tyrosine leads to direct dissociation of proximal disulfide bonds. Spectra for the tryptophan, disulfide, tryptophan/disulfide, and tyrosine/disulfide pairs are reported in the range of 250–300 nm and confirm that energy transfer occurs, followed by excited state fragmentation. The distance dependence of this process leads to loss of energy transfer by ∼15 Å, which is consistent with Dexter exchange transfer. Action spectra were acquired and fit to reveal distance constraints for the gas phase structure of the Trpcage in the +2 charge state. In combination with molecular dynamics calculations, it is demonstrated that the data is consistent with retention of the native structure for the +2 charge state.
Experimental Section
Peptides
The peptides Ac-RRWWCR-NH3, CGYGPKKKRKVGG, PHCKRM, VTCG, RGDC, EAGDDIVPCSMSYTWTGK, CQDSETRTFY, and VCYDKSFPISHVR were purchased from American Peptide Company. A series of nine polyalanine helical peptides with sequences Ac-WAAAAAAAACK, Ac-AWAAAAAAACK, Ac-AAWAAAAAACK, Ac-AAAWAAAAACK, Ac-AAAAWAAAACK, Ac-AAAAAWAAACK, Ac-AAAAAAWAACK, Ac-AAAAAAAWACK, and Ac-AAAAAAAAWCK were synthesized. Additionally, the peptides, Ac-AAAAAAAAACK, VYCG, WWCG, and VWCG were also synthesized. All peptides were synthesized according to standard solid phase peptide synthesis procedures.33 The Trpcage variants C-Trpcage, Trpcage-C, and Trpcage Y3F were also synthesized by solid phase peptide synthesis.
Disulfide Formation
A 5 μL aliquot of 3 mM peptide solution was combined with 1 μL of propylmercaptan and 10 μL of DMSO. The mixture was heated in a water bath at 37 °C for 12 h and then lyophilized to remove DMSO. Samples were resuspended in 200 μL 50:50:1 water:ACN:TFA. Disulfides between peptides were formed in the same manner using 5 μL of a 3 mM solution of each peptide.
Synthesis of Indole-3-Methanethiol
Gramine, methyl iodide, potassium thioacetate, and potassium carbonate were purchased from Sigma-Aldrich. A total of 1.2 mmol of gramine was reacted with an excess of methyl iodide (5 mmol) in 5 mL of 1:5 methanol:acetonitrile and stirred for 3 h. The precipitate was collected by vacuum filtration and rinsed with acetone. The resulting solid was then reacted with an excess of potassium thioacetate (3.6 mmol) in a 5 mL 2:2:1 mixture of tetrahydrofuran:acetonitrile:water. The mixture was stirred vigorously for 4 h. A total of 2 mL of water and chloroform were added and the organic layer was extracted and washed with water. The resulting organic layer was dried by nitrogen. The product was redissolved in methanol (4 mL) and an excess of potassium carbonate was added and the mixture was stirred vigorously for 2 h. The final product identity was confirmed by MS/MS. Disulfide modifications of indole-3-methanethiol and 1H-indole-3-thiol (purchased from Sigma) were performed in the same fashion as peptide modifications.
Molecular Modeling
All calculations and molecular modeling were performed in Maestro (Schrödinger Inc., Portland, Oregon) using the OPLS atomic force fields. To estimate disulfide-to-tryptophan distances in peptides, the cysteine and tryptophan side chains were rotated in sterically allowed conformations to find the maximum and minimum distances between the disulfide S atoms and tryptophan C4 and C6. MD runs were performed with no solvent at 270 K for 10 ns with 100 ps equilibration time and 1.5 ps time-step intervals.
Simulated annealing calculations were performed on the Trpcage variants up to temperatures of 700 or 1000 K. Each simulation included 50 cycles, each cycle beginning at 300 K, incrementally raising to 400, 500, and 700 K or 1000 K before lowering to 300 K and then 200 K.
Mass Spectrometry Disulfide Photodissociation Experiments
Peptide solutions were sprayed through an ESI source into an LTQ modified with a 266 nm Nd:YAG laser (Continuum, Santa Clara, CA) interfaced with the ion trap. Seven UV-photodissociation experiments are performed by isolating the parent ion and exposing it to a single laser pulse in the ion trap prior to scanning out. The yield of disulfide cleavage is calculated by
For the vast majority of peptides examined, only a single product ion is observed in significant yield.
A second setup utilized an Nd:YAG optical parametric oscillator (OPO) laser (Opotek Inc., Carlsbad, CA) interfaced with an LTQ. Ions are exposed to a single laser pulse per scan. This setup was used to obtain action spectra of ions in the range of 250–300 nm. Laser power levels for each wavelength are recorded for normalization.
Action spectra are compiled by calculating the PD yield for each wavelength and normalizing them according to differences in the laser power. It should be noted that the laser power of the OPO is generally one-fourth of that of the Nd:YAG at 266 nm laser, and comparing PD yields obtained between the two instruments reflects this.
Ion Mobility–Mass Spectrometry Experiments
Peptides were introduced by nanoelectrospray ionization (nESI) on a Waters Synapt G2 HDMS in 50:50 H2O:ACN with 0.1% TFA. Tetraalkyl ammonium salts served as ion mobility calibrants to convert drift times to nitrogen collision cross section (CCS) values.34−36 The N2 CCS values for the theoretically generated conformations were calculated using a modified version of the trajectory method.34
Results and Discussion
Photochemistry
It has been demonstrated previously that disulfide bonds can be homolytically cleaved very specifically in the gas phase by UV light.29,37 For example, if two peptides connected by a disulfide bond are irradiated by 266 nm light, only the disulfide bond will be broken and two radical peptides will be produced. The efficiency of this process can vary substantially as shown in Table 1, where several examples of PD yields for disulfide bond cleavage from precursor selected peptide pairs are given. The PD yields range from less than 1% to up to 37%. There are several potential factors that may influence PD yields in these experiments. For example, the peptide structure or charge state could potentially influence photon absorption. Alternatively, the presence of noncovalent interactions between the two peptides might prevent observation of the individual peptide radicals even if the disulfide bond were cleaved. In addition, it is also possible that native chromophores could enhance dissociation for some peptide pairs via EET.
Table 1. PD Yields for Selected Peptide Pairs.
| peptides | PD yield | charge state |
|---|---|---|
| Ac-RRWWCR-NH3 + EAGDDIVPCSMSYTWTGA | 37% | 4+ |
| CGYGPKKKRKVGG + VCYDKSFPISHVR | 24% | 6+ |
| VTCG + Ac-RRWWCR-NH3 | 13% | 2+ |
| CGYGPKKKRKVGG + CQDSETRTFY | 13% | 5+ |
| CGYGPKKKRKVGG + CQDSETRTFY | <1% | 4+ |
| CGYGPKKKRKVGG + CQDSETRTFY | <1% | 3+ |
| CGYGPKKKRKVGG + CQDSETRTFY | <1% | 2+ |
| RGDC + VTCG | <1% | 1+ |
| RGDC + PHCKRM | <1% | 1+ |
Close examination of the results in Table 1 suggests that some of these factors are likely influencing the observed PD yields. For example, sequences containing chromophoric residues (Trp, Tyr, and Phe) appear to undergo disulfide cleavage more efficiently than peptides that do not. This suggests that EET may occur from native chromophores to the disulfide bond. Another notable trend is that higher charge states tend to have the best PD yields for peptide pairs that are observed in multiple charge states. This is evident in CGYGPKKKRKVGG + CQDSETRTFY where only the highest observable charge state has a PD yield greater than 1%, as shown in Table 1. Although the charge state per se may not be important, it is likely for peptide pairs that charge repulsion overcomes any noncovalent interactions that otherwise inhibit dissociation. In the absence of Coulombic repulsion, two peptide radicals generated by disulfide bond cleavage can simply recombine38 to regenerate the disulfide bond if the fragments are temporarily held together by noncovalent interactions.
In order to potentially quantify the contribution of EET to disulfide PD yield, it is desirable to avoid complications arising from such noncovalent interactions. This can be accomplished in a straightforward manner by replacing one of the peptides with a small organic molecule that is not likely to form any significant noncovalent bonds. Propyl mercaptan (PM) is a small saturated hydrocarbon that cannot form hydrogen bonds or participate in any other strong intermolecular interactions. PM also readily forms disulfide bonds with peptides containing free cysteine residues. Accordingly, cleaving disulfide bonds formed with PM should give a more accurate reflection of the underlying photochemistry.
Table 2 shows the PD yields for various peptides connected to PM via a disulfide bond. The PD yields are very high for peptides which contain chromophores, as was also the case for some peptides in Table 1. Additionally, the PD yields for peptides modified with PM are higher than the PD yields obtained from related peptide pairs. For example, Ac-RRWWCR-NH3 has a yield of 53% when modified with PM, whereas the yields obtained when attached to other peptides are only 37% and 13% as seen in Table 1. Even peptides that contain no chromophores (such as RGDC or VTCG) give PD yields of 6% and 7%, respectively, whereas these peptides only have a PD yield of less than 1% if bound to each other by a disulfide bond. All of these results are consistent with the idea that peptide pairs can form strong interfering noncovalent interactions and that PM is not sticky and, thus, gives more accurate PD yields.
Table 2. PD yields for peptides tagged with PM.
| peptide bound to propyl mercaptan | PD yield | charge state |
|---|---|---|
| VWCG + PM | 58% | 1+ |
| Ac-RRWWCR-NH3 + PM | 53% | 1+ |
| CGYGPKKKRKVGG + PM | 24% | 3+ |
| CGYGPKKKRKVGG + PM | 20% | 2+ |
| CGYGPKKKRKVGG + PM | 16% | 1+ |
| PHCKRM + PM | 7% | 2+ |
| PHCKRM + PM | 9% | 1+ |
| VYCG + PM | 17% | 1+ |
| VTCG + PM | 7% | 1+ |
| RGDC + PM | 6% | 1+ |
| AcA5WA3CK + PM | 72% | 1+ |
| AcA9CK + PM | 8% | 1+ |
The results in Table 2 reveal a strong correlation between the presence of aromatic residues and high disulfide PD yield. In order to investigate whether disulfide dissociation is truly due to EET, we recorded the action spectra for tryptophan, a PM-modified cysteine-containing peptide (AcA9CK-PM), and a peptide which contains both cysteine and tryptophan (AcA5WA3CK-PM). The results are shown in Figure 1. The AcA9CK-PM spectrum is characterized by weak absorption and featureless decay in the region from 250 to 300 nm. The tryptophan spectrum shows broad absorption with a single peak at 287 nm. This spectrum is consistent with previously reported spectra for tryptophan in the gas phase.39 The peptide that contains both moieties yields a spectrum that closely resembles the tryptophan spectrum, although it is red-shifted by ∼10 nm. This spectrum was obtained solely by monitoring homolytic fragmentation of the disulfide bond, which is by far the most dominant fragment observed at all wavelengths. The resemblance of the AcA5WA3CK-PM spectrum to the spectrum for tryptophan itself and the observed red-shift are both consistent with fragmentation that occurs following EET.
Figure 1.
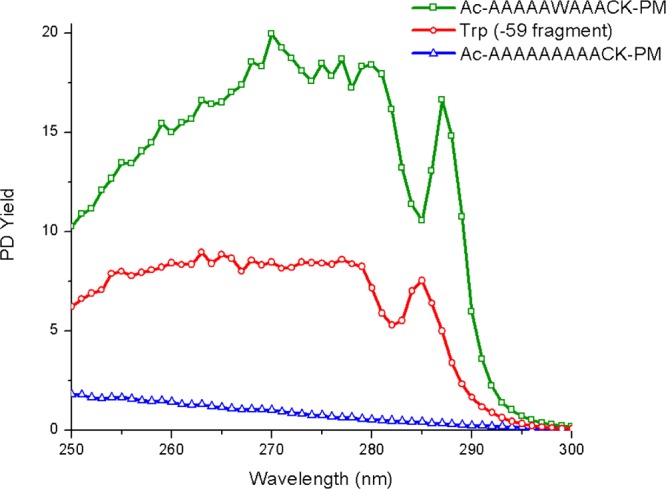
Action spectra for AcA5WA3CK-PM, tryptophan, and AcA9CK-PM. Note: PD yields here reflect the lower power of the OPO laser setup.
The results in Figure 1 unambiguously establish that PD of the disulfide bond is occurring after excitation of the tryptophan chromophore. In order to determine whether this fragmentation occurs due to EET to a dissociative excited state, we compared the PD spectrum for AcA5WA3CK-PM to that obtained by typical collision induced dissociation (CID) and the results are shown in Figure 2. The PD spectrum confirms that homolytic fragmentation of the disulfide bond yields by far the most abundant product ion, which is consistent with direct dissociation in the excited state. In contrast, more typical results consisting of a variety of b and y ions are observed by CID. Importantly, CID in an ion trap strongly favors fragmentation via the lowest energy dissociation pathways. It is clear in Figure 2b that homolytic cleavage of the disulfide bond does not occur via a low energy dissociation pathway and is not favored following vibrational excitation. Taken together, the results in Figures 1 and 2 indicate that the dominant cleavage of the disulfide bond following photoactivation most likely occurs due to excitation of a dissociative excited state. Importantly, this type of chemistry should also exhibit a distance dependence related to the underlying mode of EET.
Figure 2.
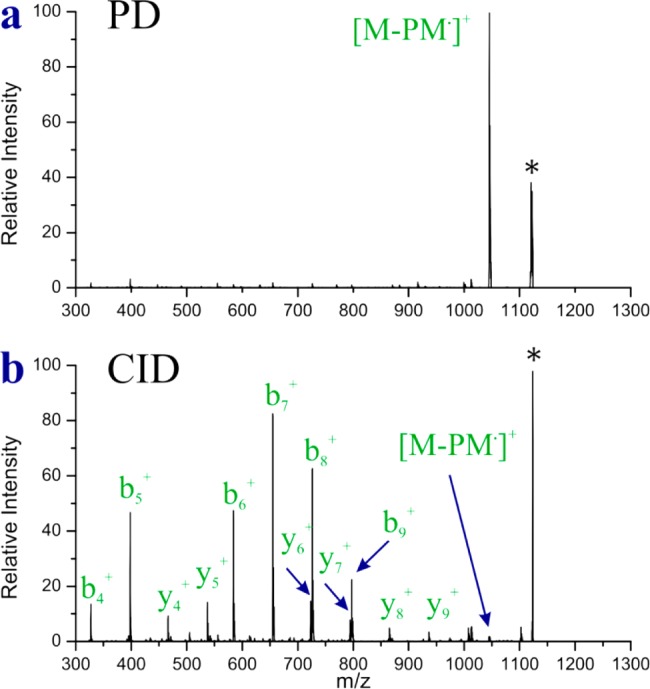
(a) PD and (b) CID spectra for AcA5WA3CK-PM. * precursor.
In order to evaluate the distance dependence for Trp/PM EET, we sought to measure the disulfide PD yield for peptides where the distances between donor and acceptor could be reasonably estimated. Previous work has indicated that peptides based on an Ac-AxK (where x ≥ 7) motif tend to form alpha helices in the gas phase.40,41 Building on this general motif, we synthesized Ac-WA8CK, which has two of the alanine residues at opposite ends of the helix replaced with PM modified cysteine and tryptophan. The collision cross section for this peptide as determined by ion mobility is consistent with an α helical structure (the predicted cross section from molecular modeling matches the experimental data within ∼3%, Supporting Information Table S1). The estimated distance between the donor and acceptor for this peptide is ∼14.5 ± 2.5 Å. The large uncertainty in the distance comes from the flexibility of the side chains, which are assumed to retain significant rotational freedom. The PD yield for Ac-WA8CK is fairly low at 24%. Nevertheless, the PD yield is still appreciably greater than the 8% obtained for Ac-A9CK, which contains no tryptophan. The results from Ac-A9CK serve as the baseline for inherent non-EET assisted disulfide bond fragmentation in a polyalanine based peptide. It is clear that even at a distance of ∼14.5 Å, some EET is occurring.
We also attempted to use the polyalanine scaffold to determine the close contact EET efficiency; however, as the distance between donor and acceptor is decreased by altering donor/acceptor positions in the polyalanine chain, the rotational freedom of the side chains becomes increasingly problematic. Furthermore, it is not clear that peptides where tryptophan replaces a central alanine are able to retain entirely helical structures (i.e., cross sections from ion mobility exceeded 3% difference relative to predicted values; see Supporting Information Table S1). Therefore, we adopted an alternative approach where we simply attached the donor and acceptor to each other via short covalent bonds. Direct attachment of the disulfide bond to the indole chromophore is possible and represents the closest distance that the two moieties can be brought together. By attachment of an amine group on the other side of the disulfide (see structure in Figure 3a), a suitable ion can be generated for examination by ESI.
Figure 3.
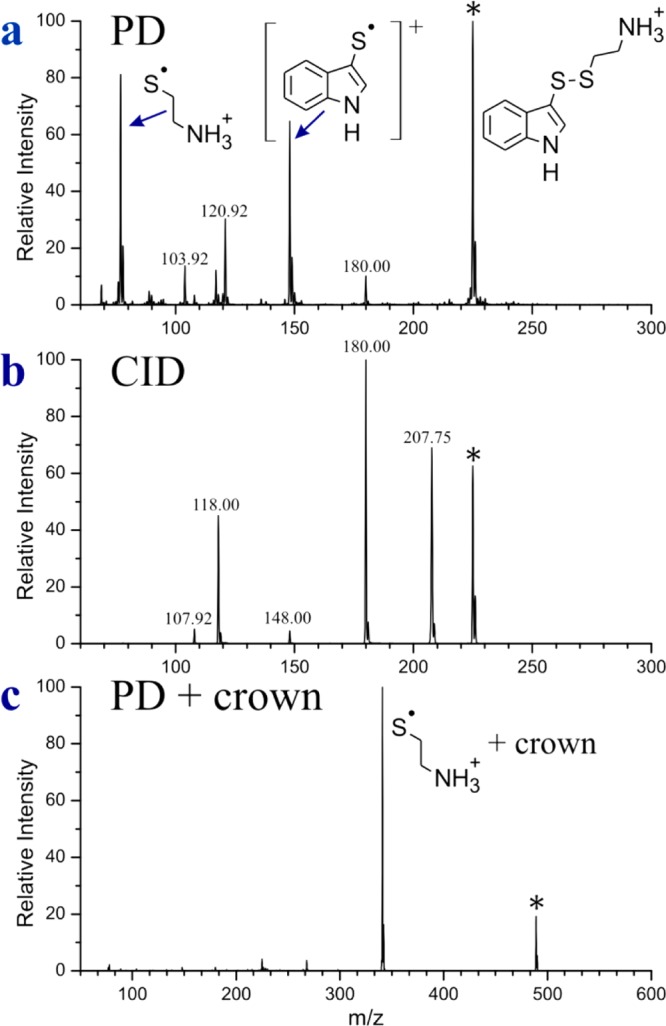
(a) PD of the molecule shown to the far right. Structures corresponding to the products of interest are also shown. (b) CID of the same molecule. (c) PD of the same molecule in complex with 18-crown-6.
The PD spectrum for the protonated molecule is shown in Figure 3a and it contains several interesting features. The expected product at 77 Da results from homolytic cleavage of the disulfide bond. The intensity of this peak is lower than might be anticipated, given the unavoidable proximity of the donor and acceptor. In addition, several unexpected fragments are observed. For example, there is a peak at 148 Da that corresponds to the radical cation of the indole chromophore. This peak is most likely generated by dissociative electron transfer from the indole ring to the disulfide bond. Indeed, the action spectra for homolytic cleavage of the disulfide bond and electron transfer to the indole are distinct (see Supporting Information Figure S1), suggesting formation via distinct chemical processes. No similar signs of electron transfer chemistry were detected in any experiments with peptides. The remaining smaller peaks likely originate from internal conversion of the photon energy to vibrational excitation as the peaks at 108, 118, and 180 Da are also observed by CID (Figure 3b). Other peaks in Figure 3a, such as 104 and 121 Da, appear to be the products of rearrangements following a combination of electronic and vibrational excitation.
The extremely close proximity of the protonated amine appears to significantly influence the fragmentation behavior of this small molecule. In order to explore this possibility further, we solvated the ammonium group by noncovalent complexation with 18-crown-6 ether (18C6), which forms three strong hydrogen bonds with protonated primary amines.42 The PD spectrum for the 18C6 complex is shown in Figure 3c. The electron transfer product is no longer observed to any significant extent, and most other dissociation channels are suppressed as well. Homolytic cleavage of the disulfide bond is enhanced significantly and the resulting PD yield is quite high (∼93%). The distance between chromophore centers for the donor and acceptor in this system is ∼4 Å, and the PD yield is likely the maximum value that can be obtained for the indole/disulfide donor/acceptor pair. Insertion of an additional CH2 group in between the chromophores results in reduced PD yield (88%), consistent with this idea.
Given the distance constraints that we have established up to this point, it is possible to narrow down the mode by which EET is likely occurring for this system. The observation that the PD yield decays significantly within 15 Å suggests that FRET is not responsible for most of the energy transfer. Instead, we propose that the indole/disulfide EET occurs primarily via Dexter exchange transfer (DET), which has an exponential distance dependence and typically occurs within 15 Å (Supporting Information Figure S2).
In proteins, Trp is not the only chromophore that may contribute to the cleavage of disulfide bonds via EET. Tyrosine also has significant absorption in the near UV, and would be expected to facilitate EET mediated disulfide bond cleavage. To explore this possibility further, we replaced tryptophan with tyrosine to create the peptide Ac-A5YA3CK-PM, which can be compared with the Trp analog from Figure 2. The results are shown in Figure 4. The comparison reveals that tyrosine yields a distinct spectrum, which is similar to action spectra acquired for tyrosine itself. The distinctive shoulder occurs at a unique wavelength for both Trp and Tyr, which should allow for contributions from the two chromophores to be distinguished. Furthermore, the Tyr spectrum loses intensity at shorter wavelengths more rapidly than Trp. The relative intensity of the Tyr spectrum is also significantly smaller than that for Trp, suggesting that Tyr will only make significant contributions to PD yield at very short distances.
Figure 4.
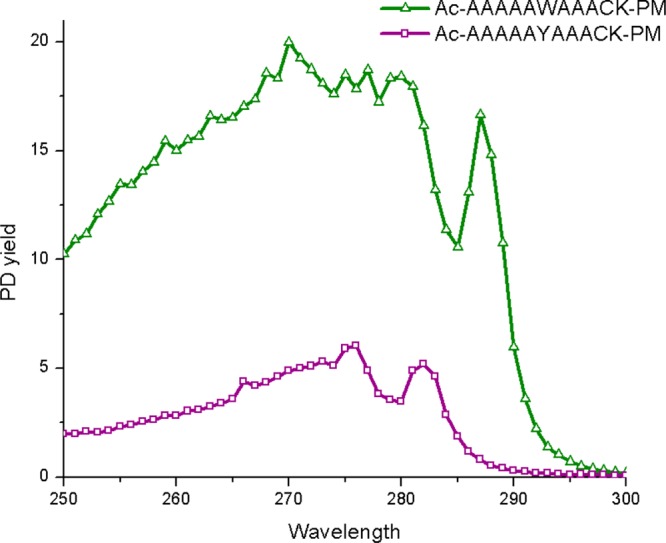
Action spectra of Ac-A5WA3CK-PM as compared with Ac-A5YA3CK-PM.
Trpcage
We now apply these tools to the examination of protein structure. The Trpcage is a very small and fast folding protein that was designed computationally.43 It is comprised of only 20 amino acids with the sequence NLYIQWLKDG GPSSGRPPPS. It contains one Tyr and one Trp. The native fold traps the Trp side chain in the center of a hydrophobic pocket created by a small α helix and a run of proline residues; this pocket is the defining feature of the structure and the source of the molecule’s name. A + – + salt bridge can form between the Lys, Asp, and Arg side chains. This salt bridge has been demonstrated to be important for the stability of the protein in solution.44,45 An additional salt bridge can form between the N- and C-termini. The stability and importance of these salt bridges in the gas phase has been questioned.46 Furthermore, previous work has demonstrated that Trpcage can adopt non-native structures in the gas phase.18 This is not surprising because the gas phase is a very different environment than the aqueous solution for which the protein was engineered. Nevertheless, other studies have suggested that native-like Trpcage structures can be stable in the gas phase under appropriate conditions.47,48
There are no cysteine residues in the standard Trpcage, therefore, we added a cysteine to both the N-terminus and C-terminus (called C-Trpcage and Trpcage-C, respectively). NMR experiments reveal that the N and C-terminal residues are fairly disordered, which suggests these additions are not in sterically confined locations and are not anticipated to significantly impact the structure or folding of the protein in solution.
Examination of the action spectra for each charge state, which are shown in Figure 5, reveals several important structural features. C-Trpcage-PM 2+ exhibits the largest PD yield and the shape of the spectrum is noticeably different from the spectra for other charge states. Importantly, the intensity for this spectrum decreases more rapidly at shorter wavelengths, which is characteristic of EET from tyrosine and indicates very close Tyr/disulfide proximity. At the same time, the shoulder at ∼287 nm is indicative of EET from Trp, suggesting that both chromophores contribute to the spectrum. In order to explore this idea further, a combination spectrum created by additively combining the Tyr and Trp spectra is shown in Figure 5b. The tyrosine spectrum from Ac-A5YA3CK-PM was combined in a 1:1 ratio with the spectrum from Ac-WA8CK-PM. This would correspond to a situation where the tyrosine residue is close at <6 Å, and the tryptophan residue is significantly farther away (>10 Å). As can be seen in Figure 5b, the combination spectrum is a reasonably good match to the experimental C-Trpcage-PM 2+ spectrum. The primary differences are due to less distinct absorption at ∼282 and ∼287 nm, which may be caused by internal solvation of the chromophores. Solvation of tyrosine has previously been demonstrated to yield less distinct spectral features.49 Alternate ratios of 2:1 or 1:2 mixtures of the same spectra yielded less satisfactory fits (see Supporting Information Figure S3).
Figure 5.
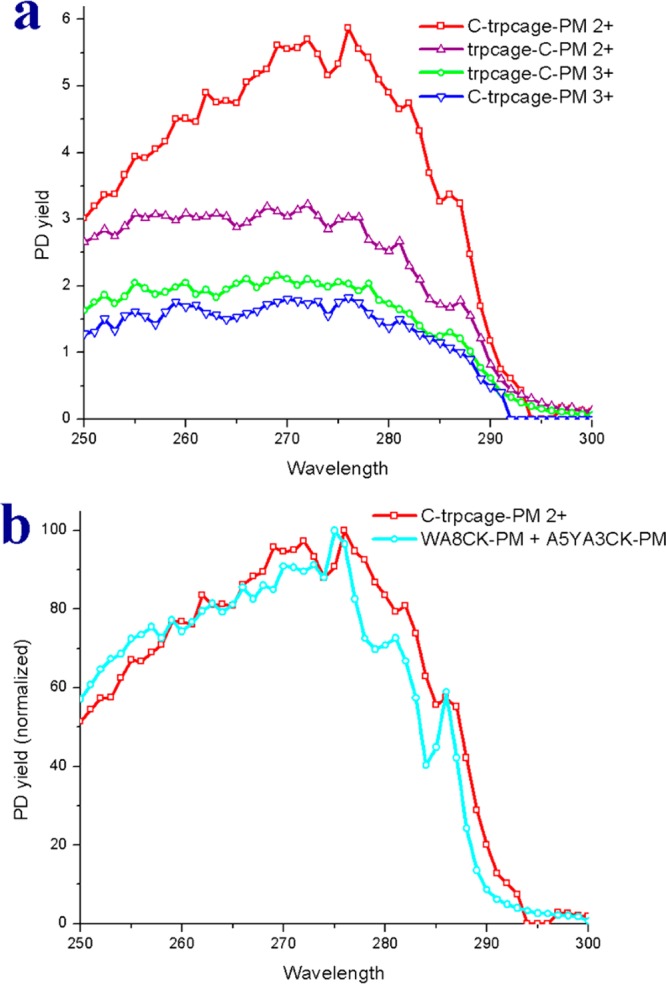
(a) Action spectra for the C-Trpcage 2+, Trpcage-C 2+ and Trpcage-C 3+. (b) Comparison of the action spectrum of the C-Trpcage (2+) and the combination spectrum from Ac-A5YA3CK-PM and Ac-WA8CK-PM.
The remaining spectra in Figure 5a are significantly flatter and consistent with minimal tyrosine character. To test this hypothesis, a variant of Trpcage-C was synthesized with phenylalanine substituted for tyrosine. Although phenylalanine is also a potential chromophore, the observed absorption in the 250–300 nm range is so poor that we do not detect any appreciable dissociation of phenylalanine containing peptides. Accordingly, spectra for the Y3F mutant of the Trpcage should reveal exclusively EET due to tryptophan. The results are shown in Supporting Information Figure S4 overlaid with the spectra collected from the tyrosine-containing version. Differences in the spectra are minimal which supports the idea that tyrosine is too far from the C-terminal cysteine to contribute substantially to EET.
The C-Trpcage-PM 2+ is the best candidate for more in depth structural characterization because distance constraint information is provided for both chromophores. Simulated annealing was carried out on the C-Trpcage-PM 2+ starting from the native structure. Two separate simulated annealing approaches were used to generate low energy gas-phase structures from the NMR structure: one reaching temperatures of 1000 K and another reaching temperatures of 700 K. High temperature simulated annealing is preferable to find the global energy minimum because significant activation may be required to overcome the energy barriers needed to reach such a structure. Accordingly, the global energy minimum structure generated from simulated annealing at 1000 K may not be the structure that is actually observed. In fact, the structure that is generated (Supporting Information Figure S5) bears no resemblance to the solution phase structure and is also distinct from those noted in previous studies of the Trpcage.47,48 In contrast, the structure generated from a more modest 700 K annealing resembles the solution-phase structure while being significantly lower in energy than structures generated from other conformational searches or simple energy minimization.
The lowest energy structure obtained from the 700 K simulated annealing is shown in Figure 6 in comparison with the native structure. Most of the structure is unperturbed, with a few interesting exceptions. Notably, the N-terminus has a different orientation that allows for extensive hydrogen bonding between the N- and C- termini. The distances between the chromophores and the disulfide bond in the structure in Figure 6 are completely consistent with the experimental data in Figure 5. It should be noted that one charge is located at the N-terminus, which remains hydrogen bonded to the C-terminus, stabilizing the native-like fold. The second charge is located at arginine, which is in a + – + salt bridge with Asp and Lys. Previous results have suggested that Trpcage does not retain salt bridges in the gas phase, and that the +2 charge state may not have a native-like structure.46 There are several possible explanations for these apparent contradictions. The conditions of our source may be gentler, facilitating the retention of the native like structure. It is also possible that the salt bridge may convert to simple hydrogen bond interactions via proton transfer without significantly influencing the remainder of the structure, though previous results with other proteins have demonstrated salt bridges are frequently critical for the retention of native structure in the gas phase.50,51 It is also possible that the additional cysteine residue may stabilize the native structure by enabling extra hydrogen bonding between the termini. Comparison of our data with previously proposed structures that differ substantially from the native fold does not yield good agreement (i.e., the Tyr and Trp distances are not consistent with our results). Taking all of the evidence into consideration, we conclude that it is possible to preserve a native like state for the Trpcage in the +2 charge state under appropriately gentle source conditions.
Figure 6.
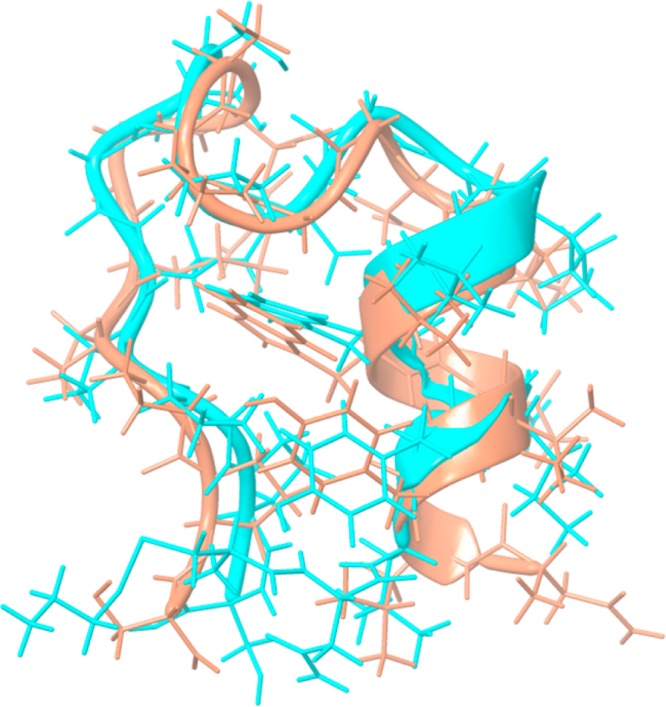
Comparison of the C-Trpcage-PM simulated annealing structure (blue) superimposed with the Trpcage NMR structure (orange).
Detailed evaluation of the remaining structures is more difficult because the data only reveal a fairly long-range interaction with Trp and absence of a close range interaction with Tyr. The data for Trpcage-C-PM 2+ are consistent with the native fold structure, although many other structures would likely be suitable as well. Activation of the C-Trpcage 2+ structure by harsher source conditions or supplemental collisional energy result in significantly lowered PD yields, whereas native MS solvent conditions (1 mM NH4OAc) do not significantly change the yield (Supporting Information Table S2). These observations for C-Trpcage 2+ are consistent with the expected behavior that would accompany a transition from the native-like structure to the lowest energy gas phase structure. For the +3 charge states, the results indicate a more unfolded structure, which results in a slightly increased Trp/disulfide interaction distance. This is consistent with previous observations by ion mobility illustrating Coulomb induced unfolding of proteins in higher charge states.52
Conclusions
We have developed an action EET system based on Trp/Tyr and disulfide bonds suitable for experiments on ions in the gas phase. Excitation of Trp/Tyr by UV light leads to EET and excited state dissociation of disulfide S–S bonds in a distance dependent fashion. Trp can facilitate homolytic fragmentation of disulfide bonds out to ∼15 Å, whereas Tyr is only active at very close proximity (<6 Å). These distance regimes suggest that the majority of the EET for these chromophores occurs via Dexter exchange transfer and that FRET is likely a minor contributor. The combination of distance information with initiating chromophore identity provides a powerful new platform for investigating protein structure in the gas phase. Furthermore, the ability to use native chromophores (i.e., Trp and Tyr) in combination with disulfide bonds generated from cysteine residues provides facile access to the powerful suite of molecular biology tools that have been developed to manipulate protein sequence. If a native sequence does not contain probes in appropriate locations, standard site-directed mutagenesis can be used to incorporate the desired residues. This presents a considerable advantage over existing methods of applying EET which require more disruptive addition of larger synthetic chromophores. This method should lead to enhancement of our understanding of protein structure in the gas phase.
Acknowledgments
The authors thank Richard Hooley, Mike Pirrung, and Chris Switzer for helpful advice on synthetic procedures. We thank the NSF (CHE-0747481) and NIH (GM103531) for financial support and Greg Beran, Arun Agarwal for stimulating conversation. Financial support for J.A.M., N.M.L., and S.M.S. was provided by the National Institutes of Health (UH2TR000491) and the Vanderbilt Chemical Biology Interface training grant (T32GM065086, N.M.L.), the Vanderbilt University College of Arts and Science, the Vanderbilt Institute of Chemical Biology, and the Vanderbilt Institute for Integrative Biosystems Research and Education.
Supporting Information Available
Additional spectra, ion mobility results, and structures for the Trpcage. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yamashita M.; Fenn J. B. J. Phys. Chem. 1984, 88, 4451. [Google Scholar]
- Tanaka K.; Waki H.; Ido Y.; Akita S.; Yoshida Y.; Yoshida T.; Matsuo T. Rapid Commun. Mass Spectrom. 1988, 2, 151. [Google Scholar]
- Aebersold R.; Mann M. Nature 2003, 422, 198. [DOI] [PubMed] [Google Scholar]
- Jurneczko E.; Barran P. E. Analyst 2011, 136, 20. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Julian R. R. Int. J. Mass Spectrom. 2011, 308, 225. [Google Scholar]
- Benesch J. L. P.; Robinson C. V. Nature 2009, 462, 576. [DOI] [PubMed] [Google Scholar]
- Ly T.; Julian R. R. J. Am. Chem. Soc. 2010, 132, 8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmer D. E.; Koeniger S. L. J. Am. Soc. Mass Spectrom. 2007, 18, 322. [DOI] [PubMed] [Google Scholar]
- Bohrer B. C.; Merenbloom S. I.; Koeniger S. L.; Hilderbrand A. E.; Clemmer D. E. Annu. Rev. Anal. Chem. 2008, 1, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Julian R. R. Phys. Chem. Chem. Phys. 2012, 14, 16243. [DOI] [PubMed] [Google Scholar]
- Winkler J. R. Science 2013, 339, 1530. [DOI] [PubMed] [Google Scholar]
- Periasamy A. J. Biomed. Opt. 2001, 6, 287. [DOI] [PubMed] [Google Scholar]
- Deuschle K.; Chaudhuri B.; Okumoto S.; Lager I.; Lalonde S.; Frommer W. B. Plant Cell 2006, 18, 2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Hohng S.; Ha T. Nat. Methods 2008, 5, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantanen T.; Järvenpää M.-L.; Vuojola J.; Kuningas K.; Soukka T. Angew. Chem. 2008, 120, 3871. [DOI] [PubMed] [Google Scholar]
- Olaya-Castro A.; Scholes G. D. Int. Rev. Phys. Chem. 2011, 30, 49. [Google Scholar]
- Danell A. S.; Parks J. H. Int. J. Mass Spectrom. 2003, 229, 35. [Google Scholar]
- Iavarone A. T.; Patriksson A.; van der Spoel D.; Parks J. H. J. Am. Chem. Soc. 2007, 129, 6726. [DOI] [PubMed] [Google Scholar]
- Danell A.; Parks J. J. Am. Soc. Mass. Spectrom. 2003, 14, 1330. [DOI] [PubMed] [Google Scholar]
- Iavarone A. T.; Meinen J.; Schulze S.; Parks J. H. Int. J. Mass Spectrom. 2006, 253, 172. [Google Scholar]
- Iavarone A. T.; Parks J. H. J. Am. Chem. Soc. 2005, 127, 8606. [DOI] [PubMed] [Google Scholar]
- Dashtiev M.; Azov V.; Frankevich V.; Scharfenberg L.; Zenobi R. J. Am. Soc. Mass. Spectrom 2005, 16, 1481. [DOI] [PubMed] [Google Scholar]
- Frankevich V.; Martinez-Lozano Sinues P.; Barylyuk K.; Zenobi R. Anal. Chem. 2012, 85, 39. [DOI] [PubMed] [Google Scholar]
- Forbes M.; Jockusch R. J. Am. Soc. Mass. Spectrom. 2011, 22, 93. [DOI] [PubMed] [Google Scholar]
- Sagoo S. K.; Jockusch R. A. J. Photochem. Photobiol., A 2011, 220, 173. [Google Scholar]
- Talbot F. O.; Rullo A.; Yao H.; Jockusch R. A. J. Am. Chem. Soc. 2010, 132, 16156. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C. Int. J. Mass Spectrom. 2000, 200, 571. [Google Scholar]
- Cheng P. Y.; Zhong D.; Zewail A. H. J. Phys. Chem. Lett. 1995, 237, 399. [Google Scholar]
- Bookwalter C.; Zoller D.; Ross P. J. Am. Soc. Mass. Spectrom. 1995, 6, 872. [DOI] [PubMed] [Google Scholar]
- Daly S.; Poussigue F.; Simon A.-L.; MacAleese L.; Bertorelle F.; Chirot F.; Antoine R.; Dugourd P. Anal. Chem. 2014, 8798–8804. [DOI] [PubMed] [Google Scholar]
- Wu L.-Z.; Sheng Y.-B.; Xie J.-B.; Wang W. J. Mol. Struct. 2008, 882, 101. [Google Scholar]
- Haywood J.; Mozziconacci O.; Allegre K. M.; Kerwin B. A.; Schöneich C. Mol. Pharmaceutics 2013, 10, 1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W. C.; White P. D.. Fmoc Solid Phase Peptide Synthesis, 1st ed.; Oxford University Press: New York, 2000. [Google Scholar]
- Campuzano I.; Bush M. F.; Robinson C. V.; Beaumont C.; Richardson K.; Kim H.; Kim H. I. Anal. Chem. 2012, 84, 1026. [DOI] [PubMed] [Google Scholar]
- May J. C.; Goodwin C. R.; Lareau N. M.; Leaptrot K. L.; Morris C. B.; Kurulugama R. T.; Mordehai A.; Klein C.; Barry W.; Darland E.; Overney G.; Imatani K.; Stafford G. C.; Fjeldsted J. C.; McLean J. A. Anal. Chem. 2014, 86, 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruotolo B. T.; Benesch J. L. P.; Sandercock A. M.; Hyung S.; Robinson C. V. Nat. Protoc. 2010, 82, 9557. [DOI] [PubMed] [Google Scholar]
- Agarwal A.; Diedrich J. K.; Julian R. R. Anal. Chem. 2011, 83, 6455. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Almaraz R. T.; Xiang F.; Hedrick J. L.; Franz A. H. J. Am. Soc. Mass Spectrom. 2009, 20, 1603–16. [DOI] [PubMed] [Google Scholar]
- Nolting D.; Marian C.; Weinkauf R. Phys. Chem. Chem. Phys. 2004, 6, 2633. [DOI] [PubMed] [Google Scholar]
- Rossi M.; Blum V.; Kupser P.; von Helden G.; Bierau F.; Pagel K.; Meijer G.; Scheffler M. J. Phys. Chem. Lett. 2010, 1, 3465. [Google Scholar]
- Hudgins R. R.; Jarrold M. F. J. Am. Chem. Soc. 1999, 121, 3494. [Google Scholar]
- Julian R. R.; Beauchamp J. L. Int. J. Mass Spectrom. 2001, 210–211, 613. [Google Scholar]
- Neidigh J. W.; Fesinmeyer R. M.; Andersen N. H. Nat. Struct. Biol. 2002, 9, 425. [DOI] [PubMed] [Google Scholar]
- Zhou R. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geney R.; Layten M.; Gomperts R.; Hornak V.; Simmerling C. J. Chem. Theory Comput. 2005, 2, 115. [DOI] [PubMed] [Google Scholar]
- Patriksson A.; Adams C. M.; Kjeldsen F.; Zubarev R. A.; van der Spoel D. J. Phys. Chem. B 2007, 111, 13147. [DOI] [PubMed] [Google Scholar]
- Adams C. M.; Kjeldsen F.; Patriksson A.; van der Spoel D.; Gräslund A.; Papadopoulos E.; Zubarev R. A. Int. J. Mass Spectrom. 2006, 253, 263. [Google Scholar]
- Patriksson A.; Marklund E.; van der Spoel D. Biochemistry 2007, 46, 933. [DOI] [PubMed] [Google Scholar]
- Kirk B. B.; Trevitt A. J.; Blanksby S. J.; Tao Y.; Moore B. N.; Julian R. R. J. Phys. Chem. A 2013, 117, 1228. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Julian R. R. Int. J. Mass Spectrom. 2011, 308, 225. [Google Scholar]
- Breuker K.; Brüschweiler S.; Tollinger M. Angew. Chem. 2011, 123, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmer D. E.; Jarrold M. F. J. Mass Spectrom. 1997, 32, 577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.