

Abstract
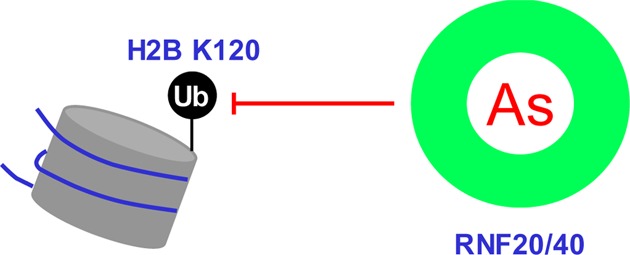
Arsenic is a widespread environmental contaminant. However, the exact molecular mechanisms underlying the carcinogenic effects of arsenic remain incompletely understood. Core histones can be ubiquitinated by RING finger E3 ubiquitin ligases, among which the RNF20-RNF40 heterodimer catalyzes the ubiquitination of histone H2B at lysine 120. This ubiquitination event is important for the formation of open and biochemically accessible chromatin fiber that is conducive for DNA repair. Herein, we found that arsenite could bind directly to the RING finger domains of RNF20 and RNF40 in vitro and in cells, and treatment with arsenite resulted in substantially impaired H2B ubiquitination in multiple cell lines. Exposure to arsenite also diminished the recruitment of BRCA1 and RAD51 to laser-induced DNA double-strand break (DSB) sites, compromised DNA DSB repair in human cells, and rendered cells sensitive toward a radiomimetic agent, neocarzinostatin. Together, the results from the present study revealed, for the first time, that arsenite may exert its carcinogenic effect by targeting cysteine residues in the RING finger domains of histone E3 ubiquitin ligase, thereby altering histone epigenetic mark and compromising DNA DSB repair. Our results also suggest arsenite as a general inhibitor for RING finger E3 ubiquitin ligases.
Owing to its natural abundance and industrial use, arsenic’s environmental impact is felt by nearly 150 million people in more than 70 countries.1 Epidemiological studies revealed that human exposure to arsenic in drinking water is significantly associated with the prevalence of skin, lung, and bladder cancers.2−4 Inhibition of DNA repair has been suggested as a major mechanism in arsenic genotoxicity.5 In this respect, humans exposed to arsenite in drinking water were found to have increased frequencies of chromosome aberrations in peripheral lymphocytes and elevated frequencies of micronuclei in exfoliated cells.5 The inhibition of DNA repair might be attributable to the formation of three-coordinate trigonal-pyramidal complexes with cellular cysteine-containing proteins,6 where arsenite was previously found to displace Zn2+ in the zinc finger motifs of some DNA repair proteins including poly(ADP-ribose) polymerase 1 (PARP1) and XPA.7−11
Aside from the carcinogenic effect of arsenic species, trivalent arsenic, in the form of arsenic trioxide, has demonstrated remarkable success in the clinical treatment of acute promyelocytic leukemia.12 In this regard, As(III) was found to bind to the RING finger domain of PML in the oncogenic PML-RARα fusion protein, and this binding ultimately results in the proteasomal degradation of the fusion protein.13 RING finger, a protein structural domain of the zinc-finger type containing a Cys3HisCys4 amino acid motif, is also present in the majority of E3 ubiquitin ligases.14 Among them, the RNF20-RNF40 heterodimer targets histone H2B lysine 120 (K120) for monoubiquitination, and this ubiquitination event is important for decompacting the 30 nm chromatin fiber15 and facilitating DNA double-strand break (DSB) repair.16,17 We reasoned that arsenite may also bind to the RING finger motifs of RNF20 and RNF40, thereby perturbing H2B ubiquitination and suppressing DNA DSB repair.
To investigate whether NaAsO2 can bind directly to the RING finger domain of RNF20 and RNF40, we performed in vitro binding assays using synthetic RING finger peptides of the two proteins. MALDI-TOF MS results revealed mass increases of 72 and 144 Da upon incubation of the peptides with arsenite (Figure 1a). The +72 Da mass shift reflects the binding of As(III) to the RING finger peptides with the release of three protons, suggesting the coordination of As(III) with three cysteines.10 The MS data, therefore, suggest that each molecule of the RING finger peptides can bind up to two As(III).
Figure 1.
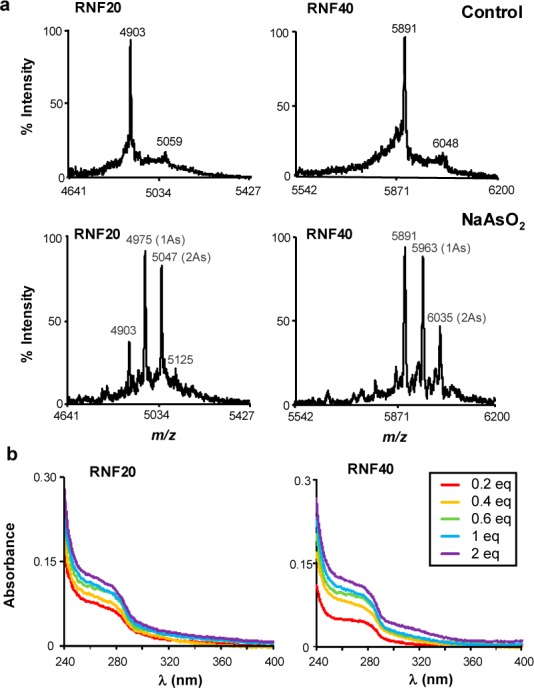
In vitro binding between NaAsO2 and RING finger peptides of RNF20 and RNF40. (a) MALDI-TOF mass spectrometry for monitoring the interaction between arsenite and the RING finger peptides of RNF20 and RNF40. The molar ratios between the RING finger peptides and arsenite were 1:2. (b) UV absorption spectra of the RING finger peptides titrated with increasing amounts of NaAsO2.
Thiol coordination to As(III) gives rise to new charge-transfer electronic transitions in the near UV range (250–320 nm) that can be monitored by UV absorbance.18 We found, from optical absorption experiments, that the two RING finger peptides displayed increased absorbance in this wavelength range upon addition with increasing amounts of NaAsO2 (Figure 1b).18 Together, these assays demonstrated that NaAsO2 can bind to sulfhydryl group of cysteine residues in the RING finger domains of RNF20 and RNF40 in vitro.
We next investigated the binding of arsenite with the RING domains of RNF20 and RNF40 in cells. To this end, we synthesized a biotin-As probe by conjugating p-aminophenylarsine oxide (PAPAO) with biotin19,20 and assessed arsenite binding by streptavidin agarose affinity assay. Our results showed that the treatment of cells with biotin-As facilitated the pull-down of ectopically expressed RNF20 and RNF40, suggesting that RNF20 and RNF40 can bind to the biotin-As probe in cells (Figures 2a and S1a). Moreover, we failed to pull down the two proteins when the cells were pretreated with NaAsO2 or PAPAO, though pretreatment with Zn2+ still led to the successful pull-down of the two proteins, demonstrating that the interactions between biotin-As and RNF20 or RNF40 are specific (Figures 2a and S1). To determine if the cysteine residues in the RING finger domains are required for such interactions, we mutated the two cysteine residues in each of the two Zn2+-binding sites of the RING finger motifs of RNF20 and RNF40 to alanines (i.e., the C922,924A and C957,960A mutants for RNF20 and the C948,950A and C983,986A mutants for RNF40) and conducted the same pull-down assays. Our results showed that the mutations abolished the pull-down of the two proteins (Figure 2a), indicating that an intact RING finger is indispensable for the interaction between biotin-As and RNF20 or RNF40. Along this line, Western analysis revealed that the mutations did not lead to decreased expression of the two proteins (Figure S1).
Figure 2.
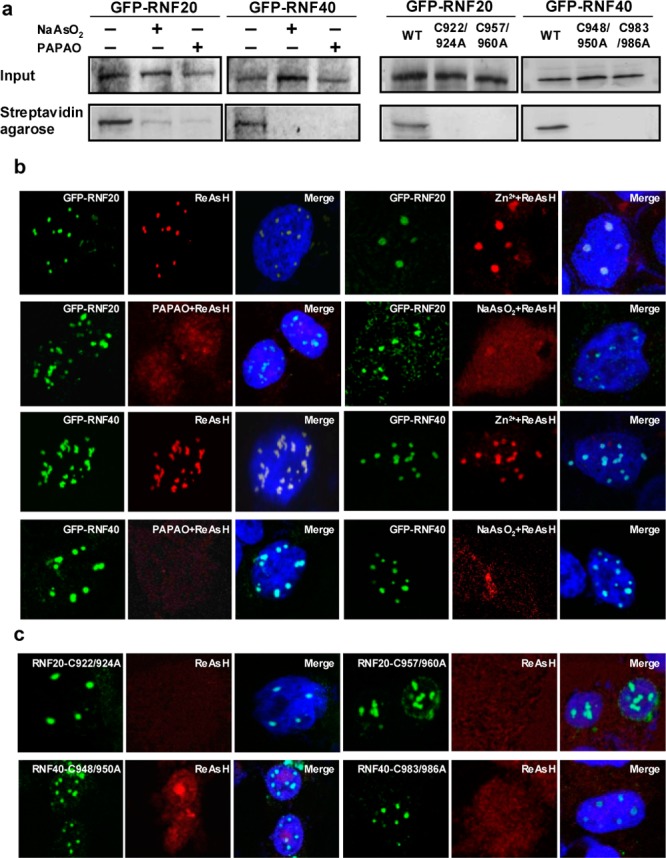
Binding between NaAsO2 and the RING finger domains of RNF20 and RNF40 in cells. (a) Streptavidin agarose affinity pull-down assay using biotin-As as a probe revealed the binding of As(III) with RNF20 or RNF40 in cells. Pretreatment of cells with 10 μM NaAsO2 or PAPAO for 1 h attenuated the binding, and mutations of RING finger cysteines to alanines abrogated the pull-down of RNF20 and RNF40 with the biotin-As probe. (b) Colocalization of As(III)-bearing ReAsH with GFP-RNF20 and GFP-RNF40 in HEK293T cells, and such colocalization is lost in cells pretreated with 10 μM NaAsO2 or PAPAO, but not Zn2+. (c) Mutations of RING finger cysteines to alanines abolished the colocalization of RNF20 or RNF40 with ReAsH.
We further examined arsenite binding by performing fluorescence microscopy experiments with the use of ReAsH, which displays no red fluorescence until its arsenic moieties bind to nearby cysteine sulfhydryl groups in proteins.21 We found that the GFP-conjugated RNF20 or RNF40 can bind to As(III) in ReAsH as manifested by the emission of red fluorescence from the dye in ReAsH-treated cells (Figures 2b and S2). Furthermore, ReAsH colocalized to nuclear foci with GFP-fused RNF20 and RNF40 in HEK293T cells. Such colocalization was abolished when the cells were pretreated with NaAsO2 or PAPAO (but not with Zn2+) or when two of the RING finger cysteine residues were mutated to alanines (Figure 2b,c). These results demonstrate that As(III) interacts directly with RNF20 and RNF40 in human cells and such interactions necessitate the intact RING finger motif. In this respect, the background red signal observed for cells pretreated with PAPAO or NaAsO2 or cells transfected with mutant RNF20/RNF40 might be attributed to the interactions between As(III) and other cellular proteins.
The RNF20-RNF40 heterodimer induces the ubiquitination of histone H2B at K120, which is important in transcription elongation.22,23 Recently this ubiquitination was also found to be required for decompacting the 30 nm chromatin fiber15 and play a crucial role in DNA DSB repair via the homologous recombination (HR) and nonhomologous end-joining (NHEJ) pathways.16,17 In this context, cells lacking RNF20 exhibit reduced recruitment of HR and NHEJ factors to DSB sites and display markedly enhanced sensitivity to ionizing radiation and neocarzinostatin (NCS), a radiomimetic drug.16,17 We reasoned that the binding of arsenite to the RING finger motifs of RNF20 and RNF40 might diminish the heterodimer’s capability in inducing ubiquitination of histone H2B at K120 thereby suppressing DNA DSB repair. To test this hypothesis, we extracted core histones from the control and arsenite-treated cells and monitored the levels of H2B K120 ubiquitination by using LC-MS/MS (Figures 3a and S3).24 It turned out that a 24 h treatment with 5 μM NaAsO2 led to a significant diminution in the levels of H2B K120 ubiquitination in multiple cell lines (Figure 3b). The decreased ubiquitination could be attributed to the aggregation of the RING finger proteins stimulated by As(III) binding. In this vein, our refolding experiment revealed that As(III), but not Zn2+, could promote the aggregation of the RING finger motif of RNF20 in vitro (Figure S4).
Figure 3.

NaAsO2 inhibits histone H2B K120 ubiquitination in cells. (a) The product-ion spectrum (MS/MS) of the [M + 2H]2+ ion of the tryptic peptide AVTKGGVTSSK from histone H2B, where K120 is modified with a diglycine remnant. An asterisk (*) indicates those ions bearing a diglycine moiety. (b) Relative levels of H2B ubiquitination in GM00637, IMR90, and HEK293T cells without or with a 24 h treatment of 5 μM NaAsO2.
We next investigated whether arsenite-induced perturbation in H2B K120 ubiquitination confers diminished recruitment of DNA repair proteins to DNA DSB sites. To this end, we employed immunofluorescence microscopy to follow the recruitment of BRCA1 and RAD51 to laser-induced DNA DSB sites. Our results revealed that treatment with arsenite led to marked decreases in the recruitment of these two proteins to laser-induced DNA DSB sites (Figure 4). Additionally, the diminished recruitment of these two proteins is not due to their decreased expression, as real-time PCR results showed that arsenite treatment did not lead to decreased expression of BRCA1, RAD51, or several other genes important in DNA DSB repair (i.e., XRCC1, Ku70, and Ku80; Figure S5).
Figure 4.
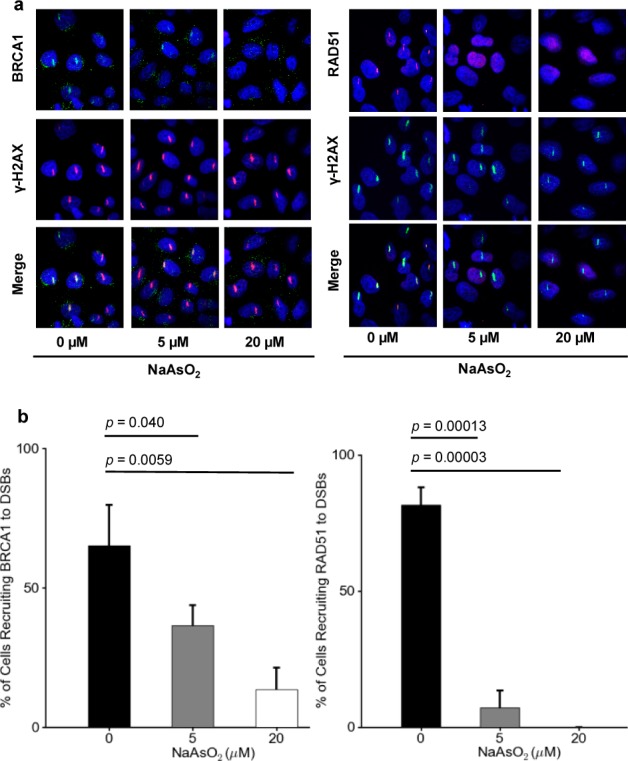
NaAsO2 suppresses the recruitment of BRCA1 and RAD51 to DSB sites. (a) Immunofluorescence displaying that the treatment of HeLa cells with NaAsO2 compromises the recruitment of BRCA1 and RAD51 to laser-induced DNA DSB sites. (b) Quantified percentages of cells recruiting BRCA1 and RAD51 to DSB sites upon treatment with 0, 5, and 20 μM of NaAsO2. The p values were calculated by using unpaired two-tailed t-test.
Diminished histone H2B ubiquitination and the ensuing compromised recruitment of DNA repair factors to DNA DSB sites may perturb DSB repair. To test this, we utilized U2OS cells with a chromosomally integrated copy of DR-GFP and EJ5-GFP reporters to examine whether arsenite treatment leads to compromised DNA DSB repair via the HR and NHEJ pathways.25 DR-GFP contains the SceGFP cassette, where the GFP coding sequence is interrupted by a single recognition site of the rare-cutting I-SceI nuclease and a 5′ and 3′ truncated fragment of GFP (iGFP).25 HR-directed repair of the I-SceI-induced DSB leads to the restoration of a functional GFP gene.25 EJ5-GFP carries the pCAGGS promoter separated from the rest of a GFP expression cassette flanked by two I-SceI sites; NHEJ of I-SceI-produced distal DSB ends results in the restoration of the GFP cassette.25 Flow cytometry-based measurement of GFP-positive U2OS cells after I-SceI transfection facilitated us to quantify the levels of HR and NHEJ in control and arsenite-treated cells. Our results revealed that arsenite exposure led to significantly diminished repair of DSBs via the HR and NHEJ pathways (Figures S6 and S7a). It is of note that treatment with NaAsO2 did not alter the transfection efficiency (Figure S7b) or interfere with the GFP-based reporter assays for monitoring HR and NHEJ activities. In the latter respect, our control experiments showed that, without I-SceI transfection, the percentages of GFP-positive U2OS cells are similar in the presence or absence of NaAsO2 treatment (Figure S7a). Moreover, consistent with the diminished DNA DSB repair, we found, from clonogenic survival assay, that treatment with arsenite renders HEK293T cells more sensitive toward treatment with NCS, a radiomimetic drug (Figure S8).
Trivalent arsenic is a known human carcinogen. Although arsenite has been shown to bind and inhibit the activity of zinc finger-harboring DNA repair proteins (i.e., XPA and PARP1),7−10 this is the first report to demonstrate that arsenite can bind to the RING finger domains of a histone E3 ubiquitin ligase, which perturbs histone epigenetic mark and compromises DNA DSB repair. In this vein, DNA DSBs can emanate from collapse of replication fork, processing of DNA interstrand cross-links, or following exposure to ionizing radiation.26,27 DNA DSBs are among the most deleterious types of DNA lesions; failure to repair DSBs can lead to cell death or promote chromosomal rearrangements, which in turn can stimulate malignant transformation.28 Thus, arsenite may exert its carcinogenic effect partly through inhibiting DNA DSB repair. In keeping with its inhibitory effect on DNA repair, we also found that arsenite sensitizes cells toward a radiomimetic drug. Considering that ionizing radiation is widely used in cancer treatment, our results suggest the potential application of arsenite as a sensitizing agent for cancer radiation therapy.
Maintaining an open and biochemically active chromatin conformation is also important for proteins involved in other DNA repair pathways to access damage sites in chromatin. Thus, compromised H2B K120 ubiquitination arising from arsenite exposure may also account for the diminished repair of other types of DNA lesions.
Our findings that arsenite may promote cancer development through binding to the RING finger domains of a histone E3 ubiquitin ligase is reminiscent of the mechanism underlying arsenite’s role as a chemotherapeutic agent for the treatment of acute promyelocytic leukemia. In the latter case, arsenite binds directly to the RING finger motif of the PML-RARα fusion protein, which triggers eventually the proteasomal degradation of the oncoprotein.13 Viewing the crucial roles of the ubiquitin-proteasome system in physiology and pathophysiology and the fact that the majority of the E3 ubiquitin-protein ligases harbor RING finger or RING finger-related motifs,14 our study suggests that arsenite may also perturb ubiquitination events mediated by other RING finger E3 ubiquitin ligases. In this context, the reduced efficiency in HR and NHEJ repair may also arise, in part, from the interaction between arsenite and other RING finger E3 ubiquitin ligases that are important in DNA damage response and repair. Along this line, we observed that As(III) can also bind to the RNF8 and RNF168 histone E3 ubiquitin ligases (Figures S9–S11). Thus, arsenite may be considered as a general inhibitor for RING finger E3 ubiquitin ligases. Together, our results support the notion that As(III) may induce the remission of acute promyelocytic leukemia and cancer development on the basis of the same chemical rationale (i.e., through binding to RING finger proteins), albeit on different molecular targets (i.e., PML-RARα oncoprotein in one case, and RING finger histone E3 ubiquitin ligases in the other).
Acknowledgments
This research was supported in part by the National Institutes of Health (R01 ES019873 to Y.W.) and the Intramural Research Program of the NIH, National Institute on Aging (AG000746-02). The authors would like to thank Profs. Jeremy M. Stark, Jiri Lukas, Gerd P. Pfeifer, Yossi Shiloh, and Alan R. Lehmann for generously providing cells and plasmids.
Supporting Information Available
Detailed experimental conditions, mass spectrometry, flow cytometry, immunofluorescence, and avidin agarose pull-down assay results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ravenscroft P.; Brammer H.; Richards K.. Arsenic pollution: a global synthesis; Wiley-Blackwell: Chichester, U.K., 2009. [Google Scholar]
- Chen C. J.; Chuang Y. C.; Lin T. M.; Wu H. Y. Cancer Res. 1985, 45, 5895–9. [PubMed] [Google Scholar]
- Smith A. H.; Goycolea M.; Haque R.; Biggs M. L. Am. J. Epidemiol. 1998, 147, 660–9. [DOI] [PubMed] [Google Scholar]
- Council N. R.Arsenic in the Drinking Water; National Academy Press: Washington, DC, 1999. [Google Scholar]
- Kitchin K. T. Toxicol. Appl. Pharmacol. 2001, 172, 249–61. [DOI] [PubMed] [Google Scholar]
- Shen S.; Li X. F.; Cullen W. R.; Weinfeld M.; Le X. C. Chem. Rev. 2013, 113, 7769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W.; Liu W.; Cooper K. L.; Qin X. J.; de Souza Bergo P. L.; Hudson L. G.; Liu K. J. J. Biol. Chem. 2009, 284, 6809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter I.; Schwerdtle T.; Thuy C.; Parsons J. L.; Dianov G. L.; Hartwig A. DNA Repair 2007, 6, 61–70. [DOI] [PubMed] [Google Scholar]
- Hartwig A.; Blessing H.; Schwerdtle T.; Walter I. Toxicology 2003, 193, 161–9. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Sun X.; Cooper K. L.; Wang F.; Liu K. J.; Hudson L. G. J. Biol. Chem. 2011, 286, 22855–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X. J.; Liu W.; Li Y. N.; Sun X.; Hai C. X.; Hudson L. G.; Liu K. J. Toxicol. Sci. 2012, 127, 120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z. X.; Chen G. Q.; Ni J. H.; Li X. S.; Xiong S. M.; Qiu Q. Y.; Zhu J.; Tang W.; Sun G. L.; Yang K. Q.; Chen Y.; Zhou L.; Fang Z. W.; Wang Y. T.; Ma J.; Zhang P.; Zhang T. D.; Chen S. J.; Chen Z.; Wang Z. Y. Blood 1997, 89, 3354–60. [PubMed] [Google Scholar]
- Zhang X. W.; Yan X. J.; Zhou Z. R.; Yang F. F.; Wu Z. Y.; Sun H. B.; Liang W. X.; Song A. X.; Lallemand-Breitenbach V.; Jeanne M.; Zhang Q. Y.; Yang H. Y.; Huang Q. H.; Zhou G. B.; Tong J. H.; Zhang Y.; Wu J. H.; Hu H. Y.; de The H.; Chen S. J.; Chen Z. Science 2010, 328, 240–243. [DOI] [PubMed] [Google Scholar]
- Lipkowitz S.; Weissman A. M. Nat. Rev. Cancer 2011, 11, 629–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierz B.; Chatterjee C.; McGinty R. K.; Bar-Dagan M.; Raleigh D. P.; Muir T. W. Nat. Chem. Biol. 2011, 7, 113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyal L.; Lerenthal Y.; Gana-Weisz M.; Mass G.; So S.; Wang S. Y.; Eppink B.; Chung Y. M.; Shalev G.; Shema E.; Shkedy D.; Smorodinsky N. I.; van Vliet N.; Kuster B.; Mann M.; Ciechanover A.; Dahm-Daphi J.; Kanaar R.; Hu M. C.; Chen D. J.; Oren M.; Shiloh Y. Mol. Cell 2011, 41, 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N.; Bekker-Jensen S.; Faustrup H.; Melander F.; Bartek J.; Lukas C.; Lukas J. Cell 2007, 131, 887–900. [DOI] [PubMed] [Google Scholar]
- Spuches A. M.; Kruszyna H. G.; Rich A. M.; Wilcox D. E. Inorg. Chem. 2005, 44, 2964–72. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yang F.; Shim J. Y.; Kirk K. L.; Anderson D. E.; Chen X. Cancer Lett. 2007, 255, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalef E.; Gitler C. Methods Enzymol. 1994, 233, 395–403. [DOI] [PubMed] [Google Scholar]
- Martin B. R.; Giepmans B. N.; Adams S. R.; Tsien R. Y. Nat. Biotechnol. 2005, 23, 1308–14. [DOI] [PubMed] [Google Scholar]
- Kim J.; Hake S. B.; Roeder R. G. Mol. Cell 2005, 20, 759–70. [DOI] [PubMed] [Google Scholar]
- Zhu B.; Zheng Y.; Pham A. D.; Mandal S. S.; Erdjument-Bromage H.; Tempst P.; Reinberg D. Mol. Cell 2005, 20, 601–11. [DOI] [PubMed] [Google Scholar]
- Darwanto A.; Curtis M. P.; Schrag M.; Kirsch W.; Liu P.; Xu G.; Neidigh J. W.; Zhang K. J. Biol. Chem. 2010, 285, 21868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A. J.; Jasin M. Methods Mol. Biol. 2005, 291, 373–84. [DOI] [PubMed] [Google Scholar]
- Ciccia A.; Elledge S. J. Mol. Cell 2010, 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price B. D.; D’Andrea A. D. Cell 2013, 152, 1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna K. K.; Jackson S. P. Nat. Genet. 2001, 27, 247–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.