

Abstract
The seco-B-ring bryostatin analogue, macrodiolide WN-1, was prepared in 17 steps (longest linear sequence) and 30 total steps with three bonds formed via hydrogen-mediated C–C coupling. This synthetic route features a palladium-catalyzed alkoxycarbonylation of a C2-symmetric diol to form the C9-deoxygenated bryostatin A-ring. WN-1 binds to PKCα (Ki = 16.1 nM) and inhibits the growth of multiple leukemia cell lines. Although structural features of the WN-1 A-ring and C-ring are shared by analogues that display bryostatin-like behavior, WN-1 displays PMA-like behavior in U937 cell attachment and proliferation assays, as well as in K562 and MV-4-11 proliferation assays. Molecular modeling studies suggest the pattern of internal hydrogen bonds evident in bryostatin 1 is preserved in WN-1, and that upon docking WN-1 into the crystal structure of the C1b domain of PKCδ, the binding mode of bryostatin 1 is reproduced. The collective data emphasize the critical contribution of the B-ring to the function of the upper portion of the molecule in conferring a bryostatin-like pattern of biological activity.
Introduction
The bryostatins are a family of marine macrolides isolated by Pettit and co-workers from the bryozoan Bugula neritina based on an assay of their anti-neoplastic activity against the P388 leukemia cell system.1 Bryostatin 1 (Figure 1), the most abundant member of this class, binds the C1 domain of protein kinase C (PKC) isozymes,2 modulating an impressive array of downstream effects, including anti-neoplastic activity, immunopotentiating activity, restoration of apoptotic function, and the ability to act synergistically with other chemotherapeutic agents.3 Indeed, preliminary data on the properties of bryostatin 1 for cancer therapy showed such promise that a Good Manufacturing Processes campaign was undertaken, wherein 18 g of bryostatin 1 was isolated from a collection of 10,000 gallons of wet bryozoan.4 This supply of material has supported dozens of phase I and phase II clinical trials against diverse cancers,3f and in addition has led to the identification of bryostatin 1 as a promising candidate for the treatment of Alzheimer’s disease5 and HIV.6
Figure 1.

PKC binding affinities (Ki) of bryostatin 1, bryostatin 7, and selected bryostatin analogues. aBinding affinity to PKCα. See ref (7h) for PKCα binding affinity of bryostatin 1 and bryostatin 7. bBinding affinity to a mixture of rat brain PKC isozymes. cRefers to U937 attachment and inhibition of proliferation assays.
These compelling biological properties, along with their daunting structural complexity and limited natural abundance, have motivated efforts to define concise routes to the bryostatins and simplified functional analogues.7−10 Such analogues are yielding critical insights into the structural features responsible for high-affinity PKC binding, as well as for bryostatin’s unique pattern of biological response, in which it paradoxically antagonizes many of the responses to phorbol 12-myristate 13-acetate (PMA), the paradigm for activators of PKC. Of equal importance, these analogues are affording tools to dissect the biochemical mechanism(s) downstream of PKC binding which are responsible for their unique biological effects. In 1988, Wender, Pettit, and Blumberg developed a pharmacaphore model for the bryostatins suggesting that more-accessible analogues possessing simplified A- and B-rings may bind effectively to PKC.8c Subsequently, the Wender group prepared numerous analogues bearing B-ring acetals that display high-affinity PKC binding (Figure 1).8 The analogues I–V and others developed in the Wender laboratory were shown to function similarly to bryostatin 1 with regard to the pattern of PKCδ-GFP translocation induced in rat basophilic leukemia cells.8h,8i,8k
The core concept of functional synthesis is to retain efficacy while effecting structural simplification. While high-affinity binding to PKC is one critical function, bryostatin 1 is distinguished from other PKC ligands by its failure to induce many typical PMA responses and by its antagonism of those same responses when it and PMA are present together. Keck and Blumberg have shown that this latter activity is in fact conferred by the A- and B-ring “spacer domain”, as evidenced by the effects of various functional group deletions and modifications of the bryostatin 1 A- and B-rings on whether a given analogue behaves like bryostatin 1 or the tumor-promoting PMA in U937 histiocytic lymphoma cells (Figure 1).9 Their studies reveal that analogues Merle 28 and Merle 30, which retain the C7-acetoxy and C8-gem-dimethyl moieties, exhibit a biological response very similar to that of bryostatin 1.9b,9d While Merle 23 and Merle 32, which both lack the C7-acetoxy group, display PMA-like behavior, the response of Merle 27 shows that the C7-acetoxy group alone does not enforce a bryostatin 1-like effect.9a,9c,9g
The A- and B-ring-modified “Merle compounds” show that the C7-acetoxy and C8-gem-dimethyl moieties are important for retaining bryostatin 1-like behavior in U937 histiocytic lymphoma cells. Hence, the question was posed as to whether further structural simplification could be availed by replacing the B-ring pyran with a simple ester linkage, as in seco-B-ring analogue WN-1 (Figure 1). In this article, we report the synthesis of WN-1 via hydrogen-mediated C–C coupling and describe preliminary data on its biological properties. Remarkably, although WN-1 incorporates A- and C-rings common to analogues that display bryostatin 1-like behavior, WN-1 elicits a PMA-like response in U937 histiocytic lymphoma cells. These data provide further evidence that the bryostatin A- and B-rings function as more than a spacer domain, and that the conformation induced by the B-ring and overall lipophilicity figure among the collection of factors that govern the partitioning between bryostatin-like and PMA-like behavior. Notably, the 17-step synthesis of WN-1 (longest linear sequence) represents the shortest route to any active bryostatin analogue reported, to date.
Research Design and Methods
Synthesis of WN-1
Retrosynthetically, a convergent assembly of WN-1 from fragments A and B via successive ester bond formation was envisioned (Figure 2). The synthesis of fragment A takes advantage of the hydrogen-mediated reductive coupling of glyoxal 1 and enyne 2.11 This transformation was utilized in our recent synthesis of bryostatin 77g and serves to construct the C20–C21 bond with control of C20 carbinol stereochemistry and C21 alkene geometry. Acylation of the C20 hydroxyl with octanoic acid using diisopropylcarbodiimide and DMAP with subsequent addition of methanol and HF·pyridine to the reaction mixture furnished fragment A in eight steps from commercial materials (Scheme 1).
Figure 2.

Retrosynthetic analysis of WN-1 illustrating C–C bonds formed via hydrogenative coupling.
Scheme 1. Synthesis of Fragment A via Hydrogen-Mediated Reductive Coupling of Glyoxal 1 and 1,3-Enyne 2.

Indicated yields are of material isolated by silica gel chromatography. See ref (7g) for the preparation of compounds 1 and 2. See Supporting Information for further experimental details.
Fragment B was formed through the asymmetric double allylation of neopentyl glycol 3 via iridium-catalyzed transfer hydrogenation.12 The resulting C2-symmetric diol 4, which forms as a single enantiomer,13 was subjected to palladium-catalyzed alkene alkoxypalladation–alkoxycarbonylation14 to form the pyran 5 as a single diastereomer. Exposure of 5 to the lithium enolate of tert-butyl acetate resulted in Claisen condensation to form the β-ketoester 6, which is converted to acetate 7. Treatment of β-ketoester 7 with sodium borohydride modified by l-tartaric acid15 enabled reduction of the C3 ketone to form the alcohol 8 as a 9:1 mixture of diastereomers. Conversion of the alcohol 8 to the TBS-ether 9 followed by oxidative cleavage of the terminal olefin16 provided fragment B (Scheme 2).
Scheme 2. Synthesis of Fragment B via Transfer Hydrogenative Double Allylation of Neopentyl Glycol 3.

Indicated yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
An efficient coupling of fragments A and B was realized using the PyBroP reagent in the presence of Hunig’s base and DMAP, forming the desired ester 10 in 96% yield.17 Treatment of 10 with an excess of TBS-triflate enabled selective hydrolysis of the tert-butyl ester.18 The resulting acid was exposed to aqueous acetic acid to effect concomitant removal of the C25/C26 acetonide and C3 TBS ether to provide a triol acid (not shown),10a which upon treatment with TES-triflate under cryogenic conditions provided the hydroxy acid 11. Formation of the macrodiolide 12 was achieved under Yamaguchi conditions.19 Conversion of diene 12 to the α,β-unsaturated acid 13 was carried out using a modified Lemieux–Johnson oxidation20 followed by Pinnick oxidation of the resulting aldehyde.21 Finally, treatment of 13 with TMS-diazomethane followed by global deprotection using HF·pyridine provided WN-1 in 17 steps (longest linear sequence, Scheme 3). A total of 20 mg of WN-1 was prepared through this route.
Scheme 3. Synthesis of the Macrodiolide WN-1.

Indicated yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Biological Evaluation of WN-1
Determination of Binding Affinity to PKCα
The biological evaluation of WN-1 began with the determination of its binding affinity (Ki) toward purified PKCα, so as to allow direct comparison of WN-1 to compounds in the “Merle” series. Despite the absence of the B-ring, WN-1 bound to human PKCα with Ki = 16.1 ± 1.1 nM.22 These results demonstrate that a seco-B-ring bryostatin analogue retains potent affinity toward PKCα in the nanomolar range but also indicate that WN-1 showed some diminution in potency, as reflected in its 44-fold weaker affinity compared to that of Merle 30, which it most closely resembles.
Activity of WN-1 in Leukemia Cell Lines That Show Differential Response to PMA and Bryostatin 1
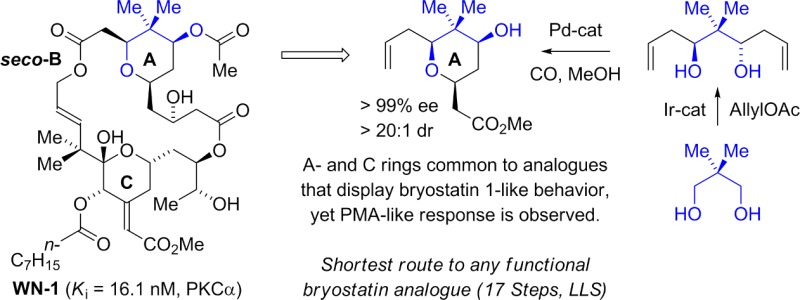
While binding affinity to PKC remains an important benchmark for the evaluation of bryostatin analogues, observing downstream biological responses is essential for determining whether the compound will elicit the unique pattern of activity associated with bryostatin 1 or, instead, that of other PKC activators such as the phorbol esters. For U937 human histiocytic lymphoma cells, PMA induces a differentiation response which is reflected by inhibition of proliferation and attachment of the cells, whereas bryostatin 1 induces a minimal response.9,23 Further, when bryostatin 1 and PMA are co-administered, the effects of bryostatin 1 dominate, inhibiting the actions of PMA.
For the U937 cell assays, WN-1 was found to inhibit cell proliferation in a pattern similar to that of PMA (Figure 3). As depicted, cell growth decreased with increasing concentrations of WN-1. When PMA was co-administered with various concentrations of WN-1, there was no change in the level of inhibition of proliferation, whereas bryostatin 1 was able to suppress the effects of PMA. Likewise, WN-1 induced cell attachment similarly to PMA, whereas bryostatin 1 had only minimal effect. Once again, when co-administered, bryostatin 1 blocked the attachment induced by PMA; WN-1 did not. From the dose–response curves, WN-1 was approximately 1000-fold less potent than PMA. K562 and MV-4-11 are two additional cell lines in which PMA inhibits cell growth, whereas bryostatin 1 does not block growth and inhibits the response to PMA.9g In both of these lines, once again WN-1 acted like PMA to inhibit growth (Supplemental Figure 1, Supporting Information).
Figure 3.
Cell proliferation and attachment assays. See Supporting Information for experimental details.
Unlike the U937, K562, and MV-4-11 cells, some leukemia cell lines are sensitive to growth inhibition by bryostatin 1 as well as PMA. Toledo cells are particularly sensitive. In the Toledo cells, WN-1 inhibited growth similarly to PMA or bryostatin 1. The maximal effect of WN-1 was achieved by 100 nM, reflecting the greater potency of all three ligands in this system.
Effect of WN-1 on TNFα Gene Expression
The biological responses of proliferation and attachment in the U937 cells were determined at 60 h. To examine whether the potency of WN-1 was appreciably affected by instability under the culture conditions, we measured the induction by WN-1 and by PMA of TNFα (tumor necrosis factor alpha) mRNA expression in the U937 cells at 2, 6, and 24 h (Figure 4). Instability did not appear to be a problem. Potencies were similar over this time range, and, as seen for the effects on cell growth or attachment, WN-1 again showed approximately 1000-fold weaker potency than PMA.
Figure 4.
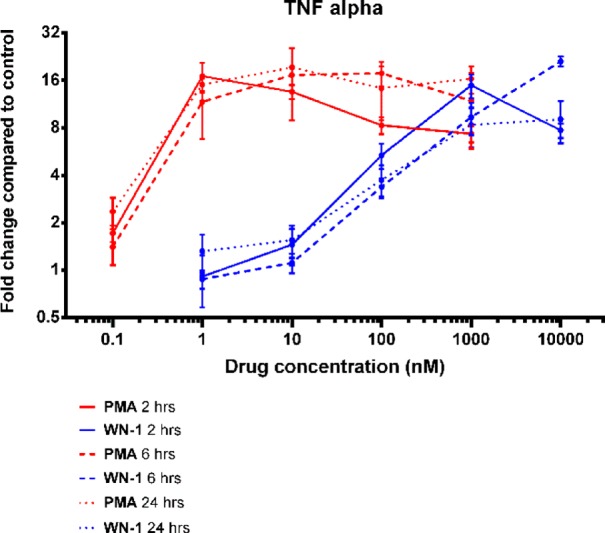
Induction of TNFα mRNA expression. See Supporting Information for experimental details.
Molecular Modeling
To evaluate the effect of replacing the B-ring with an ester linkage on the overall conformation of the macrolide ring, a thorough conformational search of WN-1 was performed in octanol solvent. The lowest-energy conformation found retained a strong similarity to the conformation observed for bryostatin 1 by single-crystal X-ray diffraction (Figure 5, left).1b The A- and C-rings are nearly completely superimposable, and the endocyclic ester oxygen of WN-1 aligns well with the pyran oxygen of the bryostatin B-ring.24 These models suggest that the pattern of internal hydrogen bonds evident in bryostatin 1 is preserved in WN-1.
Figure 5.

Left: Overlay of the crystal structure of bryostatin 1 (gray) with the lowest-energy conformer of WN-1 (magenta), based on a molecular mechanics-based conformational search and DFT geometry optimizations at the B97-D3/6-31G(d) level. Intramolecular hydrogen bonds are shown as black dashed lines. Right: Binding mode of WN-1 in the PKCδ C1b domain. Hydrogen bonds are indicated by dashed black lines. An initial model of WN-1 was constructed using the crystal structure of bryostatin 1 obtained from the Cambridge Structural Database, and then a thorough conformational search of WN-1 was performed in octanol solvent. See Supporting Information for further details.
As anticipated given the conformational homology between WN-1 and bryostatin 1, when WN-1 is docked into the crystal structure of the C1b domain of PKCδ,25 the binding mode of bryostatin 1 is reproduced: the C26 hydroxyl hydrogen-bonds to the backbone at Thr 242 and Leu 251, and the C-ring methoxycarbonyl group hydrogen-bonds to Gly 253 (Figure 5, right). The C11 carbonyl oxygen in the ester linkage of WN-1 in the docked structure is solvent exposed and does not form any interactions with the C1 domain. It should be noted that WN-1 is lacking the C9 hydroxyl of bryostatin 1, which forms an additional hydrogen bond to the backbone carbonyl of Met 239. The conformational analysis and docking results suggest that the difference in binding affinity between WN-1 and bryostatin 1 is not due to any significant change in conformation. It is possible that, in the absence of the B-ring, the C9 hydroxyl contributes much more to binding affinity than it does in the context of the native AB-ring system.9d
An analogue structurally related to WN-1 is the seco-B-ring bryostatin analogue Merle 42 (Ki = 0.75 nM), which is described in a doctoral thesis from the Keck laboratory as a minor reaction product generated upon global deprotection of their penultimate synthetic intermediate (Figure 6).26a The major reaction product formed in their synthetic route was the unexpected 21-membered macrodiolide Merle 43 (Ki = 13.8 nM). The authors suggest that the acyl migration resulting in the undesired isomer Merle 43 can be attributed to activation of the C1-carbonyl upon treatment of the precursor with LiBF4 in CH3CN/H2O at 60 °C, establishing the intrinsic instability of Merle 42 with respect to the formation of Merle 43, perhaps driven by the ring strain associated with the C15–C17 enoate. As described in the companion article, this interpretation is supported by the calculated energies of Merle 42 and Merle 43.26b
Figure 6.

Relative thermodynamic stabilities calculated for the seco-B-ring analogues of WN-1 and iso-WN-1, Merle 42 and Merle 43, and the hypothetical congeners C9-deoxy-Merle 42 and C9-hydroxy-WN-1. aBinding affinity to PKCα. bRefers to U937 attachment and inhibition of proliferation assays. The binding affinities for Merle 42 and Merle 43 are reported by Keck.26b
The reported instability of Merle 42 as well as the similarity in binding affinities between WN-1 and Merle 43 prompted us to perform a battery of experiments to probe the structural integrity of WN-1 with respect to the formation of the ring-expanded macrodiolide iso-WN-1.27 The macrodiolide WN-1 was exposed to LiBF4 in CD3CN/D2O at 60 °C for a period of 12 h without any detectable isomerization to iso-WN-1 or decomposition, as determined by 1H NMR. Similarly, WN-1 was recovered unchanged from simulated conditions for binding affinity determination. Further, the sample of WN-1 sent to the National Cancer Institute for biological characterization, which was stored in DMSO solution for over a 2 month period, was recovered unchanged, and the binding affinities determined for two different batches of WN-1 were identical within the limits of experimental error. No trace of iso-WN-1 was observed under the aforementioned experiments, demonstrating that the structural integrity of WN-1 is remarkably high.
To more quantitatively assess the relative stability of WN-1, the energies of WN-1, iso-WN-1, Merle 42, Merle 43, and the hypothetical seco-B-ring analogues C9-deoxy-Merle 42 and C9-hydroxy-WN-1 were calculated using the lowest-energy conformer for each compound. Geometry optimizations for each structure were run at the B97-D3/6-31G(d) level, and subsequent single-point energies were calculated at the ωB97X-D/6-311G(2d,2p) level. For the C9-deoxy analogues, it was found that WN-1 is 2.27 kcal/mol more stable than iso-WN-1 and 2.81 kcal/mol lower in energy than C9-deoxy-Merle 42. Conversely, for the C9-hydroxy analogues, Merle 42 was found to be 6.54 kcal/mol less stable than both Merle 43 and C9-hydroxy-WN-1. These data confirm that the rearrangement of Merle 42 and Merle 43 is energetically favorable, while the equivalent rearrangement of WN-1 and iso-WN-1 is not. Furthermore, these studies demonstrate that the transposition of the carbonyl has a significant impact on the stabilities of these seco-B-ring analogues (Figure 6). The collective data suggest that the enhanced stability of WN-1 stems from the increased conformational flexibility of the C11-ester compared to the more rigid C15-enoate moiety of Merle 42.
All three analogues, Merle 42, Merle 43, and WN-1, demonstrate PMA-like behavior in U937 cell attachment and proliferation assays, further demonstrating the critical contribution of the B-ring for bryostatin-like behavior. The significant difference in binding affinities between WN-1 and Merle 42 underscores how overall lipophilicity or conformational preferences associated with the B-ring dramatically impact the chemical stability and binding affinities of these analogues, and suggests that the magnitude of the contribution of the C9 hydroxyl to overall binding affinity may be dependent upon the presence or absence of the B-ring.
Conclusions
In summary, we report the synthesis of the seco-B-ring bryostatin analogue WN-1 via C–C bond-forming hydrogenation that features a palladium-catalyzed alkoxycarbonylation of the C2-symmetric diol 4 to form the C9-deoxygenated bryostatin A-ring. The present route delivers WN-1 in 17 steps (longest linear sequence), representing the most concise route to any active bryostatin analogue reported to date. WN-1 binds to purified human PKCα with Ki = 16.1 ± 1.1 nM. Remarkably, although structural features of the WN-1 A- and C-rings are common to analogues that display bryostatin-like behavior, WN-1 displays PMA-like behavior in U937 cell attachment and proliferation assays. These data demonstrate that B-ring characteristics of bryostatin 1, such as conformational effects or overall lipophilicity,7h play a critical role in shifting the biological response from PMA-like to bryostatin-like. Future studies will be aimed at better understanding how the interactions between bryostatin analogues, the PKCα C1 domain, and the cell membrane impact biological response.
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM093905) are acknowledged for partial support of this research. Partial support was also provided by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Z1A BC 005270). The Cancer Prevention Research Institute of Texas (RP101501) is acknowledged for Postdoctoral Fellowship support (I.P.A.). Skilled technical assistance was provided by Kim Wasik. This project was funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
Supporting Information Available
Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Pettit G. R.; Day J. F.; Hartwell J. L.; Wood H. B. Nature 1970, 227, 962. [DOI] [PubMed] [Google Scholar]; b Pettit G. R.; Herald C. L.; Doubek D. L.; Herald D. L.; Arnold E.; Clardy J. J. Am. Chem. Soc. 1982, 104, 6846. [Google Scholar]; c It is believed that the natural product actually derives from a bacterial symbiont of B. neritina:Sudek S.; Lopanik N. B.; Waggoner L. E.; Hildebrand M.; Anderson C.; Liu H.; Patel A.; Sherman D. H.; Haygood M. G. J. Nat. Prod. 2007, 70, 67. [DOI] [PubMed] [Google Scholar]
- For selected studies on the binding of bryostatin 1 to PKC isozymes, see:; a Berkow R. L.; Kraft A. S. Biochem. Biophys. Res. Commun. 1985, 131, 1109. [DOI] [PubMed] [Google Scholar]; b Kraft A. S.; Smith J. B.; Berkow R. L. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 1334. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kazanietz M. G.; Lewin N. E.; Gao F.; Pettit G. R.; Blumberg P. M. Mol. Pharmacol. 1994, 46, 374. [PubMed] [Google Scholar]
- For reviews of the chemistry and biology the bryostatins and their analogues, see:; a Mutter R.; Wills M. Bioorg. Med. Chem. 2000, 8, 1841. [DOI] [PubMed] [Google Scholar]; b Hale K. J.; Hummersone M. G.; Manaviazar S.; Frigerio M. Nat. Prod. Rep. 2002, 19, 413. [DOI] [PubMed] [Google Scholar]; c Kortmansky J.; Schwartz G. K. Cancer Invest. 2003, 21, 924. [DOI] [PubMed] [Google Scholar]; d Wender P. A.; Baryza J. L.; Hilinski M. K.; Horan J. C.; Kan C.; Verma V. A. In Drug Discovery Research: New Frontiers in the Post-Genomic Era; Huang Z., Ed.; Wiley: Hoboken, NJ, 2007; Chapter 6, p 127. [Google Scholar]; e Hale K. J.; Manaviazar S. Chem.—Asian J. 2010, 5, 704. [DOI] [PubMed] [Google Scholar]; f Wender P. A.; Loy B. A.; Schrier A. J. Isr. J. Chem. 2011, 51, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yu L.; Krische M. J. In Total Synthesis: At the Frontier of Organic Chemistry, Li J. J., Corey E. J., Eds.; Springer: Heidelberg, Germany, 2013; pp 103–130. [Google Scholar]; h For information on bryostatin 1 in clinical trials for the treatment of cancer, see: http://clinicaltrials.gov.
- Schaufelberger D. E.; Koleck M. P.; Beutler J. A.; Vatakis A. M.; Alvarado A. B.; Andrews P.; Marzo L. V.; Muschik G. M.; Roach J.; Ross J. T.; Lebherz W. B.; Reeves M. P.; Eberwein R. M.; Rodgers L. L.; Testerman R. P.; Snader K. M.; Forenza S. J. Nat. Prod. 1991, 54, 1265. [DOI] [PubMed] [Google Scholar]
- For studies related to the use of bryostatin in treatment of Alzheimer’s disease, see:; a Etcheberrigaray R.; Tan M.; Dewachter I.; Kuiperi C.; Van der Auwera I.; Wera S.; Qiao L.; Bank B.; Nelson T. J.; Kozikowski A. P.; Van Leuven F.; Alkon D. L. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11141. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sun M.-K.; Alkon D. L. Eur. J. Pharmacol. 2005, 512, 43. [DOI] [PubMed] [Google Scholar]; c Alkon D. L.; Sun M.-K.; Nelson T. J. Trends Pharmacol. Sci. 2007, 28, 51. [DOI] [PubMed] [Google Scholar]; d Hongpaisan J.; Sun M.-K.; Alkon D. L. J. Neurosci. 2011, 31, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Williams P.; Sorribas A.; Howes M.-J. R. Nat. Prod. Rep. 2011, 28, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Hongpaisan J.; Xu C.; Sen A.; Nelson T. J.; Alkon D. L. Neurobiol. Dis. 2013, 55, 44. [DOI] [PubMed] [Google Scholar]; g Xu C.; Liu Q.-Y.; Alkon D. L. Neuroscience 2014, 268, 75. [DOI] [PubMed] [Google Scholar]
- For studies related to the use of bryostatin in treatment of HIV, see:; a Perez M.; de Vinuesa A. G.; Sanchez-Duffhues G.; Marquez N.; Bellido M. L.; Munoz-Fernandez M. A.; Moreno S.; Castor T. P.; Calzado M. A.; Munoz E. Curr. HIV Res. 2010, 8, 418. [DOI] [PubMed] [Google Scholar]; b Mehla R.; Bivalkar-Mehla S.; Zhang R.; Handy I.; Albrecht H.; Giri S.; Nagarkatti P.; Nagarkatti M.; Chauhan A. PLoS One 2010, 5, e11160. [DOI] [PMC free article] [PubMed] [Google Scholar]; c DeChristopher B. A.; Loy B. A.; Marsden M. D.; Schrier A. J.; Zack J. A.; Wender P. A. Nat. Chem. 2012, 4, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Spina C. A.; Anderson J.; Archin N. M.; Bosque A.; Chan J.; Famiglietti M.; Greene W. C.; Kashuba A.; Lewin S. R.; Margolis D. M.; Mau M.; Ruelas D.; Saleh S.; Shirakawa K.; Siliciano R. F.; Singhania A.; Soto P. C.; Terry V. H.; Verdin E.; Woelk C.; Wooden S.; Xing S.; Planelles V. PLoS Pathol. 2013, 9, e1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bullen C. K.; Laird G. M.; Durand C. M.; Siliciano J. D.; Siliciano R. F. Nat. Med. 2014, 20, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Archin N. M.; Margolis D. M. Curr. Opin. Infect. Dis. 2014, 27, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For total syntheses of naturally occurring bryostatins, see the following.; a Bryostatin 1:Keck G. E.; Poudel Y. B.; Cummins T. J.; Rudra A.; Covel J. A. J. Am. Chem. Soc. 2011, 133, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bryostatin 2:Evans D. A.; Carter P. H.; Carreira E. M.; Prunet J. A.; Charette A. B.; Lautens M. Angew. Chem., Int. Ed. 1998, 37, 2354. [DOI] [PubMed] [Google Scholar]; c Evans D. A.; Carter P. H.; Carreira E. M.; Charette A. B.; Prunet J. A.; Lautens M. J. Am. Chem. Soc. 1999, 121, 7540. [Google Scholar]; d Bryostatin 3:Ohmori K.; Ogawa Y.; Obitsu T.; Ishikawa Y.; Nishiyama S.; Yamamura S. Angew. Chem., Int. Ed. 2000, 39, 2290. [PubMed] [Google Scholar]; e Ohmori K. Bull. Chem. Soc. Jpn. 2004, 77, 875. [Google Scholar]; f Bryostatin 7:Kageyama M.; Tamura T.; Nantz M. H.; Roberts J. C.; Somfai P.; Whritenour D. C.; Masamune S. J. Am. Chem. Soc. 1990, 112, 7407. [Google Scholar]; g Lu Y.; Woo S. K.; Krische M. J. J. Am. Chem. Soc. 2011, 133, 13876. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Kedei N.; Lewin N. E.; Géczy T.; Selezneva J.; Braun D. C.; Chen J.; Herrmann M. A.; Heldman M. R.; Lim L.; Mannan P.; Garfield S. H.; Poudel Y. B.; Cummins T. J.; Rudra A.; Blumberg P. M.; Keck G. E. ACS Chem. Biol. 2013, 8, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Bryostatin 9:Wender P. A.; Schrier A. J. J. Am. Chem. Soc. 2011, 133, 9228. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Bryostatin 16:Trost B. M.; Dong G. Nature 2008, 456, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected studies of bryostatin analogues prepared in the Wender laboratory, see:; a Wender P. A.; De Brabander J.; Harran P. G.; Jimenez J.-M.; Koehler M. F. T.; Lippa B.; Park C.-M.; Shiozaki M. J. Am. Chem. Soc. 1998, 120, 4534. [Google Scholar]; b Wender P. A.; De Brabander J.; Harran P. G.; Jimenez J.-M.; Koehler M. F. T.; Lippa B.; Park C.-M.; Siedenbiedel C.; Pettit G. R. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6624. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wender P. A.; Cribbs C. M.; Koehler K. F.; Sharkey N. A.; Herald C. L.; Kamano Y.; Pettit G. R.; Blumberg P. M. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 7197. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wender P. A.; De Brabander J.; Harran P. G.; Hinkle K. W.; Lippa B.; Pettit G. R. Tetrahedron Lett. 1998, 39, 8625. [Google Scholar]; e Wender P. A.; Lippa B. Tetrahedron Lett. 2000, 41, 1007. [Google Scholar]; f Wender P. A.; Hinkle K. W. Tetrahedron Lett. 2000, 41, 6725. [Google Scholar]; g Wender P. A.; Baryza J. L.; Bennett C. E.; Bi F. C.; Brenner S. E.; Clarke M. O.; Horan J. C.; Kan C.; Lacôte E.; Lippa B. S.; Nell P. G.; Turner T. M. J. Am. Chem. Soc. 2002, 124, 13648. [DOI] [PubMed] [Google Scholar]; h Baryza J. L.; Brenner S. E.; Craske M. L.; Meyer T.; Wender P. A. Chem. Biol. 2004, 11, 1261. [DOI] [PubMed] [Google Scholar]; i Wender P. A.; Baryza J. L.; Brenner S. E.; Clarke M. O.; Craske M. L.; Horan J. C.; Meyer T. Curr. Drug Discovery Technol. 2004, 1, 1. [DOI] [PubMed] [Google Scholar]; j Wender P. A.; DeChristopher B. A.; Schrier A. J. J. Am. Chem. Soc. 2008, 130, 6658. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Wender P. A.; Baryza J. L.; Brenner S. E.; DeChristopher B. A.; Loy B. A.; Schrier A. J.; Verma V. A. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6721. [DOI] [PMC free article] [PubMed] [Google Scholar]; l DeChristopher B. A.; Fan A. C.; Felsher D. W.; Wender P. A. Oncotarget 2012, 3, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected studies of bryostatin analogues prepared in the Keck laboratory, see:; a Keck G. E.; Kraft M. B.; Truong A. P.; Li W.; Sanchez C. C.; Kedei N.; Lewin N. E.; Blumberg P. M. J. Am. Chem. Soc. 2008, 130, 6660. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Keck G. E.; Poudel Y. B.; Welch D. S.; Kraft M. B.; Truong A. P.; Stephens J. C.; Kedei N.; Lewin N. E.; Blumberg P. M. Org. Lett. 2009, 11, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Keck G. E.; Li W.; Kraft M. B.; Kedei N.; Lewin N. E.; Blumberg P. M. Org. Lett. 2009, 11, 2277. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Keck G. E.; Poudel Y. B.; Rudra A.; Stephens J. C.; Kedei N.; Lewin N. E.; Peach M. L.; Blumberg P. M. Angew. Chem., Int. Ed. 2010, 49, 4580. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kedei N.; Lubart E. S.; Lewin N. E.; Telek A.; Lim L.; Mannan P.; Garfield S. H.; Kraft M. B.; Keck G. E.; Kolusheva S.; Jelinek R.; Blumberg P. M. ChemBioChem 2011, 12, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Kedei N.; Telek A.; Czap A.; Lubart E. S.; Czifra G.; Yang D.; Chen J.; Morrison T.; Goldsmith P. K.; Lim L.; Mannan P.; Garfield S. H.; Kraft M. B.; Li W.; Keck G. E.; Blumberg P. M. Biochem. Pharmacol. 2011, 81, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Keck G. E.; Poudel Y. B.; Rudra A.; Stephens J. C.; Kedei N.; Lewin N. E.; Blumberg P. M. Bioorg. Med. Chem. Lett. 2012, 22, 4084. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Kedei N.; Telek A.; Michalowski A. M.; Kraft M. B.; Li W.; Poudel Y. B.; Rudra A.; Petersen M. E.; Keck G. E.; Blumberg P. M. Biochem. Pharmacol. 2013, 85, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Also see ref (7h)
- For selected studies of bryostatin analogues prepared in the Trost laboratory, see:; a Trost B. M.; Yang H.; Thiel O. R.; Frontier A. J.; Brindle C. S. J. Am. Chem. Soc. 2007, 129, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost B. M.; Dong G. J. Am. Chem. Soc. 2010, 132, 16403. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Trost B. M.; Yang H.; Dong G. Chem.—Eur. J. 2011, 17, 9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C.-W.; Krische M. J. Org. Lett. 2006, 8, 891. [DOI] [PubMed] [Google Scholar]
- a Lu Y.; Kim I. S.; Hassan A.; Del Valle D. J.; Krische M. J. Angew. Chem., Int. Ed. 2009, 48, 5018. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Feng Y.; Jiang X.; De Brabander J. K. J. Am. Chem. Soc. 2012, 134, 17083. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Waldeck A. R.; Krische M. J. Angew. Chem., Int. Ed. 2013, 52, 4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The minor enantiomer of the mono-adduct is converted to the meso-diastereomer:; a Kogure T.; Eliel E. L. J. Org. Chem. 1984, 49, 576. [Google Scholar]; b Midland M. M.; Gabriel J. J. Org. Chem. 1985, 50, 1143. [Google Scholar]; c Poss C. S.; Schreiber S. L. Acc. Chem. Res. 1994, 27, 9. [Google Scholar]
- a Semmelhack M. F.; Bodurow C. J. Am. Chem. Soc. 1984, 106, 1496. [Google Scholar]; b Review:Semmelhack M. F.; Kim C.; Zhang N.; Bodurow C.; Sanner M.; Dobler W.; Meier M. Pure Appl. Chem. 1990, 62, 2035. [Google Scholar]; c For a closely related example, see:Yang Z.; Zhang B.; Zhao G.; Yang J.; Xie X.; She X. Org. Lett. 2011, 13, 5916. [DOI] [PubMed] [Google Scholar]
- Yotagai M.; Ohnuki T. J. Chem. Soc., Perkin Trans. 1 1990, 1826. [Google Scholar]; Use of NaBH4 in the absence of l-tartaric acid at −20 °C in THF provided 8 as a 1.7:1 mixture of diastereomers.
- a Lemieux R. U.; Von Rudloff E. Can. J. Chem. 1955, 33, 1701. [Google Scholar]; b Von Rudolff E. Can. J. Chem. 1956, 34, 1413. [Google Scholar]; c A recent example:Hanaki Y.; Kikumori M.; Ueno S.; Tokuda H.; Suzuki N.; Irie K. Tetrahedron 2013, 69, 7636. [Google Scholar]
- Coste J.; Frerot E.; Jouin P. J. Org. Chem. 1994, 59, 2437. [Google Scholar]
- a Borgulya J.; Bernauer K. Synthesis 1980, 545. [Google Scholar]; b Wender P. A.; Verma V. A. Org. Lett. 2008, 10, 3331. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wender P. A.; Reuber J. Tetrahedron 2011, 67, 9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanaga J.; Hirata K.; Saeki H.; Katsuki T.; Yamaguchi M. Bull. Chem. Soc. Jpn. 1979, 52, 1989. [Google Scholar]
- Yu W.; Mei Y.; Kang Y.; Hua Z.; Jin Z. Org. Lett. 2004, 6, 3217. [DOI] [PubMed] [Google Scholar]
- Bal B. S.; Childers W. E. Jr.; Pinnick H. W. Tetrahedron 1981, 37, 2091. [Google Scholar]
- For experimental details regarding binding affinity of WN-1 to PKCα, see Supporting Information.
- Vrana J. A.; Saunders A. M.; Chellappan S. P.; Grant S. Differentiation 1998, 63, 33. [DOI] [PubMed] [Google Scholar]
- A comparison of the chemical shifts and coupling constants of WN-1 and bryostatin 1 corroborate conformational homology in the A-ring and C-ring. See Supporting Information for further details.
- Zhang G.; Kazanietz M. G.; Blumberg P. M.; Hurley J. H. Cell 1995, 81, 917. [DOI] [PubMed] [Google Scholar]
- a Kraft M. B.Synthesis and Biological Evaluation of Structurally Simplified Bryostatin Analogues and the Total Synthesis of Swinholide A. Ph.D. Dissertation, The University of Utah, 2011. [Google Scholar]; b See companion article by Keck et al.:Kraft M. B.; Poudel Y. B.; Kedei N.; Lewin N. E.; Peach M. L.; Blumberg P. M.; Keck G. E. J. Am. Chem. Soc. 2014, 10.1021/ja5078188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structural assignment of WN-1 was determined by comparing the spectral data of bryostatin 1, WN-1, Merle 42, and Merle 43. As revealed in the Supporting Information, the chemical shift and splitting pattern in the 1H NMR of the C25 and C26 methine hydrogens enable unambiguous differentiation of the targeted 20-membered macrolactone and the ring-expanded isomers that incorporate a 21-membered macrolactone.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.