

Abstract
We introduce a previously unexplored parameter—halenium affinity (HalA)– as a quantitative descriptor of the bond strengths of various functional groups to halenium ions. The HalA scale ranks potential halenium ion acceptors based on their ability to stabilize a “free halenium ion”. Alkenes in particular but other Lewis bases as well, such as amines, amides, carbonyls, and ether oxygen atoms, etc., have been classified on the HalA scale. This indirect approach enables a rapid and straightforward prediction of chemoselectivity for systems involved in halofunctionalization reactions that have multiple nucleophilic sites. The influences of subtle electronic and steric variations, as well as the less predictable anchimeric and stereoelectronic effects, are intrinsically accounted for by HalA computations, providing quantitative assessments beyond simple “chemical intuition”. This combined theoretical–experimental approach offers an expeditious means of predicting and identifying unprecedented reactions.
As a class, halenium ion donors1 are electrophilic reagents that offer access to complex and useful molecular motifs.2 Oxidation, addition to π-bonds, halogenation of aromatics, and activation of heteroatoms are among their core reaction modes. The electronegativity, bonding flexibility, and delivery reagents for halogen cations strongly modulate their selectivity. For instance, some chlorenium donors are effective alcohol oxidants,3 whereas suitably chosen iodenium reagents achieve mild activations to form selected glycosidic bonds without oxidation of alcoholic functionalities.4 The key to this diversity of reactivity lies in the variable interactions of halenium ions with different functional groups.
Electrophilic activation of alkenes via halenium ion attack has been extensively studied for its ability to forge C–C, C–O, C–N, and C–X bonds. The past decade has witnessed explosive growth, especially in the field of stereoselective halofunctionalization of alkenes.5 Despite this rapid expansion and great synthetic utility, the field is still in its infancy and has yet to witness benchmarks analogous in generality and utility to asymmetric epoxidations, dihydroxylations, aminohydroxylations, hydrogenations, cyclopropanations, hydrometalations, oxidative cleavage, and aziridinations, among others.6 Several studies have probed the mechanistic underpinnings of halenium ion reactions,5a,7 but state of the art halofunctionalizations still rely on trial-and-error approaches to reaction discovery and development/optimization.
To expedite discovery of new reactions in this area, an organizing framework is needed. Reflecting on our own synthetic8 and mechanistic9 ventures, we note the parallels between protonation and halogenation of alkenes. The field would be well served by a scale like the familiar pKa tables, to classify and order the halenium ion affinities of various functional groups. Herein we present such a scale, computationally derived but validated by experiments, that quantitatively predicts not only the interactions of halenium ions with various Lewis base acceptors but also the processes that ensue after the capture of halenium ion by a substrate molecule.
We begin by addressing a question that is fundamental to all alkene halogenation reactions: How easily does a given alkene capture a halenium ion? More specifically, how can we quantify the propensity of an alkene to undergo halogenation using reliable and predictable means? We approached this question using theoretical means by comparing protonation and halogenation chemistry. While there is ample literature precedent for determination of proton affinities, an analogous approach with halenium ions (perhaps surprisingly) finds no such precedence. In analogy to ab initio derived proton affinities (PA),10 we have developed a scale of HalA with the aid of experiments and theoretical calculations. We have developed the HalA scale with the aid of experiments and theoretical calculations. For a wide range of halenium acceptors, HalA serves as a ruler to predict the relative ease of electrophilic halogenation.
We define the HalA value for a given Lewis base (:LB) as the DFT11 calculated energy change upon attachment of a halenium ion (X+). The B3LYP/6-31G* theoretical method was chosen largely based on its popularity, low cost, wide availability, and success in relation to our experimental studies. Likewise, the use of the SM8 solvation model11f,12 and LANL2DZ pseudopotential11h and basis set for iodine were default but effective choices available in the Spartan11i software package.
The acceptor fragment may be neutral or anionic (i.e., the X–LB complex is cationic or neutral), leading to two distinct cases:
The HalA values in kcal/mol are derived at T = 298.15 K (unless noted otherwise) as in eqs 1 and 2:
| 1 |
| 2 |
where ΔE(elec) = E(electronic)(X–LB adduct) – [E(electronic)(:LB) + E(electronic) (X+)]; zero point energy change ΔZPE = ZPE(X–LB adduct) – ZPE(:LB); ΔE′(vib) = E′(vib)(X–LB adduct) – E′(vib)(:LB), i.e., difference in temperature dependence of vibrational energy; N is Avogadro’s number, h is Planck’s constant, and νi is the ith vibrational frequency. Finally, the (5/2)RT quantity accounts for translational degrees of freedom and the ideal gas value for the change from two particles to one. The energy used for the free halenium ion is the value calculated for its (6-electron, s2p4) triplet ground state.13
Table 1 shows an illustrative set of HalA values for styrenes, taken from this study’s list of over 500 halenium acceptors with various functional groups (see Supporting Information); for reference, Table 1 lists absolute HalA values for styrene. As expected, for a given substrate, HalA drops with decreasing halogen electronegativity. Likewise, methyl substitution on styrene (entries 2–4) increases HalA via stabilization of the haloalkylcarbenium ion. Though these variations are qualitatively predictable, the HalA computations place them on a quantitative basis. Interestingly, an insightful and counterintuitive result was obtained by comparison of HalA (F) of β-methylstyrenes (entries 3 and 4, Table 1). In agreement with reports by Sauers14 and our previous findings for chlorenium ion,9a the HalA predictions for bromenium and iodenium affinities of (E)- and (Z)-β-methylstyrene also find an open carbocation minimum (see Figure 1). Evidence for this comes from the lengthened C(benzylic)–X bond distance computed for Br and I (as seen earlier for Cl), and a relatively short length for the C(homobenzylic)–X bond, which is close to the expected normal bond length for each system. Although the optimized structures do find the C–X bond aligned with the benzylic cation’s empty 2p orbital, the <3.5 kcal/mol barrier to −C(CH3)HX rotation reflects no significant preference for bridging.9a,14 This result explains the higher HalA (Cl, Br, and I) values for the E-isomer as compared to its sterically congested Z-analogue (where such an alignment of C–X bond is skewed due to allylic strain).
Table 1. Theoretically Estimated Relative HalA (Gas Phase) and PA for Some Illustrative Styrylic Systemsc.
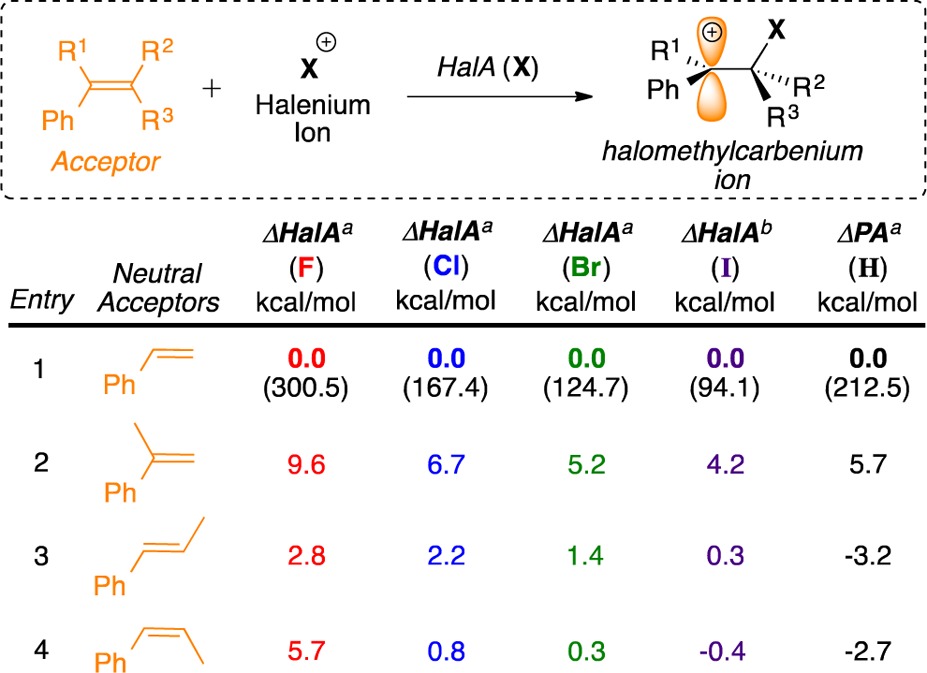
B3LYP/6-31G*.
B3LYP/6-31G*/LANL2DZ.
Numbers in parentheses are absolute HalA and PA values for styrene in kcal/mol.
Figure 1.
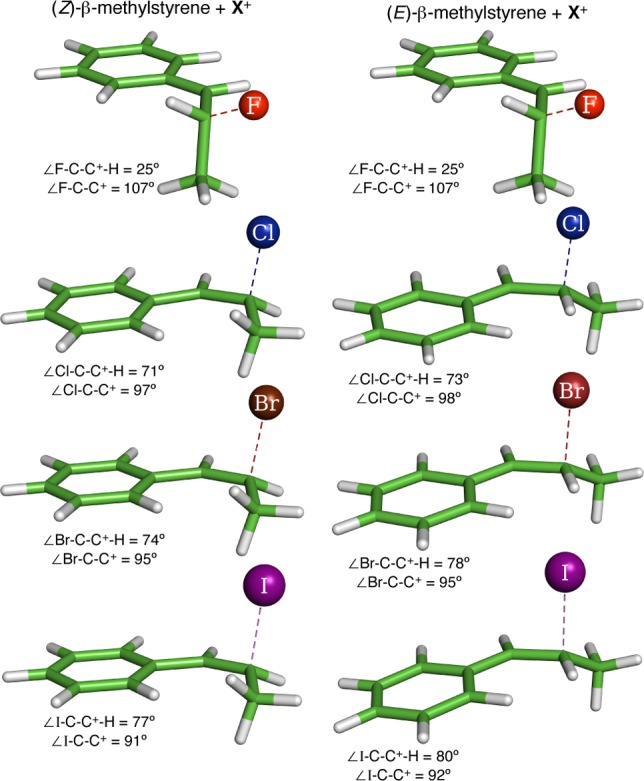
Geometry minimized structures of (E)- and (Z)-β-methylstyrene upon treatment with different halenium ions. B3LYP/6-31G* for HalA (F, Cl, and Br). B3LYP/6-31G*/LANL2DZ for HalA (I).
As often occurs among halogens, fluorine is unique in its behavior. The HalA (F) computations for the β-methylstyrenes find affinities of 303.3 kcal/mol for the E-isomer and 306.2 kcal/mol for the Z-isomer (ΔHalA = 2.9 kcal/mol in favor of Z-isomer). A closer inspection of the minimized models reveals that, regardless of initial geometry, (E)- and (Z)-β-methylstyrene gives the same 1-fluoromethyl-carbenium ion with the C–F bond orthogonal to the benzylic cation’s empty 2p orbital. This stark difference reflects the reluctancy of the C–F bond to donate into the adjacent empty 2p orbital (owing to the fluorine atom’s high electronegativity), which in turn forces the C–C bond to be aligned with the empty orbital. Thus, the difference in HalA (F) values for E- and Z-isomers arises only from their differing allylic strain that is relieved upon formation of the common fluoromethyl carbenium ion intermediate.15 As the halogens’ electronegativity decreases from F to I, the tendency of the C–X bond to align with the empty 2p orbital rises as shown by the calculated (∠X–C–C+) bond angles. Thus, though the charge (+1) is the same for each intermediate, the HalA analyses of these four styrenic acceptors serve to highlight the variations among the interaction modes of halenium ions.
Comparing halenium ions as electrophiles suggests reference to the most familiar electrophile: the proton. Is HalA simply a rehash of the familiar proton affinity (PA) scale? Clearly not. Very different conformational preferences are seen for analogous acceptor–proton vs acceptor–halenium ion complexes, with the energetic orderings even inverted in some cases. For instance, as seen in Table 1, and earlier pointed out by Kafafi and Liebman,16 the (E) and (Z) analogues of β-methylstyrene (entries 3 and 4) have lower PA values than styrene itself. Evidently, the stabilizing electron donation by the β-methyl groups is more significant in the starting olefin than in the cation formed by proton capture. In contrast, all three methylstyrene isomers show higher HalA values than the parent styrene. Thus, the PA scale is distinct from HalA. Furthermore, as Table 1 shows, significant differences are seen among the halogens, due to variations in their size, electronegativity, and polarizability. More HalA trends based on ring strain, orbital ovelap contributions, donation effects, and other secondary interactions are shown in the Supporting Information (S6–S11).
In order to determine how well experimental results agree with the theoretically predicted HalA values, we initiated studies on N-halopyridinium salts, simple systems where halenium affinity can be easily quantified via 1H NMR. These salts have been well characterized and reported as potent halenium sources. The corresponding bromenium and iodenium salts are stable even at room temperature,17 and the chlorenium analogues, though unstable to isolation, have been characterized and observed via ESI studies.18 Substituted pyridines (1) were chosen as halenium acceptors for preliminary 1H NMR studies (see Figure 2). Treatment of 1 (acceptor) with chlorenium sources (Cl+ donors) forms the chloropyridinium ion (1-Cl) with expulsion of the donor counterion. For such a transfer of halenium ion to ensue, the HalA (Cl) of 1 should be higher than that of the corresponding donor counterion.
Figure 2.
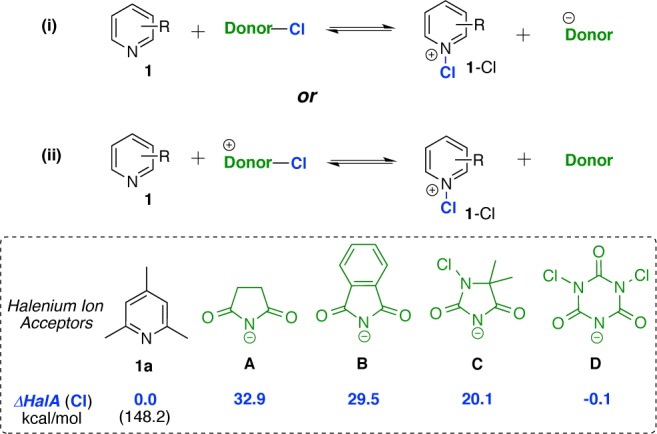
Theoretically estimated relativeHalA values for pyridine 1a in comparison to the counterions of commonly employed chlorenium sources (B3LYP/6-31G*/SM8 acetone).
Figure 2 depicts the theoretical HalA (Cl) values for counterions of commonly employed chlorenium sources in comparison to 1a. Based on these estimates, the equilibrium for reaction (i) should favor the reactant side as the anions A–C have a significantly higher HalA than 1a. Furthermore, based on the calculated HalA for the least nucleophilic anion D, the chlorenium ion is probably shared (but not completely transferred) between 1a and anion D. To verify these predictions, 1a was treated with the corresponding halenium sources and the interactions were assessed via the downfield 1H NMR shift of the C3-H resonance.
As a reference point to assess the extent of halenium ion transfer, we referred to the observed chemical shift difference between the free base-1a (C3-H at 6.82 ppm) and its protonated salt 1a-H19 (C3-H at 7.66 ppm) in acetone-d6 (Δppm = 0.84) at room temperature. As with the protonation, to ensure the formation of 1a-Cl, chlorodiethylsulfonium antimony(V) hexachloride (CDSC)20 proved to be the most effective chlorenium ion source. The resulting chloropyridinium 1a-Cl displayed a downfield shift of C3-H at 7.69 ppm (Δppm = 0.87). Figure 3 shows the experimental 1H NMR spectra of 1a upon treatment with stoichiometric amounts of different chlorenium ion sources (spectra b–f). Spectra b–d clearly show that pyridine 1a with a HalA (Cl) = 148.2 kcal/mol is incapable of abstracting a chlorenium ion from commonly employed imide based chlorenium donors (such as NCS, DCDMH, and NCP) whose counterions have higher HalA(Cl) values (i.e., >148.2 kcal/mol). Even TCCA, the most potent chlorenium source among the imide based chlorenium donors, results in only a 0.1 ppm downfield shift of C3-H of 1a indicating halogen bonding (tight van der Waals complex) rather than complete chlorenium ion transfer (see spectrum e).21 These experimental results compliment the theoretical HalA predictions.
Figure 3.
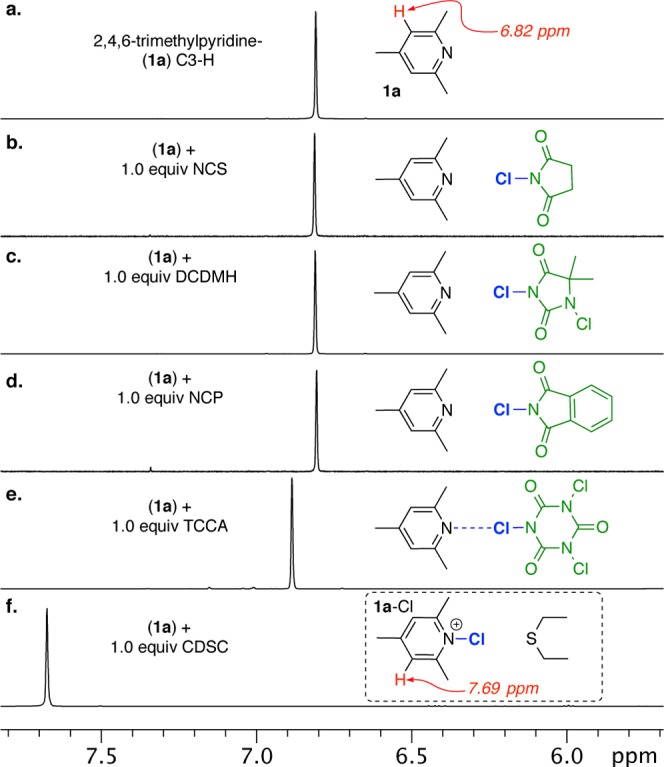
Overlay of 1H NMR (500 MHz, acetone-d6) spectra (a–f). Spectrum a represents a section of 1H NMR displaying the C3-H of 1a, whereas overlay of spectra b–f shows the effects of treatment of 1a with different chlorenium sources: NCS (N-chlorosuccinimide), DCDMH (1,3-dichloro-5,5-dimethylhydantoin), NCP (N-chlorophthalimide), TCCA (trichloroisocyanuric acid), CDSC (chlorodiethylsulfonium antimony(VI) chloride).
Since reaction of neutral species to form ionic products would always be energetically uphill in organic solvent, transfer of chlorenium ion to 1a calls for the use of a cationic chlorenium reagent (see reaction (ii), Figure 2). With this aim, CDSC, whose conjugate leaving group has a lower HalA (Cl) value (161.3 kcal/mol, gas phase) than 1a, was employed.22 Addition of 1.0 equiv of this reagent to 1a effects the formation of 1a-Cl as indicated by the 0.9 ppm downfield shift of C3-H (spectrum f), after a complete transfer of the chlorenium ion.23
Apart from 1a, several substituted pyridines with varying electronic and steric profiles were subjected to similar analysis using fluorenium, chlorenium, bromenium, and iodenium sources to further evaluate HalA estimations (see Supporting Information). In all these cases the formation of a 1:1 complex of Lewis base:halenium ion was confirmed via 1H NMR analysis; the chemical shift of C3-H of 1a-Cl (spectrum f) remained unchanged upon addition of superstoichiometric quantities of halenium source (see plot in Figure 4, dashed box).
Figure 4.
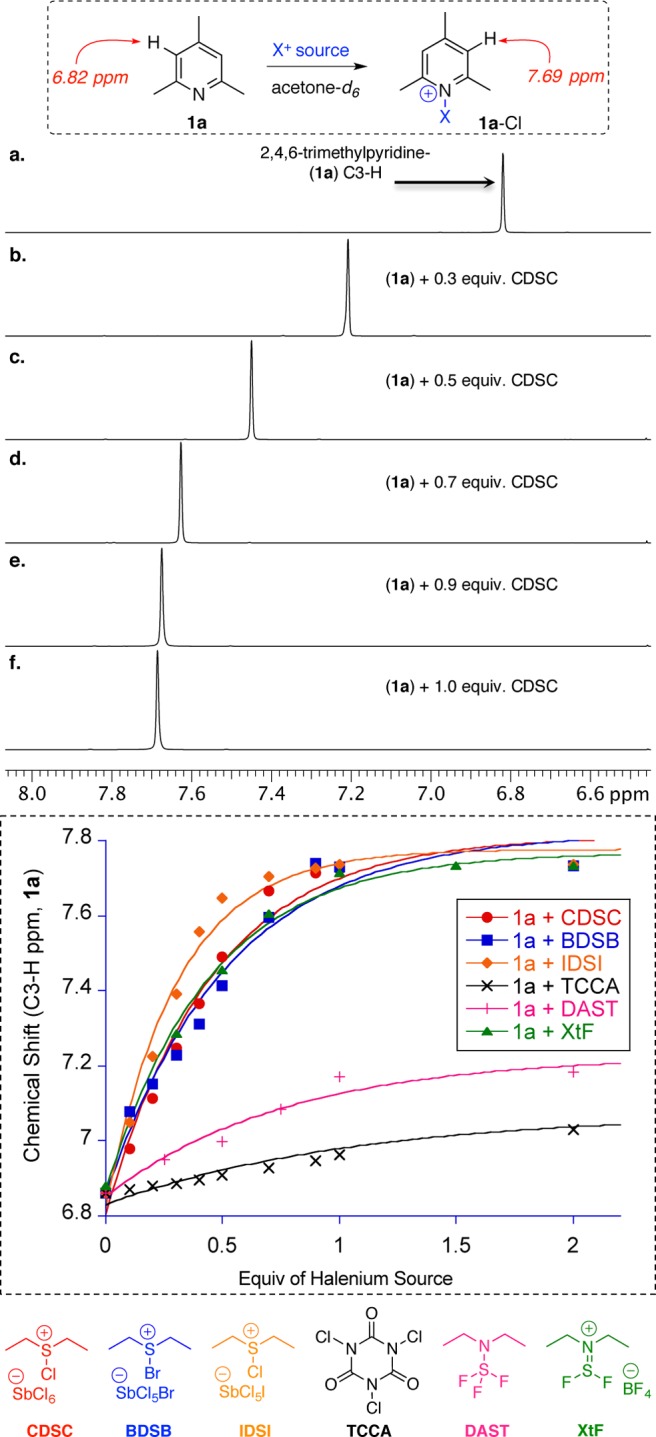
Spectra a–f depicts 1H NMR data for titration of 1a with CDSC. The plot below shows change in chemical shift of C3-H of 1a upon titration with different halenium sources. CDSC (chlorodiethylsulfonium antimony(VI) chloride), BDSB (bromodiethylsulfonium antimony(VI) halide), IDSI (iododiethylsulfonium antimony(VI) halide), XtF (Xtalfluor-E).
To rigorously validate HalA assessments on a quantitative scale, we resorted to equilibrium studies of chloropyridinium salts. Quantitative HalA determination via 1H NMR analyses was complicated by the tendency of N-halopyridinium ion 1a-Cl to undergo dimerization24 with the free base 1a when subjected to substoichiometric amounts of halenium source.17a Treatment of 1a with 0.5 equiv of BDSB (or IDSI) in CDCl3 displayed a downfield shift of C3-H to 7.2 ppm. The extent of this shift is in accordance with the reported halogenated dimers of 1a.17d Figure 4 displays the plot for the observed 1H NMR chemical shift (average of 1a and 1a-X) for C3-H when 1a was titrated with different halenium sources (see plot). As seen from the overlay of 1H NMR spectra a–f (Figure 4), due to rapid exchange and possible dimerization, 1a and 1a-Cl could not be observed as individual species on the NMR time scale. To overcome this limitation, pyridines 1b and 1c were used for equilibrium studies of 1:1 complexes with CDSC as a chlorenium source. The bulky t-butyl substituents in the ortho positions efficiently inhibit the dimerization as well as rapid intermolecular transfer of halenium ions. As a consequence, the chlorinated pyridinium (1c-Cl) and the free base (1c) were observed as distinct species at −30 °C via 1H NMR under substoichiometric amounts of the halogenating reagent (see Figure 5a).
Figure 5.
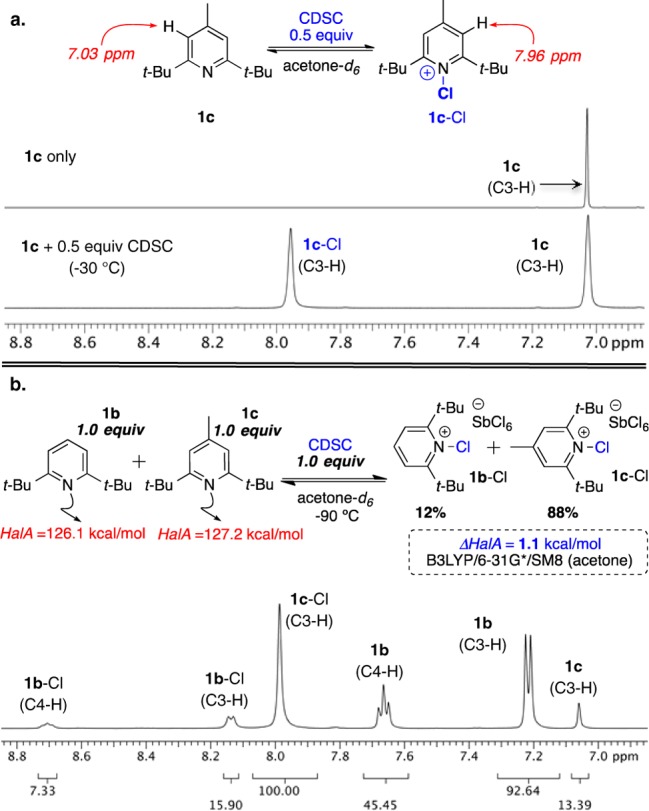
(a) Partial chlorination of 1c using 0.5 equiv of CDSC leads to distinctly observable species 1c and 1c-Cl at −30 °C by 1H NMR (500 MHz). (b) Competition study between 1b and 1c in acetone-d6 at −90 °C as quantified by 1H NMR.
Figure 5b displays a competition experiment between the two pyridines 1b and 1c, which have similar electronic and steric profiles. As anticipated, the gas phase HalA values predict 1c to have a slightly increased Lewis basicity as a result of the 4-Me substituent (ΔHalA = 2.6 kcal/mol). For a better quantitative representation, an SM8 model12 for simulated acetone was applied which attenuated the difference in their HalA values to 1.1 kcal/mol (Figure 5b). When an equimolar mixture of 1b and 1c was treated with 1.0 equiv of CDSC, an equilibrium mixture of 1c-Cl and 1b-Cl in ∼7:1 ratio was observed by 1H NMR. The experimental result is in good agreement with the theoretical HalA predictions (ΔHalA = 1.1 kcal/mol; predicting a 7:1 ratio). This study not only validates quantification via HalA but also signifies its importance in reliably predicting the outcome of reactions involving subtle steric and electronic changes.
In NMR studies of equilibria/competitions for halenium ions between pyridine 1b and a series of substituted pyridines (1a–g, Figure 6), the trend of relative halenium affinities (theoretical) was found to parallel the equilibrium ratios of the competing Lewis bases (experimental), as elucidated by 1H NMR.
Figure 6.
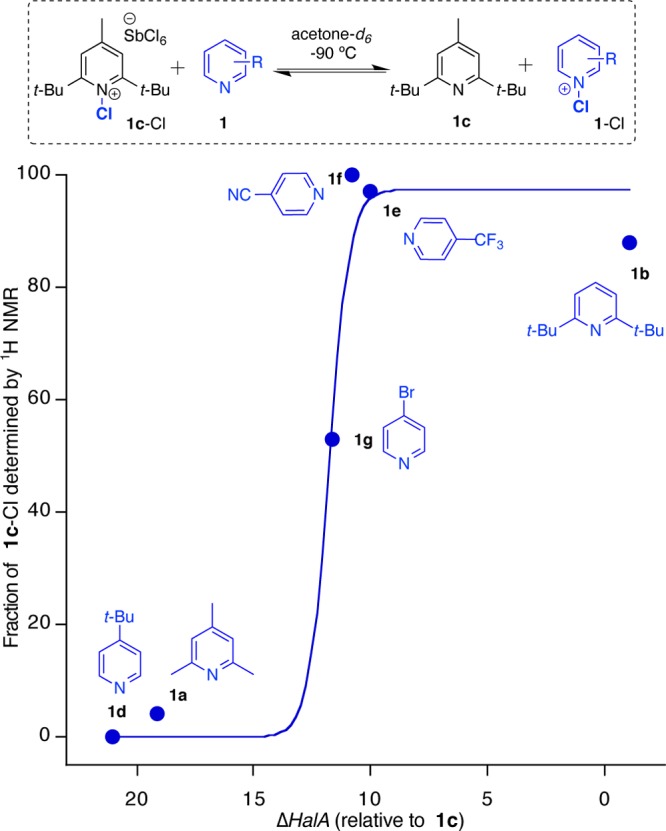
Comparison of ΔHalA (Cl) (B3LYP/6-31G*/SM8) with experimental results of equilibrium studies between 1c-Cl (prepared in situ using 1.0 equiv of CDSC) in the presence of 1.0 equiv of pyridines 1b–g. The sigmoidal curve fit (R2 = 0.974) is derived from the Henderson–Hasselbalch equation.
To explore the practical use of HalA, we applied it as a mechanistic tool to predict the stereo-, regio-, and chemoselectivity of electrophilic alkene halogenations. The following examples were chosen for proof-of-principle studies. As shown in Figure 7, a Friedel–Crafts reaction of electron rich arenes is initiated via chlorenium ion activation of a pendant alkene. Substrate 2 with a 4-MeO-C6H4 substituent cleanly affords the desired cyclized product (±)-3 upon treatment with DCDMH (Figure 7a). Under identical conditions, substrate 4 with the significantly more electron rich aromatic ring undergoes exclusively electrophilic aromatic substitution yielding product 5. This chemoselectivity is readily predicted by HalA values. The nucleophilic aryl carbon of substrate 2 is predicted to have ∼14 kcal/mol lower halenium affinity than the olefin (HalA values were calculated on truncated systems, see 6 vs 7, Figure 7b). The aryl ring of 4 on the other hand has about 2.2 kcal/mol higher halenium affinity than the olefin (see 6 vs 8, Figure 7b) despite the steric demand associated with the formation of the pentasubstituted aryl core of 5. Hence, the observed chemoselectivity may not be easily predicted or quantified. The HalA values additionally serve to quantify this selectivity and thereby enable such predictions with a greater level of confidence.
Figure 7.
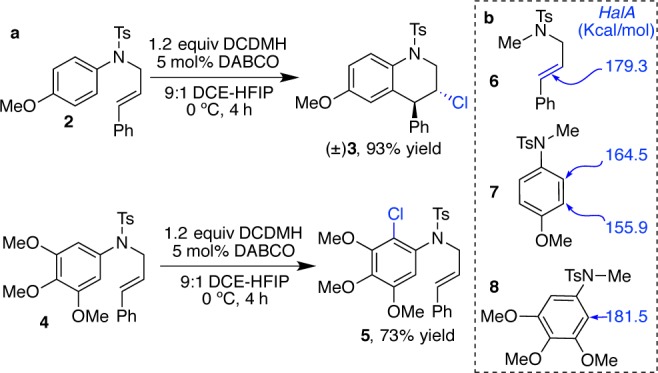
(a) Example of two reactions under identical conditions with different chemoselectivity. (b) HalA (Cl) values (B3LYP/6-31G*) for the reactive components of 2 and 4.
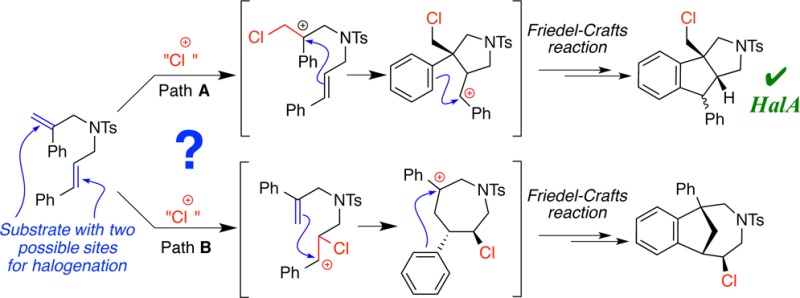
The next reaction represents an example where chemoselectivity prediction (either qualitative or quantitative) may be much less intuitive, with guesswork easily leading to the incorrect prediction. The treatment of diene 9 with a chlorenium source affords 10 as the major product (Figure 8a).25 The chlorination of the 1,1-disubstituted olefin followed by a nucleophilic attack on the resulting chlorocarbenium ion by the trans-olefin is necessary for the formation of the observed product. The HalA (Cl) values for α- and β-methylstyrene (13 and 14) reveal a 4.5 kcal/mol higher halenium affinity for α-methylstyrene (capable of forming a 3° carbenium). Interestingly, inclusion of the electron withdrawing N-sulfonyl group to better represent the structure of diene 9 greatly attenuates the difference in HalA values for the truncated structures 15 and 16 (ΔHalA = 0.2 kcal/mol). Notably, the counterintuitive increase in individual HalA (Cl) values with the introduction of the N-methyl sulfonamide results from anchimeric assistance of the neighboring sulfonamide that overrides any inductively deactivating effects (Figure 8c). The S–O bond of the N-sulfonyl group assists the stabilization of the chloromethyl carbenium ion generated from α- and β-methylstyrene. This extended delocalization (internal solvation) in the cationic intermediates 15-Cl and 16-Cl is responsible for the higher halenium affinity of 15 and 16 over 13 and 14, which lack the N-sulfonyl tether (also see Figure 1). From a practical viewpoint, this small difference suggests negligible selectivity between the two olefinic moieties in these “truncated” models. However, a detailed Boltzmann gated conformational search of the diene substrate 9 (for calculations, the N-Ms variant was employed instead of N-Ts), followed by HalA evaluation for the lowest energy conformers of the two possible chlorocarbenium ions, predicts the experimental outcome to be favored by 1.6 kcal/mol over chlorination of the trans-β-methyl styryl moiety. Moreover, the anichimeric assistance of the olefinic moiety of trans-β-methyl styryl substituent, as shown in intermediate 11 (Figure 8a), is energetically preferred over the anchimeric assistance of the S–O bond from the N-Ms group by 2.3 kcal/mol. This example underscores the utility of the HalA method for the accurate prediction of chemoselectivity in alkene halogenation reactions. The experimental confirmation of the theoretically predicted chemoselectivity suggests that even small energetic preferences (≤2 kcal/mol) can be reliably distinguished with HalA values.
Figure 8.
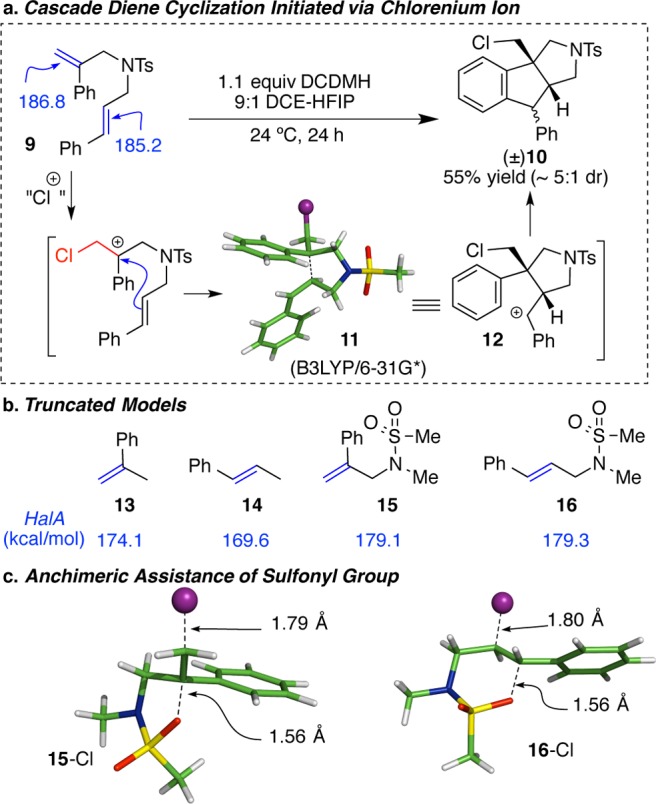
(a) Chlorocyclization of diene 9 to 10 is predicted by the higher HalA (Cl) of the 1,1-disubstituted olefin. Intermediate 11 is the computationally assessed outcome upon geometry minimization of 9 with chlorenium ion (B3LYP/6-31G*). (b) HalA (Cl) values for the reactive components in the cyclization of 9, illustrating the effect of N-Ms substitution. (c) Anchimeric assistance of the sulfonyl group toward stabilization of the chlorocarbenium ion 15-Cl and 16-Cl as predicted by HalA calculations.
Qualitative reactivity ranking of potential halogen attack sites using HalA computations can be made using the HalA table (Supporting Information) whereas quantitative comparison of affinities can be established by computing the full structures using appropriate solvation models. Figure 9 provides the HalA (Cl) scale for various functional groups to allow a qualitative comparison. As shown in Figure 9, functional groups (acceptors) that feature extended conjugation with the substituents attached span a larger range of HalA. For instance, alkenes, alkynes, amines, aromatic compounds, etc., whose HOMO can be easily perturbed by their substituents, display a wider range of HalA values in comparison to epoxides or alcohols where the attached substituents can only exert a weaker inductive effect. A comparison of halenium affinities can (a) facilitate rational selection of compatible nucleophiles (especially when the nucleophilic atom is embedded within motifs that have similar steric/electronic profiles); (b) account for the modulation of HalA values of alkenes by the anchimeric assistance of neighboring functionalities (this aspect underscores the importance of quantitatively evaluating HalA values on full structures rather than on truncated models; furthermore, subtle electronic perturbations leading to modulations of HalA values are also accounted for in the calculations); and (c) accurately predict chemoselectivity, aiding in the development of halenium initiated cascade/Domino reactions.
Figure 9.

HalA (Cl) scale based on theoretical estimates of over 500 chlorenium ion acceptors evaluated at the B3LYP/6-31G* (gas phase) level of theory.
These studies highlight the HalA scale’s use as a design/predictive tool in a field heretofore dependent on trial-and-error approaches for reaction discovery. Like the pKa scale, halenium affinity is a thermodynamic quantity and may not work to predict kinetically determined reaction outcomes. For such problems, more complete structural and energetic analyses of reaction paths and transition states26 would be necessary.
The scope of “affinity tools”, such as HalA, that broadly predict reaction chemoselectivities is certainly not limited exclusively to alkene halogenation reactions. Any electrophilic species (such as sulfenium, selenium, oxenium ions) capable of activating Lewis basic functionalities (such as olefins, alkynes, allenes, amines, etc.) can be efficiently parametrized on a similar scale to expedite the development of electrophilic functionalization reactions in general. These studies are ongoing and will be the subject of future disclosures.
Acknowledgments
Generous support was provided in part by the NIH (GM110525) and the NSF (CHE-1362812). The authors acknowledge the help of Edward Toma, Christopher Rahn, Aaron Schmidt, Thomas Chen, Ashley Burkin, and Meghan Richardson (undergraduate collaborators). We are indebted to Professor Daniel Jones (MSU) for his guidance on matters of mass spectrometry.
Supporting Information Available
Experimental details, HalA calculations, characterization data, DFT computational data, and MS Office Excel template for HalA calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Bose A.; Mal P. Tetrahedron Lett. 2014, 55, 2154. [Google Scholar]; b Chen J.; Zhou L. Synthesis 2014, 46, 586. [Google Scholar]; c de Mattos M. C. S. Curr. Org. Synth. 2013, 10, 819. [Google Scholar]; d Galligan M. J.; Akula R.; Ibrahim H. Org. Lett. 2014, 16, 600. [DOI] [PubMed] [Google Scholar]; e Getrey L.; Krieg T.; Hollmann F.; Schrader J.; Holtmann D. Green Chem. 2014, 16, 1104. [Google Scholar]; f Kamei T.; Ishibashi A.; Shimada T. Tetrahedron Lett. 2014, 55, 4245. [Google Scholar]; g Stodulski M.; Goetzinger A.; Kohlhepp S. V.; Gulder T. Chem. Commun. 2014, 50, 3435. [DOI] [PubMed] [Google Scholar]
- a Murphy C. D. J. Appl. Microbiol. 2003, 94, 539. [DOI] [PubMed] [Google Scholar]; b Neumann C. S.; Fujimori D. G.; Walsh C. T. Chem. Biol. 2008, 15, 99. [DOI] [PubMed] [Google Scholar]
- a Hiegel G. A.; Nalbandy M. Synth. Commun. 1992, 22, 1589. [Google Scholar]; b van Summeren R. P.; Romaniuk A.; Ijpeij E. G.; Alsters P. L. Catal. Sci. Technol. 2012, 2, 2052. [Google Scholar]
- a Hasty S. J.; Demchenko A. V. Chem. Heterocycl. Compd. 2012, 48, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Girard N.; Rousseau C.; Martin O. R. Tetrahedron Lett. 2003, 44, 8971. [Google Scholar]; c Fraser-Reid B.; Lopez C. J.; Gammon D. W.; Sels B. F. In Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Demchenko A. V., Ed.; Wiley: 2008; p 381. [Google Scholar]
- a In Halogen Bonding: Fundamentals and Applications; Metrangolo P., Resnati G., Eds.; Springer-Verlag Berlin: Berlin, 2008; Vol. 126, p 1. [Google Scholar]; b Chen G.; Ma S. Angew. Chem., Int. Ed. 2010, 49, 8306. [DOI] [PubMed] [Google Scholar]; c Castellanos A.; Fletcher S. P. Chem.—Eur. J. 2011, 17, 5766. [DOI] [PubMed] [Google Scholar]; d Denmark S. E.; Kuester W. E.; Burk M. T. Angew. Chem., Int. Ed. 2012, 51, 10938. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hennecke U. Chem.—Asian J. 2012, 7, 456. [DOI] [PubMed] [Google Scholar]; f Mendoza A.; Fananas F. J.; Rodriguez F. Curr. Org. Synth. 2013, 10, 384. [Google Scholar]; g Murai K.; Fujioka H. Heterocycles 2013, 87, 763. [Google Scholar]; h Tan C. K.; Yeung Y.-Y. Chem. Commun. 2013, 49, 7985. [DOI] [PubMed] [Google Scholar]; i Chen J.; Zhou L. Synthesis 2014, 46, 586. [Google Scholar]; j Zheng S.; Schienebeck C. M.; Zhang W.; Wang H.-Y.; Tang W. Asian J. Org. Chem. 2014, 3, 366. [Google Scholar]
- a Ojima I.Catalytic asymmetric synthesis, 2nd ed.; Wiley-VCH: New York, 2000. [Google Scholar]; b Sweeney J. B. Chem. Soc. Rev. 2002, 31, 247. [DOI] [PubMed] [Google Scholar]
- a Denmark S. E.; Burk M. T.; Hoover A. J. J. Am. Chem. Soc. 2010, 132, 1232. [DOI] [PubMed] [Google Scholar]; b McManus S. P.; Peterson P. E. Tetrahedron Lett. 1975, 2753. [Google Scholar]; c Ohta B. K.; Hough R. E.; Schubert J. W. Org. Lett. 2007, 9, 2317. [DOI] [PubMed] [Google Scholar]; d Olah G. A.; Bollinge J. M. J. Am. Chem. Soc. 1968, 90, 947. [Google Scholar]; e Olah G. A.; Bollinge J. M.; Brinich J. J. Am. Chem. Soc. 1968, 90, 6988. [Google Scholar]; f Olah G. A.; Peterson P. E. J. Am. Chem. Soc. 1968, 90, 4675. [Google Scholar]; g Olah G. A.; Westerma P. W.; Melby E. G.; Mo Y. K. J. Am. Chem. Soc. 1974, 96, 3565. [Google Scholar]; h Solling T. I.; Radom L. Int. J. Mass Spectrom. 1999, 185, 263. [Google Scholar]
- a Garzan A.; Jaganathan A.; Marzijarani N. S.; Yousefi R.; Whitehead D. C.; Jackson J. E.; Borhan B. Chem.—Eur. J. 2013, 19, 9015. [DOI] [PubMed] [Google Scholar]; b Jaganathan A.; Garzan A.; Whitehead D. C.; Staples R. J.; Borhan B. Angew. Chem., Int. Ed. 2011, 50, 2593. [DOI] [PubMed] [Google Scholar]; c Jaganathan A.; Staples R. J.; Borhan B. J. Am. Chem. Soc. 2013, 135, 14806. [DOI] [PubMed] [Google Scholar]; d Whitehead D. C.; Fhaner M.; Borhan B. Tetrahedron Lett. 2011, 52, 2288. [Google Scholar]; e Whitehead D. C.; Yousefi R.; Jaganathan A.; Borhan B. J. Am. Chem. Soc. 2010, 132, 3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yousefi R.; Ashtekar K. D.; Whitehead D. C.; Jackson J. E.; Borhan B. J. Am. Chem. Soc. 2013, 135, 14524. [DOI] [PubMed] [Google Scholar]; b Yousefi R.; Whitehead D. C.; Mueller J. M.; Staples R. J.; Borhan B. Org. Lett. 2011, 13, 608. [DOI] [PubMed] [Google Scholar]
- a Curtiss L. A.; Raghavachari K.; Pople J. A. J. Chem. Phys. 1993, 98, 1293. [Google Scholar]; b Delbene J. E. J. Phys. Chem. 1993, 97, 107. [Google Scholar]; c Smith B. J.; Radom L. J. Am. Chem. Soc. 1993, 115, 4885. [Google Scholar]
- a Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]; b Becke A. D. J. Chem. Phys. 1993, 98, 1372. [Google Scholar]; c Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]; d Hariharan P. C.; Pople J. A. Theor. Chim. Acta 1973, 28, 213. [Google Scholar]; e Hehre W. J.; Ditchfeld R.; Pople J. A. J. Chem. Phys. 1972, 56, 2257. [Google Scholar]; f Marenich A. V.; Olson R. M.; Kelly C. P.; Cramer C. J.; Truhlar D. G. J. Chem. Theory Comput. 2007, 3, 2011. [DOI] [PubMed] [Google Scholar]; g Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Phys. Chem. 1994, 98, 11623. [Google Scholar]; h Wadt W. R.; Hay P. J. J. Chem. Phys. 1985, 82, 284. [Google Scholar]; i Spartan ’10. Wavefunction, Inc.: Irvine, CA, 2010. [Google Scholar]
- Cramer C. J.; Truhlar D. G. Rev. Comput. Chem. 1995, 1. [Google Scholar]
- Li Y.; Wang X.; Jensen F.; Houk K. N.; Olah G. A. J. Am. Chem. Soc. 1990, 112, 3922. [Google Scholar]
- Haubenstock H.; Sauers R. R. Tetrahedron 2005, 61, 8358. [Google Scholar]
- The geometry minimized structures of (E)- and (Z)-β-methyl styrene at the B3LYP/6-31G* level of theory finds a 2.9 kcal/mol higher energy for the Z-isomer.
- Kafafi S. A.; Mautner M.; Liebman J. F. Struct. Chem. 1990, 1, 101. [Google Scholar]
- a Baruah S. K.; Baruah R. Asian J. Chem. 2004, 16, 688. [Google Scholar]; b Haque I.; Wood J. L. J. Mol. Struct. 1968, 2, 217. [Google Scholar]; c Homsi F.; Rousseau G. J. Org. Chem. 1999, 64, 81. [DOI] [PubMed] [Google Scholar]; d Homsi F.; Sylvie R.; Rousseau G. Org. Synth. 2000, 77, 206. [Google Scholar]; e Kleinberg J. J. Chem. Educ. 1946, 23, 559. [Google Scholar]; f Mendes C.; Renard S.; Rofoo M.; Roux M. C.; Rousseau G. Eur. J. Org. Chem. 2003, 463. [Google Scholar]; g Zingaro R. A.; Goodrich J. E.; Kleinberg J.; Vanderwerf C. A. J. Am. Chem. Soc. 1949, 71, 575. [Google Scholar]; h Zingaro R. A.; Tolberg W. E. J. Am. Chem. Soc. 1959, 81, 1353. [Google Scholar]
- Gozzo F. C.; Eberlin M. N. J. Mass Spectrom. 2001, 36, 1140. [DOI] [PubMed] [Google Scholar]
- For a fair comparison between the protonated and chlorinated pyridiniums, the same counteranion, SbCl6–, was employed.
- Snyder S. A.; Treitler D. S.; Brucks A. P. J. Am. Chem. Soc. 2010, 132, 14303. [DOI] [PubMed] [Google Scholar]
- It is noteworthy that 1.0 equiv of TCCA accounts for 3.0 equiv of active chlorenium ion. This NMR experiment was performed in the dark using acetone-d6 purged with O2 gas prior to TCCA addition, to avoid radical chlorination. Along with the described complex, products of benzylic chlorination (most likely via radical pathway) were observed by NMR when the same experiment was performed with the reaction mixture being exposed to light and open to atmosphere.
- B3LYP/6-31G*/SM8 is not compatible for the antimony(VI) chloride counterion associated with CDSC. Hence we resorted to comparison of gas phase HalA (Cl) values of diethyl sulfide and 1a. The gas phase HalA (Cl) value of 1a is 168.2 kcal/mol whereas diethyl sulfide has a HalA value of 161.3 kcal/mol.
- a We recognize the possibility that treatment of pyridines with halenium sources in acetone as a solvent might lead to protonation (rather than chlorination) of the pyridinium nitrogen atom yielding α-chloroacetone as a byproduct. This possibility was ruled out based on the following control experiments: (1) no change in chemical shift of the chloropyridiniums upon addition of K2CO3; (2) employing THF as a solvent to observe similar resonances of C3-H of the free base 1a and its chlorinated counterpart 1a-Cl; and (3) employing the in situ generated chloropyridinium 1a-Cl toward successfully initiating a chlorolactonization of 4-phenylpent-4-enoic acid (see Supporting Information for experimental details).; b Gil V. M. S.; Murrell J. N. Trans. Faraday Soc. 1964, 60, 248. [Google Scholar]
- Karim A.; Reitti M.; Carlsson A.-C. C.; Grafenstein J.; Erdelyi M. Chem. Sci. 2014, 5, 3226. [Google Scholar]
- Crude 1H NMR analysis indicated a 5:1 dr for 10. Although complete conversion of 9 was attained as judged by TLC and 1H NMR, the mass balance was accounted by a complex mixture of products, which were inseparable by chromatography. Identity of these products could not be assigned due to overlapping peaks in NMR. An analytically pure sample of 10 was obtained via preparative TLC.
- Crehuet R.; Bofill J. M. J. Chem. Phys. 2005, 122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.