

Abstract
Actin performs its myriad cellular functions by the growth and disassembly of its filamentous form. The hydrolysis of ATP in the actin filament has been shown to modulate properties of the filament, thus making it a pivotal regulator of the actin life cycle. Actin has evolved to selectively hydrolyze ATP in the filamentous form, F-actin, with an experimentally observed rate increase over the monomeric form, G-actin, of 4.3 × 104. The cause of this dramatic increase in rate is investigated in this paper using extensive QM/MM simulations of both G- and F-actin. To compute the free energy of hydrolysis in both systems, metadynamics is employed along two collective variables chosen to describe the reaction coordinates of hydrolysis. F-actin is modeled as a monomer with restraints applied to coarse-grained variables enforced to keep it in a filament-like conformation. The simulations reveal a barrier height reduction for ATP hydrolysis in F-actin as compared to G-actin of 8 ± 1 kcal/mol, in good agreement with the experimentally measured barrier height reduction of 7 ± 1 kcal/mol. The barrier height reduction is influenced by an enhanced rotational diffusion of water in F-actin as compared to G-actin and shorter water wires between Asp154 and the nucleophilic water in F-actin, leading to more rapid proton transport.
Introduction
The growth and disassembly of actin filaments allows cells to perform important functions such as motility and division.1 The hydrolysis of ATP modulates the dynamics of the actin filament.2 The typical pathway for actin filament growth is as follows: ATP-bound monomeric actin (G-actin) adds at the barbed end of the actin filament (F-actin), ATP is hydrolyzed in the filament, and then ADP-bound actin dissociates at the pointed end of the filament. ATP hydrolysis and inorganic phosphate (Pi) release happen in the filament and modulate the filament’s physical properties and the binding affinity of various actin-binding proteins.
Actin has evolved to achieve such dynamics by selectively hydrolyzing ATP in the filamentous form as opposed to the monomeric form. Experimentally, this rate difference has been investigated for over 25 years.3 The most recent experiments suggest a rate of ATP hydrolysis of 0.3 ± 0.1 s–1 in F-actin and 7 × 10–6 s–1 in G-actin.2,4 This rate difference equates to ATP hydrolysis (4.3 ± 2.2) × 104 times faster in the actin filament than in monomeric actin at 310 K. The cause of this dramatic increase remains unknown and is the focus of the present paper.
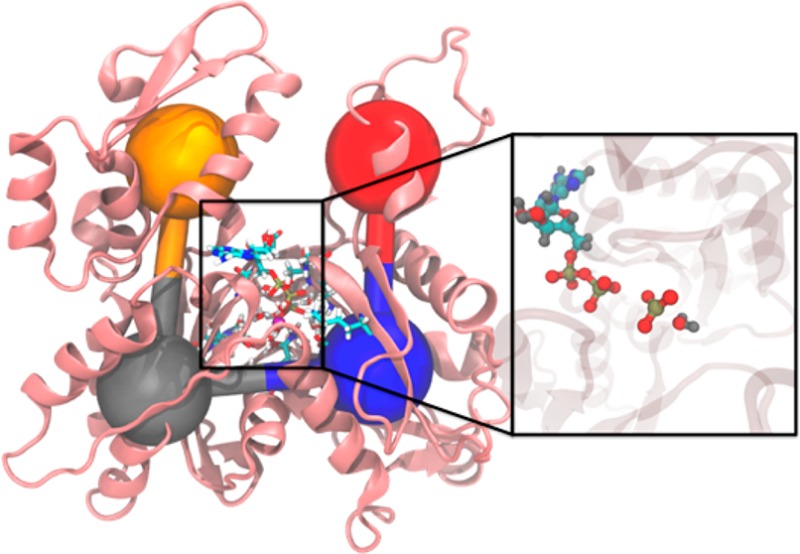
A wealth of structural data for G-actin is available from X-ray structures.2,5−7 The actin monomer consists of 375 amino acids that are parsed into four subdomains. The subdomains are arranged in a “U” shape around the centrally located nucleotide (Figure 1a). Upon polymerization, a flattening of the subdomain dihedral angle (SD2–SD1–SD3–SD4; see Figure 1b) has been observed experimentally and with simulation.8,9 Additional structural changes upon polymerization include small changes to nucleotide binding residues Gln137 and His161 which, upon mutation, can alter hydrolysis rates.10−12 Additionally, Asp11, Asp154, and Asp157 have all been implicated as possible bases in the hydrolysis reaction.12−14 While Asp157 has been ruled out as a catalytic base,13 the double mutation of Asp154 and Asp157 was found to be lethal in yeast cells.14
Figure 1.
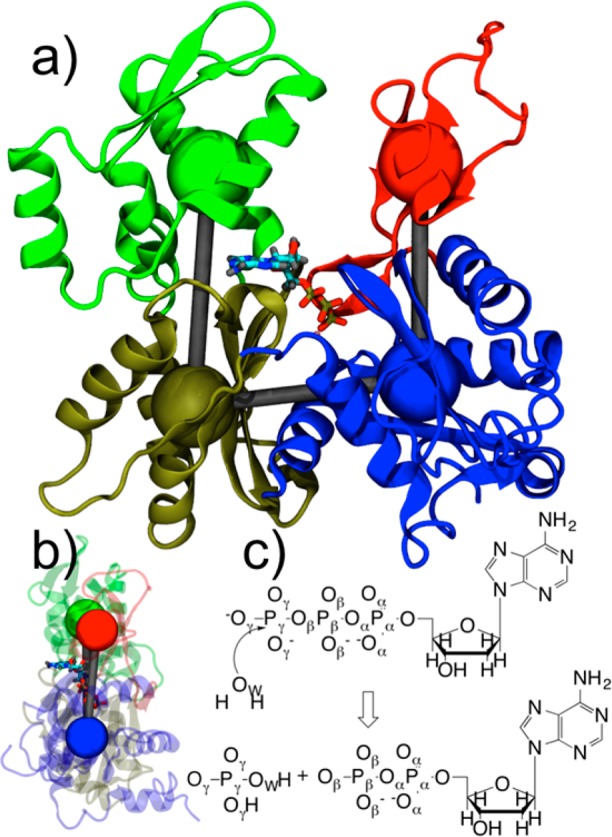
(a) Structure of G-actin with four subdomains colored differently and coarse-grained variables depicted as colored spheres. Subdomain (SD) 1 is in blue, SD2 in red, SD3 in gold, and SD4 in green. ATP and magnesium ion are also depicted. (b) Side view of actin with SD2–SD1–SD3–SD4 dihedral angle in flat, F-actin conformation. (c) ATP hydrolysis reaction with atom labels.
The mechanism of ATP hydrolysis has been studied theoretically in G-actin15,16 as well as other environments.15,17−21 ATP is hydrolyzed by cleavage of the Pγ–Oβ bond (see Figure 1c for atom labels) and the addition of a lytic water to Pγ. Most studies have found the reaction to occur via a dissociative pathway whereby the Pγ leaves prior to addition of the lytic water.15,16,22 Two recent studies, one in a protein environment20 and one in solution,23 have suggested a concerted mechanism for hydrolysis. Here, coarse-grain (CG) constrained QM/MM simulations of the hydrolysis reaction in F- and G-actin allow us to compare and contrast the mechanism in each species.
In addition to monitoring the formation and cleavage of Pγ–Oβ bonds, the proton transfer process is extremely important in hydrolysis. The lytic water must transfer a proton, either before or after nucleophilic addition, to an Oγ to form H2PO4–. This process has been suggested to go through one or two additional waters in aqueous solution.23 Carboxylate groups in the vicinity of the reaction are found to help catalyze this part of the reaction in protein environments.15,20 Asp154 in actin, in particular, is found to temporarily accept a proton during ATP hydrolysis in G-actin15 in support of experimental observations.14
While significant experimental and simulation work has shed some light on the mechanism of ATP hydrolysis in actin, the mechanism by which F-actin hydrolyzes ATP 4.3 × 104 faster than G-actin still remains unknown. To understand how polymerization accelerates ATP hydrolysis, the large-scale flattening of actin associated with polymerization (see Figure 1a and 1b) must be coupled to the quantum mechanics of chemical bond breaking and forming. Here we present a set of QM/MM simulations biased to reproduce the CG geometry observed in all-atom and CG simulations of actin and actin filaments.8,24,25 We show that the free energy barrier difference for hydrolysis predicted from these simulations agrees well with the experimentally observed rate differences. Insight into the structural changes that lead to the large rate difference is presented.
Methods
Starting Structures
The starting structures for both the G- and F-actin QM/MM systems were taken from equilibrated classical molecular dynamics simulations of the corresponding system. The details of these simulations have been published previously,8 and further details are provided in the Supporting Information (SI). For the F-actin system, a single monomer with a subdomain 2–1–3–4 dihedral angle of −0.55° (see Figure 1b) from the 13mer filament was chosen as an initial starting structure for the QM/MM simulations. This parameter was restrained by CG variables (for subdomain center-of-mass dihedral angle, see SI) in subsequent simulations with a harmonic force constant of 228.8 kcal/mol to ensure F-actin structure retention (see Figure1b). The G-actin starting structure has a subdomain dihedral angle (−27.09°) much smaller than that of F-actin. No restraints were applied to subsequent simulation of G-actin.
QM/MM Setup
The hydrolysis of ATP in actin was simulated using a QM/MM approach with density functional theory (DFT) for the level of QM. The quantum region was truncated to a methyl triphosphate, bound magnesium, surrounding waters, and 10 amino acids in the vicinity of the phosphate tail. These amino acids are Asp11, Gly13, Ser14, Gly15, Lys18, Gln137, Asp154, Gly156, Asp157, and His161 (see Figure 2). Nonsequential amino acids were truncated at the Cβ or Cγ carbon and capped with a hydrogen. The backbone atoms of sequential amino acids were included and hydrogen capped at terminal nitrogen and carbon positions. The QM region thus consisted of approximately 200 atoms, the exact number depending on the solvation environment. In the QM region, the DFT utilized the PBE functional with a TZV2P basis set. The magnesium ion was treated with GTH pseudopotentials.26,27 This setup was chosen due to its relatively low mean unsigned error for phosphate hydrolysis.28 All QM/MM simulations were performed in the CP2K software package.29
Figure 2.
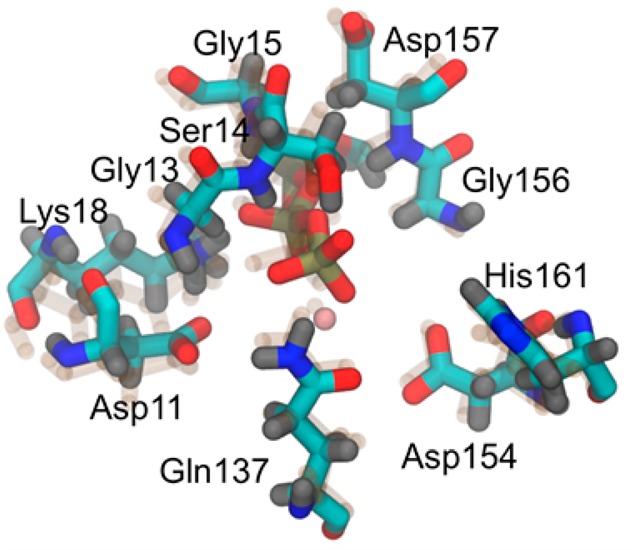
Depiction of the nucleotide binding cleft of G-actin and F-actin (transparent). The 10 amino acids in the QM region are all labeled.
The MM region was treated with the CHARMM27 force field with the omission of the CMAP correction which is not supported in CP2K.30 Periodic boundary conditions were employed in the NVT ensemble with a smoothed particle mesh Ewald treatment of the long-range electrostatics.31 For both F- and G-actin, the system consisted of 55 806 atoms total: 5831 protein atoms, 43 nucleotide atoms, 37 ions (one Mg2+, 35 K+, and 21 Cl–) and 16 625 water molecules. An integration time step of 0.5 fs was employed and total simulation time exceeded 500 ps. The temperature was maintained at 310 K with a Nose Hoover thermostat, and the dimensions of the box were 92 Å × 70 Å × 90 Å.
QM/MM Metadynamics Setup
Metadynamics was used to enhance the sampling along two collective variables chosen to represent the reaction coordinates of ATP hydrolysis.32 A hill height of 1.0 kcal/mol was chosen with a hill addition rate of 0.05 fs–1 and a width of 0.1 for each collective variable. Simulations were carried out for at least 170 ps each.
Two collective variables were chosen to model the ATP hydrolysis reaction. The coordination number between Pγ and Oβ (for atom labels, see Figure 1c) describes the making and breaking of the Pγ–Oβ bond and allows for recombination with any of the Oβ atoms. The coordination number between Pγ and both Oβ and QM water oxygens describes the associative pathway of hydrolysis in which the lytic water adds to the gamma phosphate. Coordination number is chosen instead of distance to allow any QM water to act as the lytic water. These collective variables are similar to the ones chosen by Marx et al.22 Additional explanation of these coordinates can be found in the SI.
Classical Molecular Dynamics
Classical MD simulations were carried out from the starting structures of G- and F-actin to monitor the dynamics of water in the nucleotide binding cleft. Simulations were carried out using the NAMD package33 in procedures very similar to those published previously.8 The systems were equilibrated for 20 ns followed by productions runs of 2 ns in which coordinates were written and analyzed every 200 fs. The integration time step used was 2 fs. The system sizes, box sizes, and temperature were the same as the QM/MM simulations.
Results and Discussion
Comparison of Starting Structures
As mentioned in Methods, the subdomain 2–1–3–4 dihedral angle is the primary large-scale structural difference between G- and F-actin (see Figure 1b). At the atomsitic scale, the conformations of the amino acids in the QM region differed between G- and F-actin. These are depicted in Figure 2. The residues with the largest differences between the two species are Ser14 and His161.
To quantify differences in the starting structures, we calculated the RMSD per residue (SI, Figure S2a). Deviations of up to 9 Å indicate that the starting structures for F- and G-actin differ significantly. If the crystal structure of G-actin is used as a baseline (SI, Figure S2b), F-actin shows deviations larger than that for G-actin, suggesting that these differences represent more than just thermal fluctuations. One of the two areas of largest deviation between G- and F-actin is residues 51–70, which corresponds to the small α helix in SD2 just to the N-terminal side of the D-loop. The second area with large deviation between G- and F-actin is the flap region of SD4 (residues 236–250). It should be noted that these changes are most likely a direct reflection of the change in dihedral angle because a global alignment was used to calculate RMSD.
Mechanism of ATP Hydrolysis in G- and F-Actin
The free energy of ATP hydrolysis in G- and F-actin was computed along the two collective variables using QM/MM metadynamics simulations as described in Methods. The two-dimensional free energy plots are shown in Figure 3. The x-axis of both plots gives the Pγ–Oβ coordination number which goes from a value of one in the ATP state to a value of zero in the ADP+Pi state. The y-axis of both plots gives the coordination number between Pγ and lytic water oxygens plus gamma oxygens. This value ranges from three (corresponding to coordination with the three gamma oxygens) in the ATP state to four in the ADP+Pi state with the addition of the lytic water oxygen. Values of greater than four are observed due to hydrogen bonding with additional waters in the vicinity of the gamma phosphate.
Figure 3.

Two-dimensional free energy surfaces calculated for ATP hydrolysis in (a) F-actin and (b) G-actin. These were computed using metadynamics in QM/MM simulations. The dark blue region in panel a and the purple region in panel b represent unsampled areas.
The free energy computed along the two collective variables for F-actin is depicted in Figure 3a. The basin in the lower right of the plot is the ATP basin (reactant), and the basin in the upper left is the ADP+Pi basin (product). The path the metadynamics simulation took, and the lowest free energy pathway, is neither dissociative nor associative as is evident by the transition state region in the center of the graph. Instead, the simulation has a concerted pathway for the hydrolysis of ATP in F-actin. The hydrolysis reaction in F-actin along this path, and prior to full release of Pi, is exergonic by 5 kcal/mol.
The mechanism of hydrolysis seen in G-actin is comparable to that of F-actin, and the same locally stable basins are observed. The energetics, however, are quite different (Figure 3b). While the reaction is still concerted, the transition state region in between the two basins is much narrower for G-actin than it is for F-actin. The reaction in G-actin is found to be more exergonic than in F-actin with the ADP+Pi basin being approximately 10 kcal/mol lower in free energy than the ATP basin, but the barrier is significantly higher (see next paragraph).
The one-dimensional minimum free energy paths computed from Figure 3 are depicted in Figure 4. Here, the x-axis simply depicts the reaction progress, with “0” representing the ATP state and “1” representing the ADP+Pi state. From this plot we can see that F-actin has a free energy barrier for hydrolysis (22 kcal/mol) significantly lower than that of G-actin (30 kcal/mol). The barrier height is 8 ± 1 kcal/mol lower in F-actin than in G-actin, which is in very good agreement with the experimentally observed 7 ± 1 kcal/mol barrier height difference at 310 K.2,4
Figure 4.
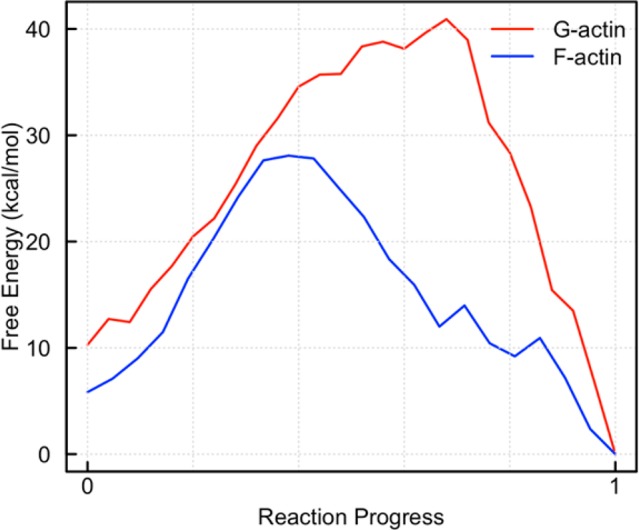
Minimum free energy path from 2D free energy surfaces for G- and F-actin. The x-axis denotes reaction progress, with “0” being ATP and “1” being ADP+Pi. The position along the x-axis at which the transition state occurs is meaningless, but the barrier heights and relative energies are meaningful. The error analysis for metadynamics is not straightforward, but the error at each position is on the order of the hill height, which is 1.0 kcal/mol.
Proton Transfer during Hydrolysis
While not biased in our metadynamics simulations, the proton transfer coordinate is important for the completion of the hydrolysis reaction. This coordinate was found to be strongly coupled to CV1 and CV2, thus supporting our choice of collective variables (see SI for additional discussion). As the lytic water adds to Pγ, the additional proton must leave the water oxygen and ultimately end up on an Oγ to create H2PO4–. The process by which this occurs is found to be different in G- and F-actin. In particular, the proton transfer from lytic water to gamma oxygen is found to be almost instantaneous in F-actin while being markedly slower in G-actin. The progress of this transfer can be monitored by looking at the coordination number of oxygens and hydrogens involved in the reaction. Specifically, the coordination number of the lytic water oxygen, gamma phosphate oxygens, and Asp154 oxygens with surrounding hydrogens was monitored (Figure 5).
Figure 5.
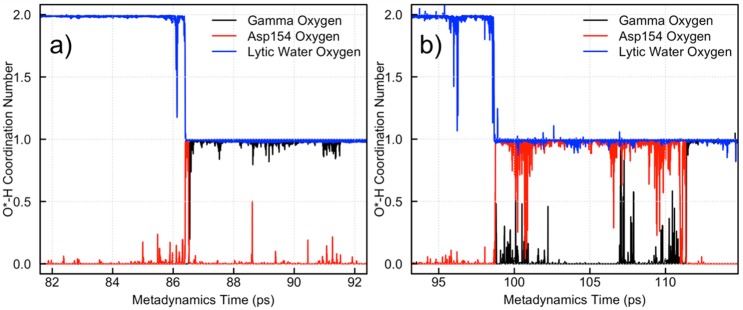
Monitoring of proton transport in (a) F-actin and (b) G-actin during ATP hydrolysis.
The F-actin hydrolysis event happens at around 86.5 ps of metadynamics simulation time indicated by the drop in protonation state of the lytic water from two to one (blue line in Figure 5a). The Asp154 carboxylate oxygen simultaneously accepts a proton, indicated by the coordination of the Asp154 oxygen (red line) going from zero to one. The carboxylic acid species is very short-lived (∼0.2 ps), with a gamma oxygen rapidly getting protonated to form H2PO4–. In contrast, the protonated Asp154 species in G-actin is relatively long-lived. In Figure 5b, a proton leaves the lytic water at approximately 99 ps, and this is directly coupled, again, with the addition of a proton to Asp154 (red curve going from zero to one in Figure 5b). The carboxylic acid species lives for over 10 ps prior to the transfer of the proton to a gamma oxygen. This behavior suggests that the flattening of actin upon polymerization lowers the energy barrier for hydrolysis by creating an environment in the nucleotide binding pocket that is more favorable for proton transport in water.
Water Dynamics in the ATP Binding Pocket
To investigate the structure of water in the nucleotide binding pocket further, various dynamic properties of water were investigated. Due to the short time-scale of the QM/MM simulations, classical MD was run from the QM/MM starting structures. The first two properties investigated are the O–H bond autocorrelation and the mean square displacement (MSD) as a function of time. The O–H bond autocorrelation provides insight into the rotational dynamics of water while the MSD gives a measure of the translational diffusion of water. The nucleotide binding pocket was defined as within 6 Å of the Pγ atom. This cutoff was chosen because it contained the first two solvation shells of water as seen in the gamma phosphorus to water oxygen g(r) (radial distribution function). The hydrolysis reaction involves not only waters in the first solvation shell (the lytic water) but also waters in the second solvation shell as they contribute to the proton transfer process.
The results of the MSD calculation for 2 ns classical MD trajectories of G- and F-actin are plotted in Figure 6a. The G-actin (red) and F-actin (blue) curves follow each other rather closely prior to 225 ps. The translational diffusion of pocket waters in G- and F-actin is therefore similar over the times of the QM/MM simulations performed in this study. The rotational diffusion, however, shows a significant difference between G- and F-actin (Figure 6b). The autocorrelation of the O–H bond vectors shows an initial rapid decline in both G-actin (red) and F-actin (blue) followed by a significant rebound of G-actin from an autocorrelation of 0.5 to 0.7 (Figure 6b). F-actin, on the other hand, plateaus at an autocorrelation of slightly less than 0.5. The inset in Figure 6b shows that differences in G- and F-actin are evident even at short (<20 ps) time scales. Because the waters in the nucleotide binding pocket of F-actin are more freely able to rotate than in G-actin they can more easily align into specific proton transport wires, hence lowering the free energy barrier of that process.
Figure 6.
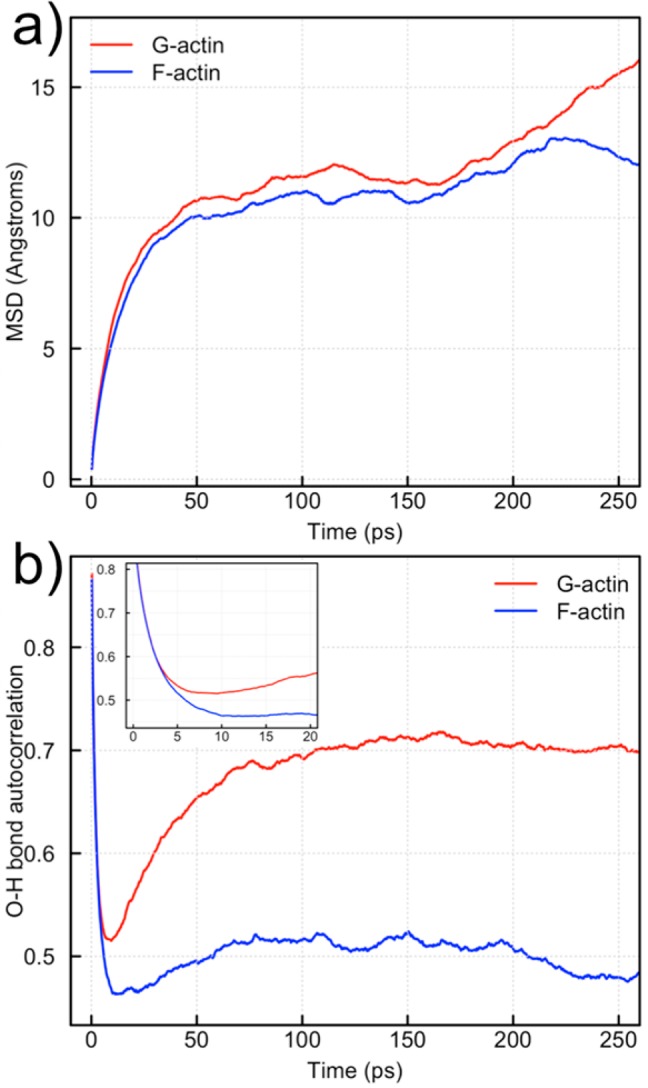
Quantification of water dynamics in the nucleotide binding pockets of G- and F-actin. (a) The mean squared displacement (MSD) of waters within 6 Å of the Pγ atom in both G- and F-actin. (b) The O–H bond autocorrelation for waters within 6 Å of the Pγ atom for both G- and F-actin. The inset is a blow-up of the time scales relevant to proton transport.
The types of water wires formed during the classical MD simulations was investigated using an analysis similar to Maupin et al.34 In the current work, water wires were defined by a continuous chain of water molecules with their oxygen atoms within 3.0 Å of one another. Of particular interest are the water wires connecting Asp154 to a putative lytic water. A histogram of these for both G- and F-actin is plotted in Figure 7. Snapshots of the most probable length water wires for F- and G-actin are plotted in Figure 8. F-actin has a larger population of wires of lengths three and four than does G-actin, suggesting that while Asp154 is not protonated for very long in F-actin it still plays an important role in the proton transfer process during ATP hydrolysis. The shorter length of the water wires to Asp154 in F-actin is also indicative of how a rapid return of the excess proton to the inorganic phosphate is achieved. The increased entropy cost of the longer water wires seen in G-actin could also be an explanation for the narrower transition path seen in the PMF of G-actin as compared to F-actin (Figure 3).
Figure 7.
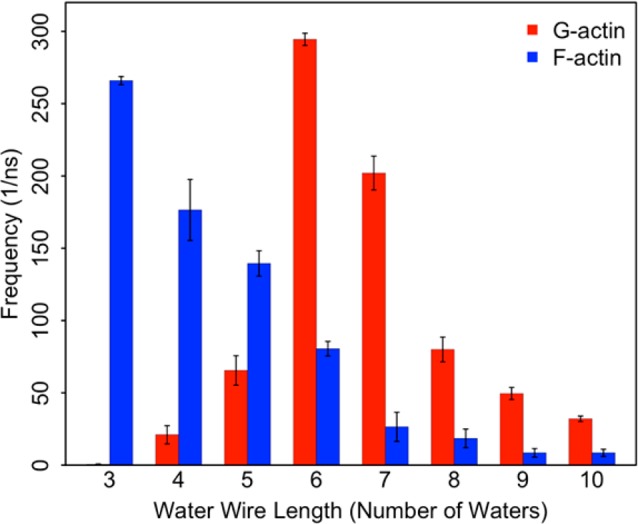
A histogram of the water wire length observed between a putative lytic water and Asp154 in both G- and F-actin. The error bars are computed using bootstrapping.
Figure 8.
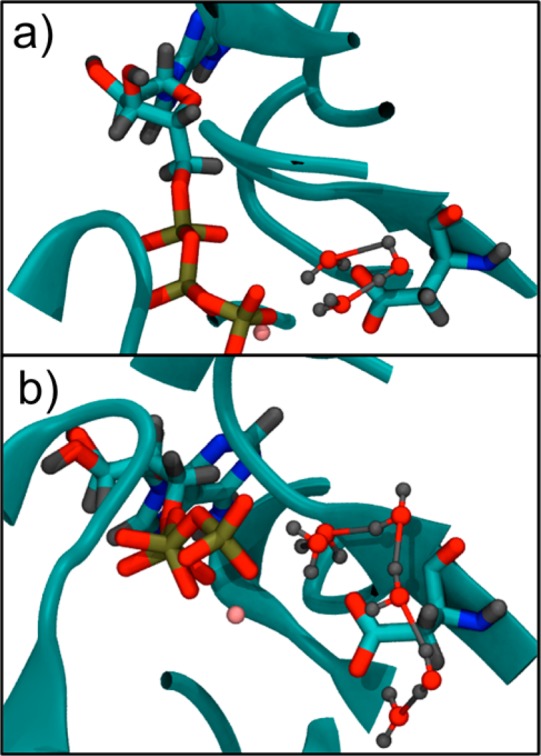
Snapshots of water wires leading to Asp154 from a putative lytic water from classical MD simulations of (a) F-actin and (b) G-actin. The F-actin snapshot (a) has a three-water wire, and the G-actin snapshot (b) has a six-water wire.
Conclusions
The hydrolysis of ATP in actin and actin filament growth are coupled in a multiscale fashion; the hydrolysis of ATP affects the ability of actin to polymerize, and the polymerization of actin affects the rate of ATP hydrolysis.3,35 The coupling of these length and time scales was investigated in this work with extensive CG structurally guided QM/MM simulations in G- and F-actin. Previous results from actin filament simulations were utilized to employ restraints on CG variables, allowing the replication of the filament environment with a reduced system size. The free energy of ATP hydrolysis was then computed for both G- and F-actin using metadynamics.
Hydrolysis in G- and F-actin occurred via a concerted mechanism in which the gamma phosphate dissociates concurrently with lytic water addition. The ∼8 kcal/mol difference in barrier height between G- and F-actin agrees well with the experimentally measured value of ∼7 kcal/mol at 310 K. Our simulations suggest that upon polymerization small changes in the positions of key amino acids in the active site promote the rearrangement of nearby water molecules, facilitating the formation of shorter water wires in F-actin to shuttle the proton from Asp154 to the phosphate of the now-dissociated gamma phosphate. The reduction in barrier height is thus attributed, at least in part, to the favorable proton transport environment seen in the F-actin nucleotide binding pocket.
Acknowledgments
Funding for this work was provided in part by the National Science Foundation through the Center for Multiscale Theory and Simulation (CHE-1136709). M.M. acknowledges funding from the National Institutes of Health under Ruth L. Kirschstein National Research Service Award (F32-GM101848) from the NIGMS. The authors acknowledge useful insight from Prof. Isaiah Sumner.
Supporting Information Available
Additional simulation details as well as RMSD analysis of the starting structures for G- and F-actin. Additional discussion of the errors associated with PBE and DFT methods employed in this study are discussed. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Carlier M. F. In Actin Based Motility: Cellular, Molecular and Physical Aspects; Carlier M. F., Ed.; Springer: Dordrecht, Netherlands, 2010. [Google Scholar]
- Rould M. A.; Wan Q.; Joel P. B.; Lowey S.; Trybus K. M. J. Biol. Chem. 2006, 281, 31909. [DOI] [PubMed] [Google Scholar]
- Pollard T. D. J. Cell Biol. 1986, 103, 2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L.; Pollard T. D. Biochemistry 2002, 41, 597. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Mannherz H. G.; Suck D.; Pai E. F.; Holmes K. C. Nature 1990, 347, 37. [DOI] [PubMed] [Google Scholar]
- Graceffa P.; Dominguez R. J. Biol. Chem. 2003, 278, 34172. [DOI] [PubMed] [Google Scholar]
- Graceffa P.; Dominguez R. Science 2001, 293, 708. [DOI] [PubMed] [Google Scholar]
- Saunders M. G.; Voth G. A. J. Mol. Biol. 2011, 413, 279. [DOI] [PubMed] [Google Scholar]
- Oda T.; Iwasa M.; Aihara T.; Maéda Y.; Narita A. Nature 2009, 457, 441. [DOI] [PubMed] [Google Scholar]
- Iwasa M.; Maeda K.; Narita A.; Maeda Y.; Oda T. J. Biol. Chem. 2008, 283, 21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobiev S.; Strokopytov B. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüler H. Biochim. Biophys. Acta 2001, 1549, 137. [DOI] [PubMed] [Google Scholar]
- Schüler H.; Korenbaum E.; Schutt C. E.; Lindberg U.; Karlsson R. Eur. J. Biochem. 1999, 265, 210. [DOI] [PubMed] [Google Scholar]
- Wertman K. F.; Drubin D. G.; Botstein D. Genetics 1992, 132, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman H.; Laino T.; Curioni A. J. Chem. Theory Comput. 2012, 8, 3373. [DOI] [PubMed] [Google Scholar]
- Akola J.; Jones R. O. J. Phys. Chem. B 2006, 110, 8121. [DOI] [PubMed] [Google Scholar]
- Akola J.; Jones R. O. J. Phys. Chem. B 2003, 107, 11774. [Google Scholar]
- Akola J.; Jones R. O. J. Phys. Chem. B 2006, 110, 8110. [DOI] [PubMed] [Google Scholar]
- Harrison C. B.; Schulten K. J. Chem. Theory Comput. 2012, 8, 2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath M. J.; Kuo I. F. W.; Hayashi S.; Takada S. J. Am. Chem. Soc. 2013, 135, 8908. [DOI] [PubMed] [Google Scholar]
- Prasad B. R.; Plotnikov N. V.; Warshel A. J. Phys. Chem. B 2013, 117, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaves R.; Mathias G.; Marx D. J. Am. Chem. Soc. 2012, 134, 6995. [DOI] [PubMed] [Google Scholar]
- Plotnikov N. V.; Prasad B. R.; Chakrabarty S.; Chu Z. T.; Warshel A. J. Phys. Chem. B 2013, 117, 12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Saunders M. G.; Voth G. A. Biophys. J. 2012, 103, 1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M. G.; Voth G. A. Structure 2012, 20, 641. [DOI] [PubMed] [Google Scholar]
- Goedecker S.; Teter M.; Hutter J. Phys. Rev. B 1996, 54, 1703. [DOI] [PubMed] [Google Scholar]
- Hartwigsen C.; Goedecker S.; Hutter J. Phys. Rev. B 1998, 58, 3641. [Google Scholar]
- Ribeiro A. n. J. M.; Ramos M. J.; Fernandes P. A. J. Chem. Theory Comput. 2010, 6, 2281. [DOI] [PubMed] [Google Scholar]
- http://cp2k.org; 2.4 ed., 2013.
- MacKerell A. D.; Bashford D.; Bellott; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. J. Phys. Chem. B 1998, 102, 3586. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103, 8577. [Google Scholar]
- Laio A.; Parrinello M. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. J. Comput. Chem. 2005, 26, 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupin C. M.; Saunders M. G.; Thorpe I. F.; McKenna R.; Silverman D. N.; Voth G. A. J. Am. Chem. Soc. 2008, 130, 11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I.; Vavylonis D.; Pollard T. D. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.