

Abstract
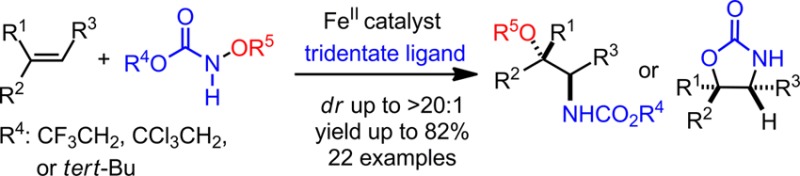
An iron-catalyzed diastereoselective intermolecular olefin amino-oxygenation reaction is reported, which proceeds via an iron-nitrenoid generated by the N–O bond cleavage of a functionalized hydroxylamine. In this reaction, a bench-stable hydroxylamine derivative is used as the amination reagent and oxidant. This method tolerates a range of synthetically valuable substrates that have been all incompatible with existing amino-oxygenation methods. It can also provide amino alcohol derivatives with regio- and stereochemical arrays complementary to known amino-oxygenation methods.
Selective olefin difunctionalization with an amino- and an oxygen-based group is an important transformation for organic synthesis because vicinal amino alcohol derivatives are widely present in synthetically valuable molecules. The osmium-based Sharpless aminohydroxylation continues to be a prevalent stereospecific method for olefin amino-oxygenation.1 This pioneering method has also inspired extensive efforts for the development of alternative approaches for a broader substrate scope and better regioselectivity.2,3 Among these approaches, nonprecious metal-catalyzed processes emerge with increasing interest: Chemler developed Cu-catalyzed methods for olefin amino-oxygenation and other difunctionalizations;2a−2d Yoon developed Cu- and Fe-catalyzed sulfonyl oxaziridine based methods.2e−2h Despite these and other excellent discoveries, new nonprecious metal-catalyzed olefin amino-oxygenation methods which achieve a broader substrate scope and regio- and stereoselectivity complementary to known methods are greatly desirable. In particular, the intermolecular olefin amino-oxygenation mediated by an iron nitrenoid has not been reported.
Unlike the N-atom transfer mediated by a rhodium nitrenoid,3,4 the iron nitrenoid mediated process is more prone to proceed through radical pathways.5 Therefore, new strategies are required to control an iron nitrenoid’s reactivity in an olefin amino-oxygenation reaction. We have previously discovered iron-catalyzed intramolecular olefin amino-oxygenation and amino-fluorination reactions (Scheme 1A).6 Our studies suggested that an iron nitrenoid is a possible intermediate in these stereoconvergent transformations and that the stereoselectivity can be modulated by N-based bidentate ligands.7
Scheme 1. Iron-Catalyzed Olefin Amino-Oxygenation with Functionalized Hydroxylamines.
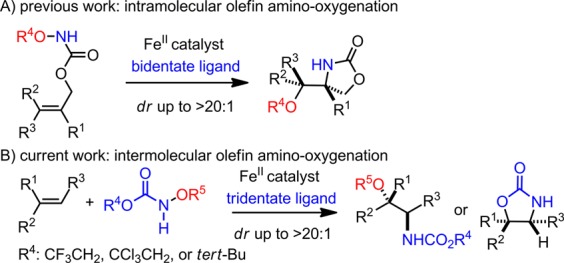
Our initial attempts to develop an intermolecular olefin amino-oxygenation with the catalyst previously identified to be effective for intramolecular amino-oxygenation failed due to the lack of reactivity. To modulate the reactivity of the iron nitrenoid to achieve a fine balance between reactivity, stability, and selectivity, we explored a variety of new amination reagents, iron catalysts, and ligands. Herein, we disclose an iron-catalyzed intermolecular olefin amino-oxygenation that proceeds through the N–O bond cleavage of a functionalized hydroxylamine. In this transformation, a bench-stable hydroxylamine derivative is applied as the amination reagent and oxidant (Scheme 1B).
This method has a few unique features that complement the existing iron-catalyzed olefin amino-oxygenation method with sulfonyl oxaziridines.2g,2h First, this method allows significant asymmetric induction with internal olefinic substrates, while the oxaziridine-based asymmetric approach is only effective for terminal olefins. Second, this method tolerates a broad range of synthetically valuable substrates, including allyl silanes, cyclopentadienes, enol ethers, glycals, indene, and silyl dienols, which are all incompatible with the iron-catalyzed olefin amino-oxygenation method with sulfonyl oxaziridines. Furthermore, this method can effectively afford amino alcohol derivatives with regio- and stereochemical arrays complementary to existing amino-oxygenation methods, especially osmium-based approaches. Therefore, we envision that this discovery will be a valuable tool for selective olefin amino-oxygenation.
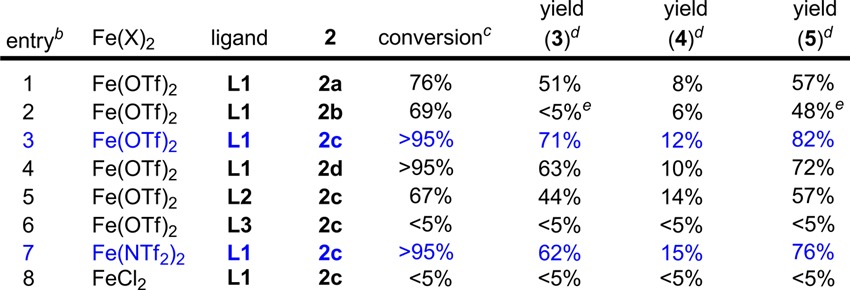
Styrene 1 was selected as a model substrate for catalyst discovery (Table 1). Our initial attempts with Fe(OTf)2–N,N′-bidentate ligands failed due to the lack of reactivity. Inspection of a range of ligands revealed that the N-based tridentate ligands are necessary for the proposed reactivity and that an achiral bisoxazoline PyBOX ligand L1 is uniquely effective:8 the Fe(OTf)2–L1 complex catalyzes the styrene amino-oxygenation with a range of functionalized hydroxyl amines (2a–2d, entries 1–4), affording both an alkoxyl oxazoline 3 and a protected amino alcohol 4 with good to excellent combined yields and regioselectivity complementary to the osmium-based methods.1a We also discovered that both 3 and 4 can be easily converted to oxazolidinone 5 with high yield through a same hydrolytic procedure. Furthermore, we noted that more electrophilic reagents lead to higher reactivity (entries 3–4 vs 1–2). Additionally, ligand screening revealed that an Fe(OTf)2–L2 complex8 is less reactive and an Fe(OTf)2–L3 complex is inactive (entries 5–6). Since we have observed a strong counterion effect in the intramolecular olefin amino-oxygenation,6 we also examined various iron salts and concluded that Fe(NTf2)2 is equally reactive compared to Fe(OTf)2 and FeCl2 is inactive (entries 7–8).
Table 1. Catalyst Discovery for the Iron-Catalyzed Intermolecular Olefin Amino-Oxygenation.
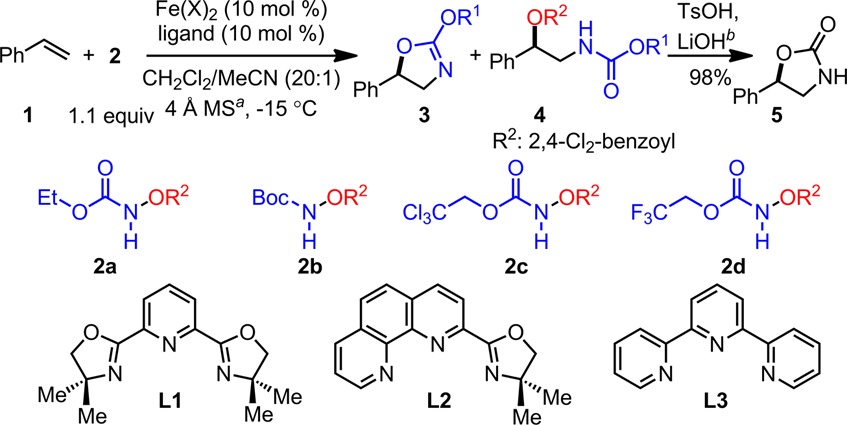
Molecular sieves were used to remove deleterious moisture.
Reactions were carried out under N2 in 1 h and then quenched with saturated NaHCO3 solution, unless stated otherwise. The crude mixture was first subjected to acidic conditions with TsOH (1.0 equiv) and then to basic conditions with LiOH (2.0 equiv) to afford 5.
Conversion was measured by GC.
Isolated yield.
An oxazolidinone was isolated directly without the additional step (41% yield); see Supporting Information.
To explore the scope and limitations of this method, a variety of olefins and dienes were evaluated under the optimized conditions (Table 2). We observed that α-methylstyrene is an excellent substrate (entry 2, 75% yield). Subsequently, we examined olefins that have been problematic for the existing amino-oxygenation methods (entries 3–8). First, an allyl silane, a substrate that is incompatible with other iron-based methods, can be efficiently amino-oxygenated with 2d (entry 3, 78% yield). Further exploration revealed that cyclopentadiene with a labile C–H bond can smoothly participate in the iron-catalyzed reaction with 2b, directly affording an oxazolidinone with a decent yield and excellent dr (entry 4, dr > 20:1). We further observed that cyclohexadiene can be converted to an oxazolidinone with a complementary regioselectivity compared with the osmium-based method (entry 5).1a Although enol ethers have been challenging substrates for existing amino-oxygenation methods, they are excellent substrates for the iron-catalyzed syn-amino-oxygenation which delivers protected amino alcohols with a good yield and dr (entries 6–7, yield up to 77% and dr up to >20:1).9 Importantly, a protected glycal can also participate in the amino-oxygenation with 2b, affording a 2-amino-α-sugar with a decent yield and excellent dr (entry 8, 63% yield, dr > 20:1 at both C1 and C2 positions).9,10 Additionally, indene can be efficiently amino-oxygenated and this reaction affords a protected cis-2-amino indanol, a valuable building block that is difficult to obtain directly with existing amino-oxygenation methods (entry 9, 71% yield, dr > 20:1).11
Table 2. Substrate Scope for the Iron-Catalyzed Olefin Amino-Oxygenation.
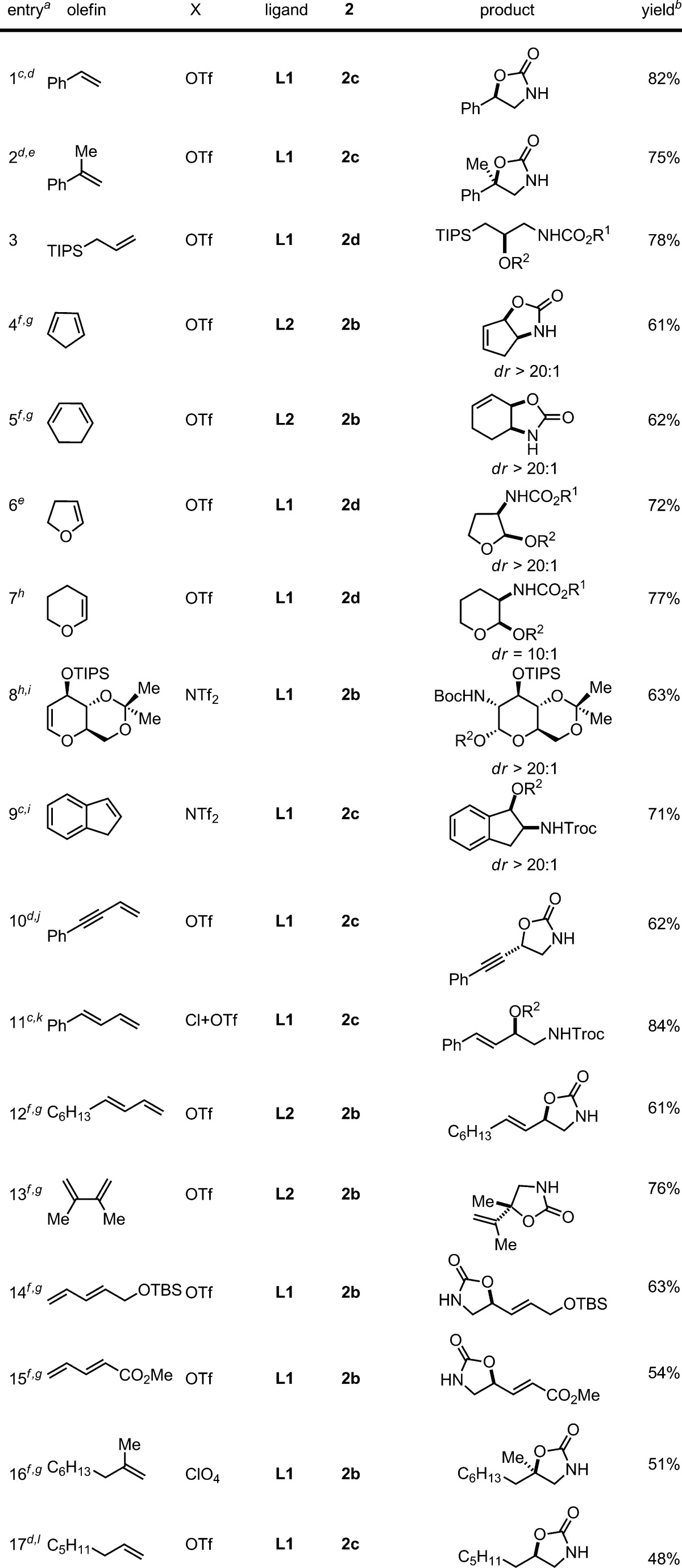
Reactions were carried out under N2 in 2 h, unless stated otherwise.
Isolated yield.
Reaction time: 1 h.
The crude mixture was treated with TsOH and then LiOH.
Reaction temp: −40 °C.
Catalyst loading: 20 mol %; reaction temp: 0 °C.
Reaction time: 12 h.
Reaction temp: −30 °C.
Fe(NTf2)2 (15 mol %), L1 (15 mol %).
Catalyst loading: 15 mol %.
Fe(OTf)2 (2.5 mol %) and FeCl2 (2.5 mol %) were used.
Catalyst loading: 30 mol %; reaction temp: 0 °C; reaction time: 24 h.
We also explored conjugated ene-ynes and dienes (entries 10–15). An ene-yne is an excellent substrate for the amino-oxygenation (entry 10, 62% overall yield). Conjugated dienes with either aliphatic or aromatic substituents can also be efficiently transformed into protected 1,2-amino alcohols with excellent regioselectivity (entries 11–13).12 To our delight, this method is also compatible with a silyl dienol with a labile C–H bond (entry 14, 63% yield). Also, a dienoate proves to be an acceptable substrate for this transformation (entry 15, 54% yield).
Additionally, we applied this method to isolated olefins. The Fe(ClO4)2–L1 complex can catalyze the amino-oxygenation of a 1,1-disubstituted olefin with 2b, affording an oxazolidinone (entry 16, 51% yield).13 We further observed that the Fe(OTf)2–L1 complex catalyzes the reaction of a monosubstituted olefin with 2c to afford the oxazolidinone with a fair yield (entry 17, 48% yield).
The catalytic asymmetric amino-oxygenation of indene 6 has been a challenge in synthetic chemistry, and osmium-based protocols deliver a mixture of racemic 1- and 2-amino indanols.11 In order to fill this gap, we have explored the asymmetric induction for the indene amino-oxygenation and discovered that an iron–chiral ligand L4(14) complex is uniquely effective to deliver a 2-amino indanol derivative 7 with a significant ee (Scheme 2A, 81% ee, dr > 20:1). Facile transformation converts 7 to 8 without erosion of its ee and dr. The asymmetric enol ether amino-oxygenation has also been unprecedented, and we observed that L4 is effective for asymmetric induction with dihydrofuran 9 as well (57% ee, dr > 20:1).
Scheme 2. Iron-Catalyzed Asymmetric Olefin Amino-Oxygenation and Control Experiments To Probe Reaction Mechanisms.
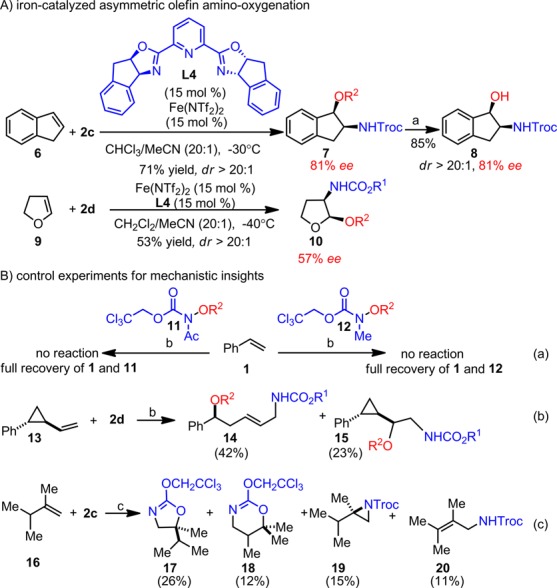
LAH, THF, −20 °C, 85%. b Fe(OTf)2 (10 mol %), L1 (10 mol %), CH2Cl2/MeCN (15:1), −15 °C, 1 h. c Fe(ClO4)2 (20 mol %), L1 (20 mol %), CH2Cl2/MeCN (15:1), −15 °C, 2 h. d R1: CF3CH2; R2: 2,4-Cl2-benzoyl.
In order to gather evidence for a mechanistic working hypothesis, we have carried out several control experiments (Scheme 2B). First, two analogues (11 and 12) of reagent 2c were prepared, such that the N–H group was masked by either an acetyl or a methyl group. Both were evaluated for the model reaction and neither was found to be reactive (eq a). These experiments suggest that the N–H group in 2c is critical for its activation. Next, we evaluated a cyclopropyl-substituted olefin 13 as a radical clock probe under the reaction conditions and observed the presence of both the ring-opening product 14 and the 1,2-amino-oxygenation product 15 (eq b). This result suggests that the reaction proceeds through a stepwise process that includes a radical amination step.
To probe the mechanism beyond the radical amination step, we further evaluated an isopropyl-substituted terminal olefin 16 (eq c). If a carbocation is generated after the radical amination, 1,2-hydride shift products may be observed. The amino-oxygenation with 16 afforded four products: in addition to the standard 1,2-amino-oxygenation product 17, an 1,3-amino-oxygenation product 18, aziridine 19, and allylic amine 20 were isolated (eq c). Importantly, 19 cannot be converted to any of the three other products under thereaction conditions. These results suggest that a carbocation may be involved in the olefin amino-oxygenation and that the corresponding aziridine is unlikely an intermediate along this pathway.
Furthermore, we studied cis/trans β-methyl styrenes as mechanistic probes and the experimental results corroborate that the amino-oxygenation occurs in a stepwise fashion and they also suggest that the C–N bond formation is likely the rate-determining step.15 Finally, we evaluated the electronic effect on styrene amino-oxygenation and concluded that amino alcohol formation is favored with substrates that can stabilize electrophilic radical species.15
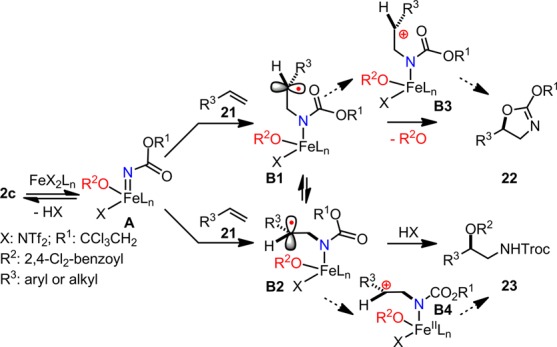
Based upon the collective evidence, a mechanistic working hypothesis of olefin amino-oxygenation that best corroborates the experimental data is presented in Scheme 3. First, the iron–ligand complex may reductively cleave the N–O bond in 2c, possibly converting it to an iron-nitrenoid A. A may then initiate radical amination with olefin 21 to afford radical species B1 together with its conformer B2 in equilibrium. Presumably, B1 can be oxidized by the iron center to a carbocation B3,16,17 which will be rapidly captured by the neighboring carbamate group, thereby affording 22. Alternatively, oxidative carboxylate ligand transfer16 may directly occur with B2 to afford the protected amino alcohol 31. We still cannot completely rule out the possibility that electron transfer from B2 to the iron center occurs first and that the oxidation product B4 will then be captured by a carboxylate to deliver 23. When the substituent (R3) has a less significant radical-stabilizing effect, B1 and B2 are relatively short-lived high energy species; therefore, the oxidative neighboring group participation through B1 may be favored to afford 22. However, when the substituent has a strong radical-stabilizing effect and both species are relatively long-lived, the ligand transfer from the iron center through B2 may become dominant to deliver 23.
Scheme 3. Mechanistic Working Hypothesis for the Iron-Catalyzed Olefin Amino-Oxygenation.
In conclusion, we have discovered a new iron-catalyzed stereoselective olefin amino-oxygenation method. This method tolerates a broad range of synthetically valuable olefins including those that are incompatible with existing amino-oxygenation methods. Our preliminary mechanistic studies revealed that an iron nitrenoid is a possible intermediate and its enantioselectivity can be controlled by chiral ligands. This discovery demonstrates the feasibility of developing a unique approach for iron-catalyzed selective olefin difunctionalization. Our ongoing efforts focus on understanding the mechanism of this new reaction and its applications in organic synthesis.
Acknowledgments
This work was supported by the NIH (GM110382), Georgia State University, and the ACS Petroleum Research Fund (ACS PRF 51571-DNI1). We thank Jeffrey Sears for synthesis of a few substrates.
Supporting Information Available
Experimental procedure, characterization data for all new compounds, selected NMR spectra, and HPLC traces. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Sharpless K. B.; Chong A. O.; Oshima K. J. Org. Chem. 1976, 41, 177. [Google Scholar]; b Li G. G.; Chang H. T.; Sharpless K. B. Angew. Chem., Int. Ed. 1996, 35, 451. [Google Scholar]
- For selected references of olefin amino-oxygenation and related transformations, see:; a Fuller P. H.; Kim J.-W.; Chemler S. R. J. Am. Chem. Soc. 2008, 130, 17638. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paderes M. C.; Chemler S. R. Org. Lett. 2009, 11, 1915. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sequeira F. C.; Chemler S. R. Org. Lett. 2012, 14, 4482. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Turnpenny B. W.; Chemler S. R. Chem. Sci. 2014, 5, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Michaelis D. J.; Shaffer C. J.; Yoon T. P. J. Am. Chem. Soc. 2007, 129, 1866. [DOI] [PubMed] [Google Scholar]; f Benkovics T.; Guzei I. A.; Yoon T. P. Angew. Chem., Int. Ed. 2010, 49, 9153. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Williamson K. S.; Yoon T. P. J. Am. Chem. Soc. 2010, 132, 4570. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Williamson K. S.; Yoon T. P. J. Am. Chem. Soc. 2012, 134, 12370. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Alexanian E. J.; Lee C.; Sorensen E. J. J. Am. Chem. Soc. 2005, 127, 7690. [DOI] [PubMed] [Google Scholar]; j Liu G.; Stahl S. S. J. Am. Chem. Soc. 2006, 128, 7179. [DOI] [PubMed] [Google Scholar]; k Desai L. V.; Sanford M. S. Angew. Chem., Int. Ed. 2007, 46, 5737. [DOI] [PubMed] [Google Scholar]; l Weinstein A. B.; Schuman D. P.; Tan Z. X.; Stahl S. S. Angew. Chem., Int. Ed. 2013, 52, 11867. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Zhu H.; Chen P.; Liu G. J. Am. Chem. Soc. 2014, 136, 1766. [DOI] [PubMed] [Google Scholar]; n Muñiz K.; Iglesias A.; Fang Y. Chem. Commun. 2009, 5591. [DOI] [PubMed] [Google Scholar]; o de Haro T.; Nevado C. Angew. Chem., Int. Ed. 2011, 50, 906. [DOI] [PubMed] [Google Scholar]; p Wardrop D. J.; Bowen E. G.; Forslund R. E.; Sussman A. D.; Weerasekera S. L. J. Am. Chem. Soc. 2010, 132, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Lovick H. M.; Michael F. E. J. Am. Chem. Soc. 2010, 132, 1249. [DOI] [PubMed] [Google Scholar]; r Schmidt V. A.; Alexanian E. J. J. Am. Chem. Soc. 2011, 133, 11402. [DOI] [PubMed] [Google Scholar]; s Xu H.-C.; Moeller K. D. J. Am. Chem. Soc. 2008, 130, 13542. [DOI] [PubMed] [Google Scholar]; t Donohoe T. J.; Johnson P. D.; Cowley A.; Keenan M. J. Am. Chem. Soc. 2002, 124, 12934. [DOI] [PubMed] [Google Scholar]; u Harris L.; Mee S. P. H.; Furneaux R. H.; Gainsford G. J.; Luxenburger A. J. Org. Chem. 2011, 76, 358. [DOI] [PubMed] [Google Scholar]; v Farid U.; Wirth T. Angew. Chem., Int. Ed. 2012, 51, 3462. [DOI] [PubMed] [Google Scholar]; w Mahoney J. M.; Smith C. R.; Johnston J. N. J. Am. Chem. Soc. 2005, 127, 1354. [DOI] [PubMed] [Google Scholar]; x Zhang B.; Studer A. Org. Lett. 2013, 15, 4548. [DOI] [PubMed] [Google Scholar]
- For selected references of rhodium nitrenoid-mediated amino-acetoxylation, see:; a Levites-Agababa E.; Menhaji E.; Perlson L. N.; Rojas C. M. Org. Lett. 2002, 4, 863. [DOI] [PubMed] [Google Scholar]; b Padwa A.; Stengel T. Org. Lett. 2002, 4, 2137. [DOI] [PubMed] [Google Scholar]; c Mulcahy J. V.; Du Bois J. J. Am. Chem. Soc. 2008, 130, 12630. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Beaumont S.; Pons V.; Retailleau P.; Dodd R. H.; Dauban P. Angew. Chem., Int. Ed. 2010, 49, 1634. [DOI] [PubMed] [Google Scholar]; e Gigant N.; Dequirez G.; Retailleau P.; Gillaizeau I.; Dauban P. Chem.—Eur. J. 2012, 18, 90. [DOI] [PubMed] [Google Scholar]; f Dequirez G.; Ciesielski J.; Retailleau P.; Dauban P. Chem.—Eur. J. 2014, 20, 8929. [DOI] [PubMed] [Google Scholar]; For a manganese-mediated stoichiometric method, see:; g Du Bois J.; Tomooka C. S.; Hong J.; Carreira E. M. J. Am. Chem. Soc. 1997, 119, 3179. [Google Scholar]
- For selected references of rhodium nitrenoid-mediated sp3 C–H amination, see:; a Espino C. G.; Du Bois J. Angew. Chem., Int. Ed. 2001, 40, 598. [DOI] [PubMed] [Google Scholar]; b Zalatan D. N.; Du Bois J. J. Am. Chem. Soc. 2008, 130, 9220. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nguyen Q.; Sun K.; Driver T. G. J. Am. Chem. Soc. 2012, 134, 7262. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected references of olefin aziridination through Cu and Rh metallonitrenes, see:; d Li Z.; Conser K. R.; Jacobsen E. N. J. Am. Chem. Soc. 1993, 115, 5326. [Google Scholar]; e Evans D. A.; Faul M. M.; Bilodeau M. T.; Anderson B. A.; Barnes D. M. J. Am. Chem. Soc. 1993, 115, 5328. [Google Scholar]; f Guthikonda K.; Du Bois J. J. Am. Chem. Soc. 2002, 124, 13672. [DOI] [PubMed] [Google Scholar]; g Lebel H.; Lectard S.; Parmentier M. Org. Lett. 2007, 9, 4797. [DOI] [PubMed] [Google Scholar]; h Jat J. L.; Paudyal M. P.; Gao H.; Xu Q.-L.; Yousufuddin M.; Devarajan D.; Ess D. H.; Kürti L.; Falck J. R. Science 2014, 343, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected references of iron-nitrenoid mediated N-atom transfer, see:; a Breslow R.; Gellman S. H. J. Chem. Soc., Chem. Commun. 1982, 1400. [Google Scholar]; b Mahy J. P.; Battioni P.; Mansuy D. J. Am. Chem. Soc. 1986, 108, 1079. [Google Scholar]; c Nakanishi M.; Salit A. F.; Bolm C. Adv. Synth. Catal. 2008, 350, 1835. [Google Scholar]; d Paradine S. M.; White M. C. J. Am. Chem. Soc. 2012, 134, 2036. [DOI] [PubMed] [Google Scholar]; e Hennessy E. T.; Betley T. A. Science 2013, 340, 591. [DOI] [PubMed] [Google Scholar]; f Liu Y.; Guan X.; Wong E. L.-M.; Liu P.; Huang J.-S.; Che C.-M. J. Am. Chem. Soc. 2013, 135, 7194. [DOI] [PubMed] [Google Scholar]; g Bach T.; Schlummer B.; Harms K. Chem. Commun. 2000, 287. [Google Scholar]; h Churchill D. G.; Rojas C. M. Tetrahedron Lett. 2002, 43, 7225. [Google Scholar]
- a Liu G.-S.; Zhang Y.-Q.; Yuan Y.-A.; Xu H. J. Am. Chem. Soc. 2013, 135, 3343. [DOI] [PubMed] [Google Scholar]; b Zhang Y.-Q.; Yuan Y.-A.; Liu G.-S.; Xu H. Org. Lett. 2013, 15, 3910. [DOI] [PubMed] [Google Scholar]; c Lu D.-F.; Liu G.-S.; Zhu C.-L.; Yuan B.; Xu H. Org. Lett. 2014, 16, 2912. [DOI] [PubMed] [Google Scholar]
- For leading references of chiral BOX and relevant ligands, see:; a Evans D. A.; Woerpel K. A.; Hinman M. M.; Faul M. M. J. Am. Chem. Soc. 1991, 113, 726. [Google Scholar]; b Nishiyama H.; Itoh Y.; Matsumoto H.; Park S.-B.; Itoh K. J. Am. Chem. Soc. 1994, 116, 2223. [Google Scholar]; c Nishikawa Y.; Yamamoto H. J. Am. Chem. Soc. 2011, 133, 8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For details of ligand synthesis, see Supporting Information (SI).
- For stereochemistry determination, see SI.
- For a manganese-mediated glycal aziridination for 2-amino sugar synthesis, see: Reference (3g). For a selected indirect method for 2-amino sugar synthesis, see:Griffith D. A.; Danishefsky S. J. J. Am. Chem. Soc. 1990, 112, 5811. [Google Scholar]
- Osmium-based aminohydroxylation of indene affords racemic mixtures of 1- and 2-amino indanols; see:; a Masruri; Willis A. C.; McLeod M. D. J. Org. Chem. 2012, 77, 8480. [DOI] [PubMed] [Google Scholar]; For an indirect method of 2-amino indanol synthesis, see:; b Corey E. J.; Roper T. D.; Ishihara K.; Sarakinos G. Tetrahedron Lett. 1993, 34, 8399. [Google Scholar]
- Fe(OTf)2/FeCl2 mixed salts were applied as the catalyst in entry 11. Fe(OTf)2 led to rapid decomposition of the diene; FeCl2 was inactive. See SI for details.
- Other Fe salts led to rapid allylic amination products. See SI for details.
- Davies I. W.; Gerena L.; Lu N.; Larsen R. D.; Reider P. J. J. Org. Chem. 1996, 61, 9629. [Google Scholar]
- Cis/trans isomeric olefins have been observed to present different reactivity and selectivity in atom transfer reactions that proceed via a stepwise mechanism. For a selected reference, see:Lee N. H.; Jacobsen E. N. Tetrahedron Lett. 1991, 32, 6533. [Google Scholar]; See SI for experimental details.
- For the oxidation of a radical species by a high-valent metal through ligand transfer or electron transfer, see:; a Kharasch M. S.; Sosnovsky G. J. Am. Chem. Soc. 1958, 80, 756. [Google Scholar]; b Kochi J. K. Science 1967, 155, 415. [DOI] [PubMed] [Google Scholar]
- For evidence for involvement of a possible carbocation intermediate, see eq c in Scheme 2B. For control experiments that exclude the aziridine as a possible intermediate, see SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.