

Abstract
Addition of anionic donors to the manganese(V)–oxo corrolazine complex MnV(O)(TBP8Cz) has a dramatic influence on oxygen-atom transfer (OAT) reactivity with thioether substrates. The six-coordinate anionic [MnV(O)(TBP8Cz)(X)]− complexes (X = F–, N3–, OCN–) exhibit a ∼5 cm–1 downshift of the Mn–O vibrational mode relative to the parent MnV(O)(TBP8Cz) complex as seen by resonance Raman spectroscopy. Product analysis shows that the oxidation of thioether substrates gives sulfoxide product, consistent with single OAT. A wide range of OAT reactivity is seen for the different axial ligands, with the following trend determined from a comparison of their second-order rate constants for sulfoxidation: five-coordinate ≈ thiocyanate ≈ nitrate < cyanate < azide < fluoride ≪ cyanide. This trend correlates with DFT calculations on the binding of the axial donors to the parent MnV(O)(TBP8Cz) complex. A Hammett study was performed with p-X-C6H4SCH3 derivatives and [MnV(O)(TBP8Cz)(X)]− (X = CN– or F–) as the oxidant, and unusual “V-shaped” Hammett plots were obtained. These results are rationalized based upon a change in mechanism that hinges on the ability of the [MnV(O)(TBP8Cz)(X)]− complexes to function as either an electrophilic or weak nucleophilic oxidant depending upon the nature of the para-X substituents. For comparison, the one-electron-oxidized cationic MnV(O)(TBP8Cz•+) complex yielded a linear Hammett relationship for all substrates (ρ = −1.40), consistent with a straightforward electrophilic mechanism. This study provides new, fundamental insights regarding the influence of axial donors on high-valent MnV(O) porphyrinoid complexes.
Introduction
A key structural factor that contributes to metal-oxo reactivity in heme enzymes is the nature of the axial ligands coordinated trans to the oxo group. The concept of axial ligand tuning is exemplified in the powerful oxygenating enzyme Cytochrome P450, in which the anionic, deprotonated Cys ligand is believed to play a critical role. It has been suggested that the Cys in Cyt-P450 plays an essential role in modulating the reactivity of Compound-I (Cpd-I), FeIV(O)(porph•+)(Cys), (porph = porphyrin) which is the key intermediate prior to substrate oxidation.1−7 Further evidence for the importance of the axial donor lies in the fact that it is the major feature that distinguishes broad classes of heme enzymes, with Cyt-P450 and chloroperoxidase (CPO) containing an axial Cys, peroxidase containing an axial His,8−10 and catalase containing an axial Tyr ligand.11,12
The preparation of synthetic analogs of heme enzymes has provided a means to test mechanistic hypotheses regarding the influence of axial donors. Synthetic models of heme active sites are particularly useful for examining complexes that are analogous to the highly unstable intermediates in enzyme systems, including high-valent metal-oxo species. Regarding the influence of axial donors on these species, a potential advantage of model systems is the ability to rationally tune the nature of the axial ligand by varying its structural and electronic properties in a systematic fashion. The influence of axial ligands on high-valent, iron-oxo porphyrin models has been investigated, including the examination of the effects of axial donors on oxygen-atom transfer (OAT) reactivity. For example, axial ligands were shown to enhance the reactivity of Fe(O)(porph) in epoxidations.13−21 A recent study on the effects of axial ligation on Cpd-I analogs, FeIV(O)(porph•+)(X), provided a detailed thermodynamic analysis which led to a possible explanation for the trend in OAT reactivity versus axial donor.15 The mechanisms of these reactions and especially the exact role played by the axial ligands are still not well understood.
In contrast to Fe(O) species, the influence of axial donors on analogous Mn(O) porphyrinoid complexes has been much less studied, with very few systematic investigations for a wide range of axial ligands reported to date.22−30 One of the few examples comes from Chang and co-workers, who showed that the reactivity of an MnV(O) corrole toward styrene can be enhanced by 10-fold with an imidazole as an axial ligand.23 The effects of axial donors on nonheme Mn(O) complexes such as Mn(O)(salen)(X) have been reported.31−34 However, direct structural, spectroscopic, or reactivity information on these complexes is in general quite limited because of the transient nature of these species.31
Previously, we took advantage of our ability to isolate and definitively characterize a relatively stable high-valent MnV(O) complex MnV(O)(TBP8Cz) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3–), which contains a ring-contracted porphyrinoid ligand, in order to examine the influence of cyanide and fluoride ions on C–H activation. Indirect evidence was obtained that suggested CN– and F– were weakly bound trans to the terminal oxo ligand and caused significant increases in H-atom abstraction rates.35 More recently, we used X-ray absorption spectroscopy to characterize one of these complexes, [MnV(O)(TBP8Cz)(CN)]−, and showed that CN– was indeed coordinated to the metal while leaving the short MnV–O bond intact.36 Addition of the CN– donor caused a large rate enhancement in OAT to thioanisole, giving the predicted two-electron reduced MnIII complex and sulfoxide product.
Herein we describe a comprehensive study regarding the influence of a wide range of anionic axial ligands on the OAT reactivity of an MnV(O) porphyrinoid complex. Significant rate enhancements for OAT to thioether substrates are seen for different axial donors. The use of a series of p-X-C6H4SCH3 substrates provides a range of OAT rates for the cyanide- and fluoride-ligated MnV(O) complexes, revealing unexpected “V-shaped” Hammett plots. These results point to a dramatic switch in mechanism for OAT, depending upon the nature of the substrate. This work provides new, fundamental insights regarding the influence of axial donors on the OAT reactivity of rarely observed MnV(O) porphyrinoid complexes.
Experimental Section
Materials
All reactions were performed under an argon atmosphere using dry solvents and standard Schlenk techniques. The MnIII(TBP8Cz) and MnV(O)(TBP8Cz) complexes were synthesized according to published methods.37 Solvents were purified via a Pure-Solv solvent purification system from Innovative Technologies, Inc. H218O (97% 18O) was purchased from Cambridge Isotopes, Inc. All other reagents were purchased from Sigma-Aldrich at the highest level of purity and used as received.
Instrumentation
UV–vis spectroscopy was performed on a Hewlett-Packard 8542 diode-array equipped with HP Chemstation software. A filter was placed between the spectrometer light and the sample to prevent bleaching of [MnV(O)(TBP8Cz)(X)]− from UV light (<400 nm). The temperature-dependent kinetics were performed on a Hewlett-Packard 8453 diode-array spectrophotometer equipped with an Unisoku thermostat cell holder. Gas chromatography (GC) was performed on an Agilent 6850 gas chromatograph fitted with a DB-5 5% phenylmethyl siloxane capillary column (30 m × 0.32 mm × 0.25 μm) and equipped with a flame-ionization detector. GC mass spectrometry (GC-MS) was performed on an Agilent 6850 gas chromatograph fitted with a DB-5 5% phenylmethyl siloxane capillary column and equipped with a mass spectrometer. LDI-TOF was conducted on a Bruker Autoflex III TOF/TOF instrument equipped with a nitrogen laser at 335 nm using an MTP 384 ground steel target plate. 31P NMR spectra were recorded on a Bruker Avance 400 MHz NMR spectrometer at 298 K. All spectra were taken in 5 mm o.d. NMR tubes, and chemical shifts were reported as δ values from standard peaks.
Resonance Raman Sample Preparation
A solution of MnV(O)(TBP8Cz) (1 mM) in CH2Cl2 (250 μL) at 25 °C was combined with Bu4N+X– (X = F, N3, OCN at 1 M). Samples were loaded into an EPR tube, immediately frozen in liquid nitrogen, and stored at 77 K until measurements were performed.
Resonance Raman Spectroscopy
Resonance Raman spectra were recorded on a triVista 555 triple monochromator (900/900/2400 gr/mm) equipped with a CCD camera (1340 × 100 pixels). A 413.2 nm line of a krypton-ion laser was used for excitation. The laser power was <15 mW at the sample to reduce the risk of photoreduction. 900/900/2400 gr/mm gratings provided a resolution of 0.69 cm–1 per CCD pixel at 413.2 nm. Samples were held in an EPR finger-dewar (77 K) in a 135° backscattering arrangement. Raw spectra were analyzed using the program Igor Pro for background subtraction. No smoothing procedures were performed on the raw data.
Product Analysis
In a typical reaction, a stirring solution of MnV(O)(TBP8Cz) (0.2 or 5 mM) in CH2Cl2 (3 mL) was combined with an anionic donor Bu4N+F– (1.3 or 12 mM) at 25 °C. To initiate the reaction, dimethyl sulfide (DMS): 11 M or triphenylphosphine (PPh3): 5 mM was added. Isosbestic conversion of [MnV(O)(TBP8Cz)(F)]− (λmax = 419, 634 nm) to [MnIII(TBP8Cz)(F)]− (λmax = 428, 471, 680 nm) was observed by UV–vis spectroscopy. Upon completion of the reaction, the solution was concentrated to ∼50 μL under vacuum. The phosphine reaction was dissolved in CDCl3 and analyzed by 31P NMR. The yield was obtained by comparing the integration of the product peak, triphenylphosphine oxide, to that of the reactant peak (PPh3). To analyze the DMS reaction, n-decane was added as an internal standard, and the solution was immediately analyzed by GC-FID. The product, dimethyl sulfoxide (DMSO), was identified by comparison with a standard sample. Yields were obtained by comparing the integration of the product peak with the integration of the internal standard peak. All yields are an average of two runs.
OAT Reaction of [MnV(O)(TBP8Cz)(X)]− with DMS and Dibutyl Sulfide
In a typical reaction, a solution of MnV(O)(TBP8Cz) (11 μM) in CH2Cl2 (2 mL) at 25 °C was combined with Bu4N+X– (11 mM) (X = N3, OCN, SCN, NO3) or Bu4N+F–·3H2O (0.03 M). Excess DMS or dibutyl sulfide (DBS) was added to begin the reaction. The changes in absorbance were monitored by UV–vis spectroscopy showing the isosbestic decay of [MnV(O)(TBP8Cz)(X)]− (λmax = 419, 634 nm) and formation the of [MnIII(TBP8Cz)(X)]−. A filter was placed between the spectrometer light and the sample to prevent bleaching of [MnV(O)(TBP8Cz)(X)]− from UV light (<400 nm). The pseudo-first-order rate constants, kobs, were obtained by nonlinear least-squares fitting (eq 1) of the growth in absorbance of [MnIII(TBP8Cz)(X)]− at 680–693 nm or decay of [MnV(O)(TBP8Cz)(X)]− at 634 nm versus time:
| 1 |
where Absf = final absorbance, Abs0 = initial absorbance, and Abst = absorbance at time (t). Second-order rate constants were obtained from the slope of the best-fit line of the linear plot of kobs versus substrate concentration.
Measurement of the Binding Constant for Fluoride to MnV(O)(TBP8Cz)
A solution of MnV(O)(TBP8Cz) (10 μM) in CH2Cl2 (2 mL) at 25 °C was combined with DBS (2.7 mM). The reaction was initiated by the addition of Bu4N+F–·3H2O (1.7–59 mM). The changes in absorbance were monitored by UV–vis spectroscopy showing the isosbestic decay of [MnV(O)(TBP8Cz)(F)]− (λmax = 419, 634 nm) and formation of [MnIII(TBP8Cz)(F)]− (λmax = 469, 680 nm). The pseudo-first-order rate constants, kobs, were obtained by nonlinear least-squares fitting (eq 1) of the growth in absorbance of [MnIII(TBP8Cz)(F)]− at 680 nm versus time. A plot of kobs versus [F–] showed saturation behavior.
The data were modeled for a 1:1 binding event as shown in eq 2, where the association constant K = k1/k–1, k2 is the pseudo-first-order rate constant for a given concentration of DMS, and [F–]total is the total concentration of TBAF.38 A fit of 1/kobs and 1/[F–]total plot is given in eq 3.
| 2 |
| 3 |
Linear Free Energy Correlations for OAT of [MnV(O)(TBP8Cz)(X)]− to para-Substituted Thioanisoles
A solution of MnV(O)(TBP8Cz) (12 μM for CN–; 15 μM for F–) in toluene (X = CN–) or CH2Cl2 (X = F–) (2 mL) at 25 °C was combined with a para-substituted thioanisole (0.13 M). The reaction was initiated by the addition of Bu4N+X– (12 mM, X = CN–; 15 mM, X = F–). The changes in absorbance were monitored by UV–vis spectroscopy showing the isosbestic decay of [MnV(O)(TBP8Cz)(X)]− (λmax = 419, 634 nm) and formation of [MnIII(TBP8Cz)(X)]− (λmax = 443, 492, 694 nm, X = CN–; λmax = 470, 679 nm, X = F–). The pseudo-first-order (kobs) rate constants were obtained as described above. The log(kX/kH) for each para-substituted thioanisole was graphed versus its Hammett constant (σ).39
Linear Free Energy Correlations for OAT of MnV(O)(TBP8Cz•+) to para-Substituted Thioanisoles
Following an earlier report,40 a solution of MnV(O)(TBP8Cz) (11 μM) in CH2Cl2 (2 mL) at 25 °C was combined with a one-electron oxidant [(4-BrC6H4)3N•+](SbCl6–) (11 μM) and monitored by UV–vis spectroscopy which showed the isosbestic conversion of MnV(O)(TBP8Cz) (λmax = 419, 634 nm) to [MnV(O)(TBP8Cz•+)] (λmax = 410, 780 nm). Once complete formation of the MnV(O)(TBP8Cz•+) complex was observed the OAT reaction was initiated by the addition of a para-substituted thioanisole (0.13 M). The changes in absorbance were monitored by UV–vis spectroscopy showing the isosbestic decay of [MnV(O)(TBP8Cz•+)] (λmax = 410, 780 nm) and formation of [MnIV(TBP8Cz)]+ (λmax = 433, 669, and 722 nm). The pseudo-first-order (kobs) rate constants were obtained by nonlinear least-squares fitting (eq 1) of the growth in absorbance of [MnIV(TBP8Cz)]+ at 722 nm versus time. The log(kX/kH) for each para-substituted thioanisole was graphed versus its Hammet constant (σ).39
Results and Discussion
OAT Reactivity of [MnV(O)(TBP8Cz)(F)]− with Thioether and Phosphine Substrates
Kinetic analysis showed that the oxidation of thioether substrates by MnV(O)(TBP8Cz) was relatively slow, with second-order rate constants of 2.0 × 10–3 M–1 s–1 for DMS and 3.8 × 10–4 M–1 s–1 for DBS.40 The addition of CN– to MnV(O)(TBP8Cz) dramatically increased the rates of sulfoxidation, giving a second-order rate constant of 9.2 M–1 s–1 for DBS.36 We next sought to extend our study on the influence of anionic donors and determine whether a correlation with reactivity and the nature of the axial donor could be made. Investigations were initiated by examining the reactivity of the fluoride anion. Addition of Bu4N+F– (TBAF) to MnV(O)(TBP8Cz) in the presence of excess DMS as substrate was monitored in CH2Cl2 at 25 °C (Scheme 1). The UV–vis spectra are shown in Figure 1a and reveal isosbestic conversion of the MnV-oxo complex to the axially ligated complex [MnIII(TBP8Cz)(F)]−, with a split Soret band at 428 and 471 nm. As previously observed, there is little change in the UV–vis spectrum for MnV(O)(TBP8Cz) upon addition of F–, but splitting in the Soret region of the corresponding [MnIII(TBP8Cz)(F)]− complex is clearly seen in Figure 1b.35,36 A related complex (Et4N)[MnIII(TBP8Cz)(Cl)] was crystallographically characterized and exhibited a similar splitting in the Soret band.37,41 With the addition of excess TBAF, the OAT reaction was complete in 1.5 h, and analysis by GC-FID revealed DMSO as the major oxidation product (85%). In comparison, the oxidation of DMS by the five-coordinate MnV(O)(TBP8Cz) is much slower, taking over 17 h for completion.
Scheme 1. Oxidation of Thioether and Phosphine Substrates by [MnV(O)(TBP8Cz)(F)]−.
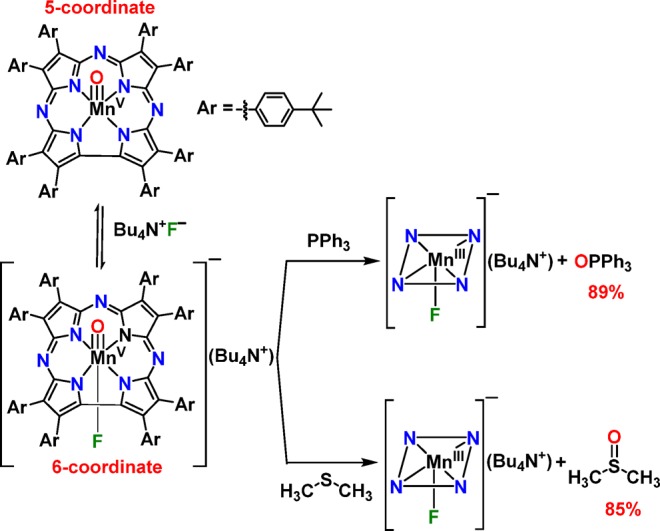
Figure 1.
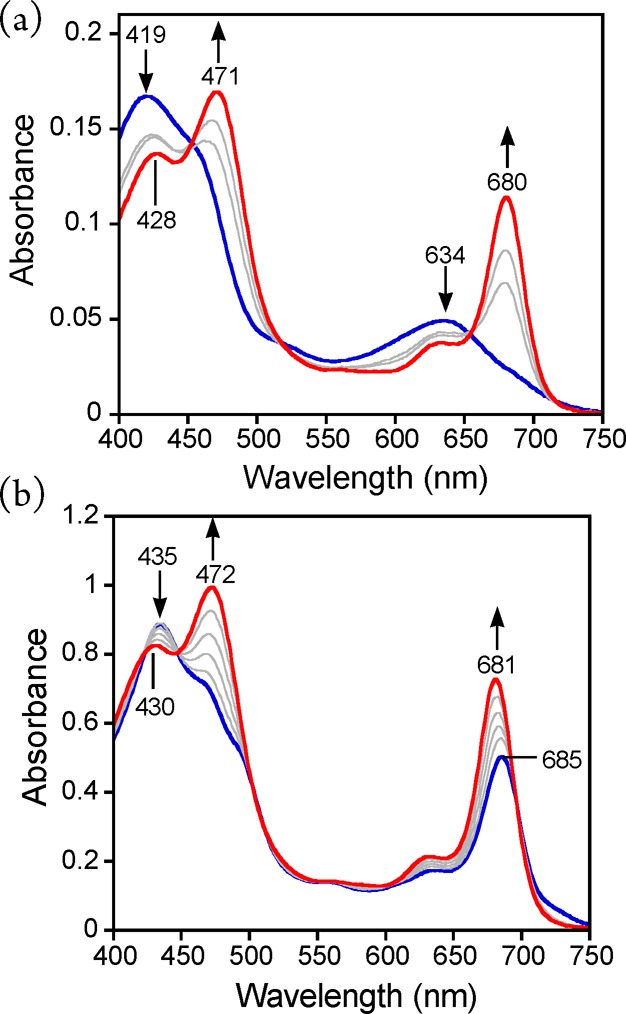
(a) UV–vis spectral changes (0–1.5 h) for the reaction of MnV(O)(TBP8Cz) + Bu4N+F– (6 equiv) (419, 634 nm) with excess DMS to give [MnIII(TBP8Cz)(F)]− (428, 471, 680 nm) in CH2Cl2 at 25 °C. (b) UV–vis spectral titration of MnIII(TBP8Cz) (435, 685 nm) (blue line) with Bu4N+F– (0–1 equiv) to give [MnIII(TBP8Cz)(F)]− (430, 472, 681 nm) in CH2Cl2 at 25 °C.
An alternate O-atom acceptor, PPh3, showed similar reactivity, resulting in the rapid conversion of [MnV(O)(TBP8Cz)(F)]− to [MnIII(TBP8Cz)(F)]−. The 31P NMR spectrum (Figure S1) of the reaction mixture identified OPPh3 as the major product in 89% yield (Scheme 1). Thus, the MnV(O) complex, in the presence of TBAF, reacts rapidly and quantitatively with both thioether and phosphine O-atom acceptors to give sulfoxide or phosphine oxide products via a formal OAT process.
Dependence of OAT Rates on Fluoride Concentration
In earlier work we showed that the rates of H-atom abstraction by MnV(O)(TBP8Cz) with C–H substrates depended on the concentration of added TBAF.35 A plot of pseudo-first-order rate constants (kobs) versus [TBAF] for two different C–H substrates gave a saturation curve that was successfully modeled as a 1:1 binding equilibrium.35 In the present study, the rates of sulfoxidation of DBS under pseudo-first-order conditions were similarly monitored by UV–vis, and the concentration of TBAF was varied. Plots of the absorbance associated with the decay and growth of the MnV and MnIII complexes, respectively, were well-fit to a single exponential kinetic model (Figure S2). The resulting kobs values were plotted versus [F–] (Figure 2a) and exhibit saturation behavior, indicative of a rapid pre-equilibrium for the binding of F– prior to the rate-determining step. A double reciprocal plot (1/kobs versus 1/[F–]total) (Figure 2b) reveals good linearity, with a best fit that gives a binding constant of K(F–) = 115 ± 4 M–1. This value is in good agreement with the binding constants found previously for fluoride anion and the MnV(O) complex in reactions with C–H substrates (163 ± 7 and 191 ± 10 M–1).35 These results provide strong evidence that addition of excess F– leads to the formation of a six-coordinate MnV(O) complex with the fluoride ligated relatively weakly trans to the oxo group.
Figure 2.
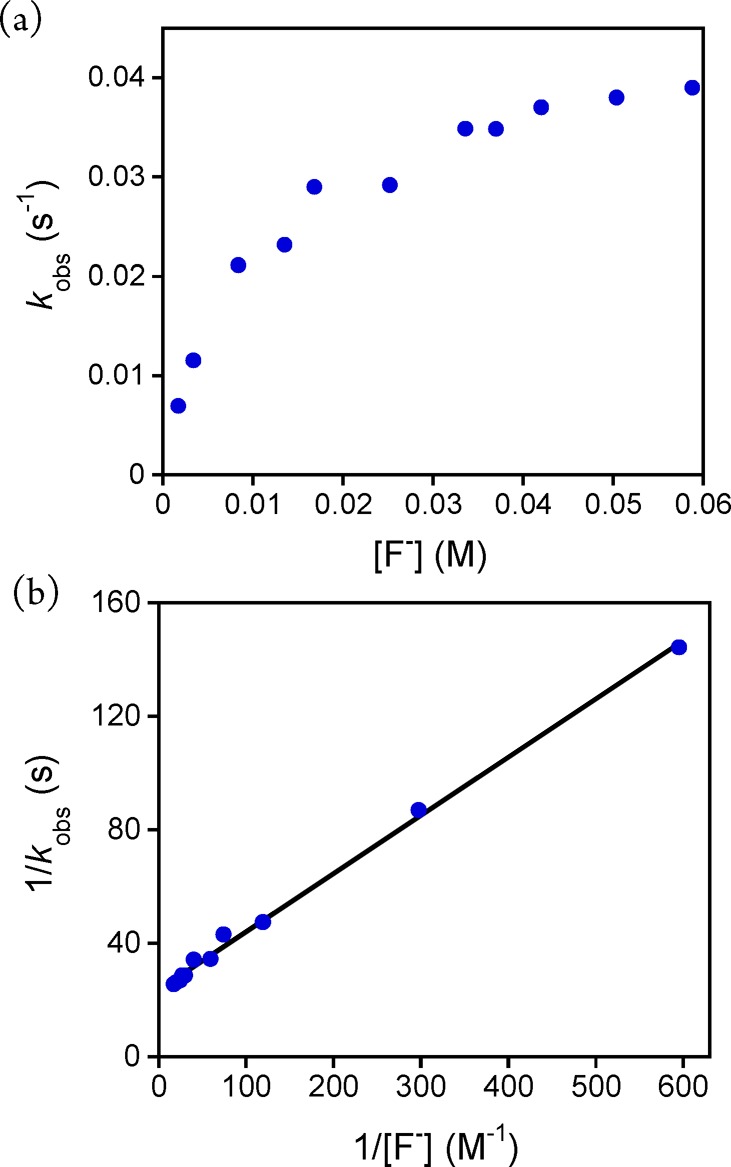
(a) Dependence of the kobs values on the concentration of Bu4N+F– for the reaction of MnV(O)(TBP8Cz) + DBS (270 equiv) in CH2Cl2 at 25 °C. (b) Double-reciprocal plot of 1/kobs versus 1/[F–] (blue circles) and best fit line (black).
Resonance Raman Spectroscopy of the Six-coordinate MnV(O) Complexes
The binding of X– to MnV(O)(TBP8Cz) can be described according to the simple equilibrium in eq 4:
| 4 |
The six-coordinate complexes were examined by resonance Raman spectroscopy for X = F–, N3–, and OCN–. Excess X– was added to push the equilibrium in eq 4 to favor the six-coordinate complexes in CH2Cl2. Frozen solution samples were excited with the 413.2 nm krypton-ion excitation line at 77 K. A Mn–O vibrational mode for the five-coordinate MnV(O)(TBP8Cz) starting complex was identified at 981 cm–1, which downshifts to 939 cm–1 upon 18O substitution (Figure S3). This 42 cm–1 shift is in good agreement with diatomic harmonic oscillator predictions as well as previously published results.42 The spectra for the F–, N3–, and OCN– complexes are shown in Figure 3, together with the five-coordinate starting material. The Mn–O mode clearly downshifts by 5 cm−1 to 976 cm–1 relative to the starting complex. The identity of the peak at 976 cm–1 was verified using 18O labeling for the F– and N3– complexes (Figures S4 and S5). These results provide strong evidence that the anionic donors are coordinated trans to the terminal oxo ligand, thereby inducing a subtle weakening of the Mn–O vibrational mode. Similar effects on the metal-oxo stretch were seen upon variation of trans axial donors in both heme and nonheme FeIV(O) complexes.15,43,44
Figure 3.
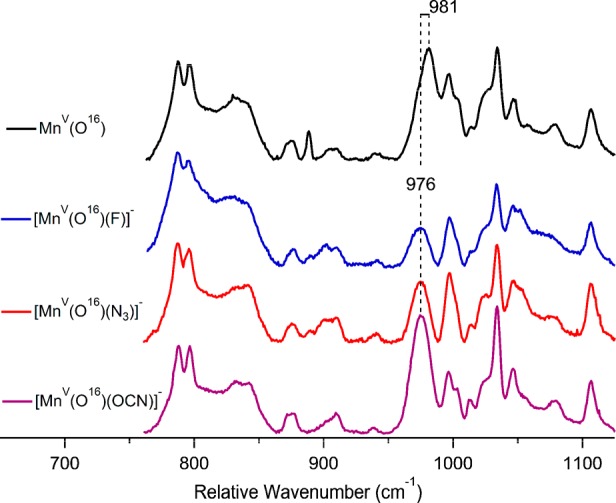
Low-frequency resonance Raman spectra of 16O [MnV(O)(TBP8Cz)(X)]− where X = no ligand, F−, N3−, or OCN−. The resonances near 900 cm–1 were used to scale the spectra. Data were collected using a 413.2 nm krypton-ion laser line.
Kinetic Studies of OAT for a Series of Anionic Axial Donors
A broad series of anionic axial ligands (X−), in conjunction with two thioether substrates (DMS and DBS), were employed for kinetic studies (Scheme 2). An example of the UV–vis spectral changes observed for these OAT reactions is shown in Figure 4, where X = N3–. Good isosbestic behavior is seen, and the final spectrum matches that for [MnIII(TBP8Cz)(N3)]−. Plots of absorbance versus time for the decay of the MnV(O) complex (634 nm) and the appearance of the MnIII(N3–) complex (685 nm) are shown in the inset, Figure 4. These plots, along with the plots for the other axial donors, were well fit by a single exponential model, consistent with the mechanism shown in Scheme 2. The resulting fits yielded pseudo-first-order rate constants (kobs), which were found to be linearly dependent on substrate concentration (Figure 5). The best-fit lines of kobs versus [substrate] yielded second-order rate constants (k) (Table 1) for the different axial donors.
Scheme 2. Kinetic Model for OAT to Thioether Substrates.
Figure 4.
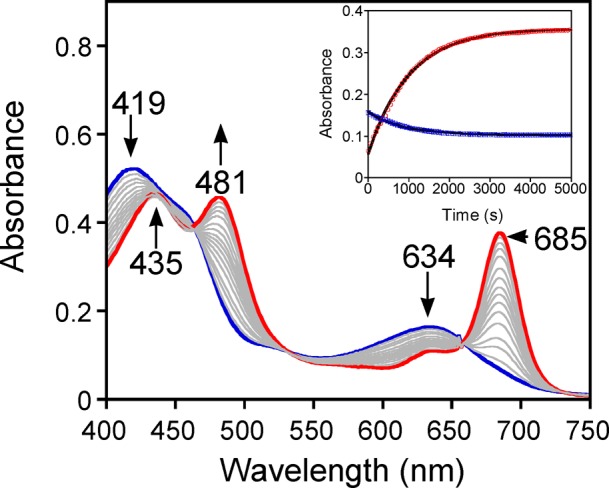
UV–vis spectral changes for the reaction of [MnV(O)(TBP8Cz)(N3)]− (9 μM) (419, 634 nm) with DMS (4.0 mM) in CH2Cl2 at 25 °C. Inset: changes in absorbance versus time for the growth of [MnIII(TBP8Cz)(N3)]− (685 nm) (open red circle) and the decay of [MnV(O)(TBP8Cz)(N3)]− (634 nm) (open blue square) together with best fits (solid black line).
Figure 5.
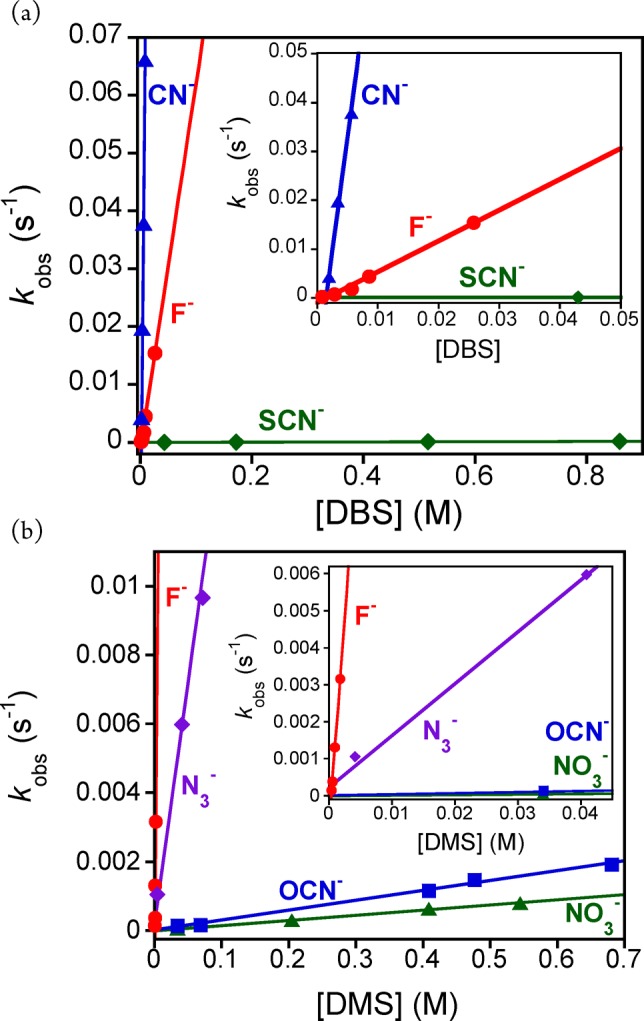
(a) Plots of kobs versus concentration of DBS for X– = CN– (solid blue triangle), F– (solid red circle), and SCN– (solid green diamond). Inset: expanded region from 0 to 0.05 M. (b) Plots of kobs versus concentration of DMS for X– = F– (solid red circle), N3– (solid purple diamond), OCN– (solid blue square) and NO3– (solid green triangle). Inset: expanded region from 0 to 0.045 M.
Table 1. Second-Order Rate Constants for OAT to Thioether Substrates.
As can be seen from Table 1, there is a remarkable enhancement in OAT reactivity for the MnV(O) complex in the presence of anionic donors. The second-order rate constant for F– reveals a dramatic rate enhancement of 1100-fold in the oxidation of DMS compared to the five-coordinate MnV(O) complex. A similar rate enhancement (1700) is seen for the oxidation of DBS. Addition of CN– as the axial donor has an even more dramatic effect, revealing a rate enhancement of 24,000 for DBS oxidation by the [MnV(O)(TBP8Cz)(CN)]− complex.
Further examination of the other anionic axial donors in Table 1 reveals some important trends. The addition of azide (N3–) leads to a 70-fold rate enhancement over the five-coordinate complex in the sulfoxidation of DMS. The cyanate (OCN–) anion gives a more modest increase of only 1.5 times for reaction with DMS, and the nitrate (NO3–) anion showed essentially no influence. The thiocyanate (SCN–) anion also has little effect over the reaction rate with DBS as substrate. Taken together, the kinetic data indicate the following trend regarding the influence of the anionic donors on the rate of OAT: none ≈ SCN– ≈ NO3– < OCN– < N3– < F– ≪ CN–. This trend is supported by our DFT calculations and suggests a possible structural explanation for the relative influence of X– on the reactivity of MnV(O)(TBP8Cz) (vide infra).
Influence of para-Substituents on the Reactivity of Thioanisole Derivatives
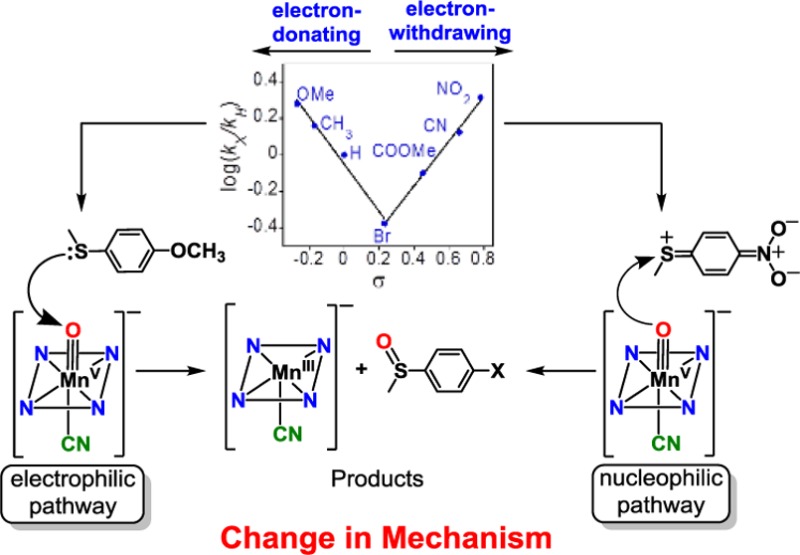
The reactive cyanide-ligated [MnV(O)(TBP8Cz)(CN)]− complex was chosen for further study with a series of p-X-C6H4SCH3 derivatives (Scheme 3). The oxidation of C6H5SCH3 proceeds in good yield (84%) to give the mono-oxygenated sulfoxide product, with 71% of the 18O label in [MnV(18O)(TBP8Cz)(CN)]− transferring to the sulfur atom.36 The reactions between the p-X-substituted thioanisole derivatives and [MnV(O)(TBP8Cz)(CN)]− were monitored by UV–vis spectroscopy, and good isosbestic behavior for the conversion of the MnV starting material to the [MnIII(CN)]− product was seen for all substrates. The resulting pseudo-first-order rate constants (kX) for the different para-substituted derivatives were used to construct the Hammett plot shown in Figure 6. For the electron-donating substituents, there is a good linear correlation with a clear negative slope of rho (ρ) = −1.29. This trend is easily explained by a mechanism involving electrophilic attack of the oxo group of the MnV(O) complex on the sulfur center of the thioanisole derivatives. A negative ρ value was also seen for H-atom abstraction by the five-coordinate MnV(O) complex with a series of p-substituted phenols.45 These data indicate that the anionic, six-coordinate MnV(O) complex maintains significant electrophilic character. However, the electron-withdrawing p-COOMe, p-CN, and p-NO2 substituents lead to a substantial increase in reaction rate and a positive rho value of ρ = +1.22. Such a “V-shaped” Hammett plot suggests a distinct change in mechanism for the electron-poor substrates. For comparison, a V-shaped Hammett plot was observed for a related MnV(imido)(corrole) complex, also implicating a mechanistic switch.46 The generality of this change in mechanism for the six-coordinate anionic MnV(O) complexes was confirmed by examining the fluoride-ligated [MnV(O)(TBP8Cz)(F)]−. Reaction of this complex with p-X-C6H4SCH3 derivatives also revealed a clear V-shaped Hammett plot (Figure S14).
Scheme 3. OAT to para-Substituted Thioanisole Derivatives.
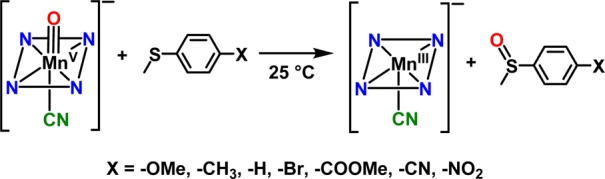
Figure 6.
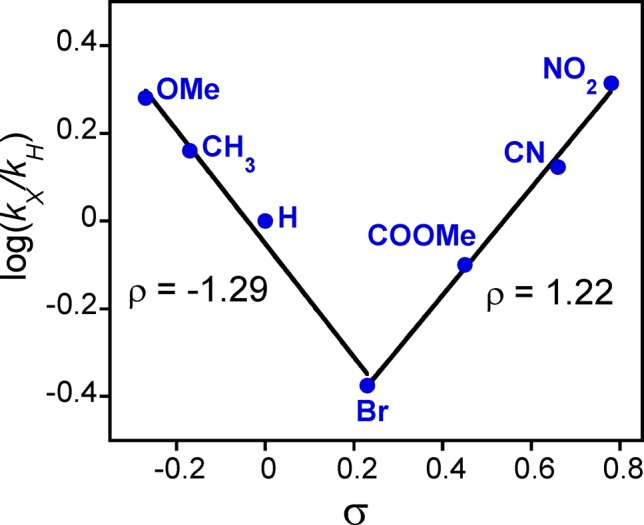
Hammett plot for the reaction of [MnV(O)(TBP8Cz)(CN)]− and para-X-substituted thioanisole derivatives.
A possible explanation for the V-shaped Hammett plots comes from consideration of the contributing resonance structures shown in Figure 7. The quinoid-type resonance forms may be partly stabilized by the electron-withdrawing p-COOMe, p-CN, and p-NO2 substituents. Evidence for the contribution of a similar quinoid-type resonance form in a p-nitrophenylthiolate–NiII complex was observed by X-ray crystallography.47 The quinoid-type resonance form shown in Figure 7 generates partial positive charge on the sulfur center, making these relatively electron-poor substrates potentially electrophilic in nature. A different mechanism for sulfoxidation could then result for the p-COOMe, p-CN, and p-NO2 substrates.
Figure 7.

Resonance structures for electron-withdrawing para-subsituted thioanisole derivatives.
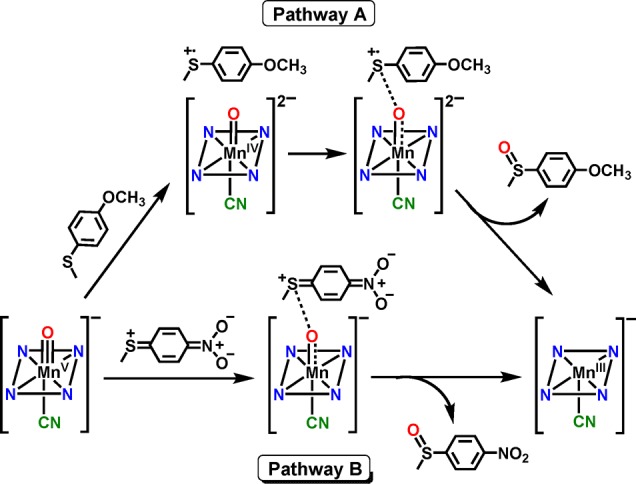
The two possible mechanistic scenarios are shown in Scheme 4. Pathway A corresponds to the electron-donating para-X substituents and shows the sulfoxidation reaction formally broken into electron-transfer and O–S bond formation steps. The redox potentials of the thioether substrates (E°ox = 1.2–1.6 V vs SCE) as compared to the parent five-coordinate MnV(O) complex (E1/2 = −0.05 V vs SCE)37 indicate that pure outer-sphere ET between ArSR and the six-coordinate, anionic MnV(O) complexes is likely to be thermodynamically highly disfavored. Thus, a more reasonable mechanism, which we favor, involves a single transition state with contributions from both the ET and S–O bond formation steps formally depicted in Pathway A. The rates for the electron-donating substituents correlate with the trend expected from a significant contribution of the ET process, which should get slower as the redox potential of the thioether increases. In contrast, the electron-withdrawing substituents may exhibit negligible contribution from electron-transfer because of their very high redox potentials. For these substrates, the quinoid-type resonance structure can enhance the rate of S–O bond formation, taking advantage of the ability of the anionic [MnV(O)(TBP8Cz)(X)]− (X = CN– or F–) complex to serve as a potential weak nucleophile.48,49 Thus, for pathway B, the transition state is dominated by S–O bond formation. This barrier would be lower for more electron-withdrawing substrates, providing an explanation for the positive ρ value and the V-shaped Hammett plot seen in Figure 6.
Scheme 4. Possible Mechanistic Pathways for Electron-Donating (Pathway A) and Electron-Withdrawing (Pathway B) para-Substituted Thioanisole Derivatives.
The cationic complex MnV(O)(TBP8Cz•+) was previously prepared by some of us and, to our knowledge, is the only example of an MnV(O)(π-radical cation) complex. This cationic complex functioned as an electrophilic oxidant toward phosphine and thioether substrates and was an ideal candidate for comparison of its OAT reactivity with the anionic axially ligated MnV(O) complexes. The MnV(O)(π-radical cation) complex was generated by chemical oxidation of the neutral MnV(O) starting material with the one-electron oxidant [(4-BrC6H4)3N•+](SbCl6–) (Scheme 5). Isosbestic behavior was observed for the reactions with the p-X-substituted thioanisole derivatives, and in this case the two-electron reduced product of MnV(O)(TBP8Cz•+) is the MnIV complex [MnIV(TBP8Cz)]+, as expected following OAT. The resulting Hammett plot of log(kX/kH) versus σ values for the para-X derivatives is shown in Figure 8. A strong linear correlation with a slope ρ = −1.40 is found throughout the range of para-X derivatives, in stark contrast to the V-shaped Hammett plot seen for the anionic [MnV(O)(TBP8Cz)(X)]− (X = CN– or F–). A single mechanism involving electrophilic attack of MnV(O)(TBP8Cz•+) (Pathway A) is inferred. These findings provide strong support for the conclusion that there is a mechanistic switch for the CN– and F– complexes and the electron-poor substrates (Pathway B), and it is the anionic nature of the CN– and F– species that likely facilitates the opening of this unique reaction channel.
Scheme 5. Generation and OAT Reaction of the Cationic [MnV(O)(TBP8Cz+•)] Complex.
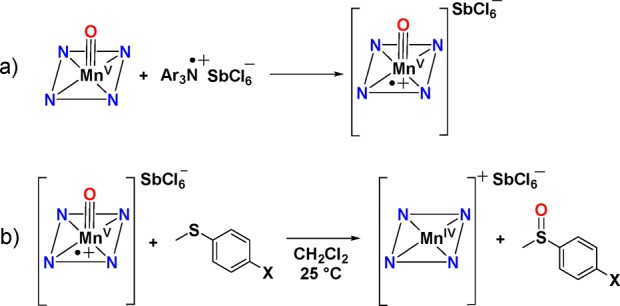
Figure 8.
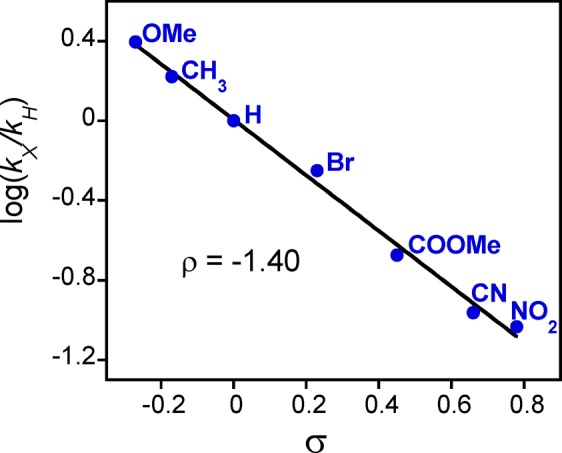
Hammett plot for the reaction of MnV(O)(TBP8Cz+•) with para-X-substituted thioanisole derivatives in CH2Cl2.
Density Functional Theory (DFT) Calculations
Computational studies were undertaken to help explain the trend in reactivity for the different anionic axial ligands. Calculations were performed at RIJCOSX-B3LYP/LANLDZ/6-31G level of theory, and the optimized geometries revealed a variation in MnV–X bond distances that were used for a qualitative comparison of the binding interactions for the various axial (X–) donors. The TBP groups were replaced with H-atoms on the Cz ligand to facilitate the computations. The calculations were performed on the singlet state only for the MnV ion, which has been shown to be the ground state in previous calculations35,40 and which is consistent with XAS measurements for the CN– complex.36 The optimized geometries for the six-coordinate F– and CN– structures were calculated previously35 but were repeated here for comparative purposes. The calculated structures for 1[MnV(O)(H8Cz)(X)]− (X = F–, CN–) revealed Mn–X distances (Table 2) indicating a binding interaction. The azide and cyanate anions also remained bound to the metal following optimization but exhibit longer bond distances. Interestingly, the nitrate anion binds only very weakly, and thiocyanate (not shown in Table 2) dissociates completely from the metal upon geometry optimization. Both NO3– and SCN– anions showed no influence on the rate of OAT. The computational results show that the MnV–X distance correlates well with the trend in reactivity for the axial donors, in which the shorter distances seen for CN– and F– correspond to large rate enhancements, whereas the other ligands exhibit either weak or no influence on OAT rates.
Table 2. Select Bond Distances (Å) from DFT Calculations.
| none | CN– | F– | N3– | OCN– | NO3– | |
|---|---|---|---|---|---|---|
| MnV–O | 1.549 | 1.568 | 1.568 | 1.568 | 1.561 | 1.557 |
| MnV–X | – | 2.146 | 1.931 | 2.354 | 2.420 | 2.518 |
Conclusions
Addition of anionic axial donors (X–) to an MnV(O) corrolazine results in binding to the MnV center. Resonance Raman spectroscopy confirms a subtle weakening of the MnV–O triple bond upon addition of the X– ligand, providing strong evidence for coordination of X– in a position trans to the terminal oxo group. The axial donors CN– and F– lead to dramatic increases in OAT reactivity to thioether substrates, and N3– also shows a strong effect. The OCN–, NO3–, and SCN– anions, however, exhibit little or no influence on OAT. These trends in reactivity correlate well with the trend in the DFT-derived MnV–X distances. A Hammett analysis with a range of p-X-C6H4SCH3 derivatives revealed an unusual V-shaped Hammett plot for [MnV(O)(TBP8Cz)(X)]− (X = CN– or F–) complexes, indicating a distinct change in mechanism for OAT to the thioether substrates. This Hammett behavior strongly contrasts the linear plot seen for the cationic complex MnV(O)(TBP8Cz•+). These findings suggest a novel bifurcated mechanistic scenario in which the axially ligated anionic complexes can function in either a purely electrophilic capacity or in an initial nucleophilic manner when strong electron-withdrawing substituents are present on the thioanisole derivatives. Our results suggest that axial ligation is a key factor in the OAT reactivity of MnV(O) complexes, and the addition of the appropriate axial donor can be a good strategy for dramatically increasing OAT reactivity. Even though the axially ligated complexes bear an overall negative charge, they function as more powerful, electrophilic oxidizing agents as compared to the nonligated, neutral MnV(O) precursor complex. With the strong electron-withdrawing p-X substituents on the thioanisole derivatives, the nucleophilic properties of the anionic MnV(O) complexes provide a further boost to their reactivity. These new mechanistic insights should help in our understanding of axial ligand effects in heme enzymes as well as in the potential design of new synthetic oxidation catalysts.
Acknowledgments
This work was supported by the NIH (GM101153 to D.P.G). The Kirin cluster at Johns Hopkins University School of Arts and Sciences is thanked for CPU time to T.Y. The National Service of Computational Chemistry Software is acknowledged for providing CPU time. M.G.Q. thanks the BBSRC for a studentship.
Supporting Information Available
Computational methods, Table S1, Figures S1−S15, and cartesian coordinates for the computational studies. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Green M. T. Curr. Opin. Chem. Biol. 2009, 13, 84–88. [DOI] [PubMed] [Google Scholar]
- Groves J. T. Nat. Chem. 2014, 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Chem. Rev. 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos T. L.; Finzel B. C.; Howard A. J. J. Mol. Biol. 1987, 195, 687–700. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Shaik S.; Sharma P. K.; Kumar D.; Thiel W. J. Am. Chem. Soc. 2003, 125, 15779–15788. [DOI] [PubMed] [Google Scholar]
- Poulos T. L. In Handbook of Porphyrin Science; Kadish K. M., Smith K. M.; Guilard R., Eds.; World Scientific Publishing Co.: Singapore, 2012; Vol. 19, p 45–109. [Google Scholar]
- Battistuzzi G.; Bellei M.; Bortolotti C. A.; Sola M. Arch. Biochem. Biophys. 2010, 500, 21–36. [DOI] [PubMed] [Google Scholar]
- Bewley K. D.; Ellis K. E.; Firer-Sherwood M. A.; Elliott S. J. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 938–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A.; Loewen P. C.; Fita I.; Carpena X. Arch. Biochem. Biophys. 2012, 525, 102–110. [DOI] [PubMed] [Google Scholar]
- Alfonso-Prieto M.; Vidossich P.; Rovira C. Arch. Biochem. Biophys. 2012, 525, 121–130. [DOI] [PubMed] [Google Scholar]
- Gross Z. J. Biol. Inorg. Chem. 1996, 1, 368–371. [Google Scholar]
- Kang Y.; Chen H.; Jeong Y. J.; Lai W.; Bae E. H.; Shaik S.; Nam W. Chem.—Eur. J. 2009, 15, 10039–10046. [DOI] [PubMed] [Google Scholar]
- Takahashi A.; Yamaki D.; Ikemura K.; Kurahashi T.; Ogura T.; Hada M.; Fujii H. Inorg. Chem. 2012, 51, 7296–7305. [DOI] [PubMed] [Google Scholar]
- Takahashi A.; Kurahashi T.; Fujii H. Inorg. Chem. 2009, 48, 2614–2625. [DOI] [PubMed] [Google Scholar]
- Song W. J.; Ryu Y. O.; Song R.; Nam W. J. Biol. Inorg. Chem. 2005, 10, 294–304. [DOI] [PubMed] [Google Scholar]
- Pan Z. Z.; Zhang R.; Newcomb M. J. Inorg. Biochem. 2006, 100, 524–532. [DOI] [PubMed] [Google Scholar]
- Hessenauer-Ilicheva N.; Franke A.; Meyer D.; Woggon W. D.; van Eldik R. J. Am. Chem. Soc. 2007, 129, 12473–12479. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Karamzadeh B.; Sastry G. N.; de Visser S. P. J. Am. Chem. Soc. 2010, 132, 7656–7667. [DOI] [PubMed] [Google Scholar]
- Ohno T.; Suzuki N.; Dokoh T.; Urano Y.; Kikuchi K.; Hirobe M.; Higuchi T.; Nagano T. J. Inorg. Biochem. 2000, 82, 123–125. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Goldberg I.; Botoshansky M.; Buchman Y.; Gross Z. J. Am. Chem. Soc. 2010, 132, 15233–15245. [DOI] [PubMed] [Google Scholar]
- Liu H. Y.; Zhou H.; Liu L. Y.; Ying X.; Jiang H. F.; Chang C. K. Chem. Lett. 2007, 36, 274–275. [Google Scholar]
- Solati Z.; Hashemi M.; Hashemnia S.; Shahsevani E.; Karmand Z. J. Mol. Catal. A: Chem. 2013, 374, 27–31. [Google Scholar]
- Jin N.; Ibrahim M.; Spiro T. G.; Groves J. T. J. Am. Chem. Soc. 2007, 129, 12416–12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunkumar C.; Lee Y. M.; Lee J. Y.; Fukuzumi S.; Nam W. Chem.—Eur. J. 2009, 15, 11482–11489. [DOI] [PubMed] [Google Scholar]
- Song W. J.; Seo M. S.; George S. D.; Ohta T.; Song R.; Kang M. J.; Tosha T.; Kitagawa T.; Solomon E. I.; Nam W. J. Am. Chem. Soc. 2007, 129, 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifi R.; Tahsini L.; Karamzadeh B.; Safari N.; Nam W.; de Visser S. P. Arch. Biochem. Biophys. 2011, 507, 4–13. [DOI] [PubMed] [Google Scholar]
- Park S. E.; Song W. J.; Ryu Y. O.; Lim M. H.; Song R.; Kim K. M.; Nam W. J. Inorg. Biochem. 2005, 99, 424–431. [DOI] [PubMed] [Google Scholar]
- Jin N.; Lahaye D. E.; Groves J. T. Inorg. Chem. 2010, 49, 11516–11524. [DOI] [PubMed] [Google Scholar]
- Wang C.; Kurahashi T.; Inomata K.; Hada M.; Fujii H. Inorg. Chem. 2013, 52, 9557–9566. [DOI] [PubMed] [Google Scholar]
- Kurahashi T.; Kikuchi A.; Shiro Y.; Hada M.; Fujii H. Inorg. Chem. 2010, 49, 6664–6672. [DOI] [PubMed] [Google Scholar]
- Kurahashi T.; Fujii H. Bull. Chem. Soc. Jpn. 2012, 85, 940–947. [Google Scholar]
- Collman J. P.; Zeng L.; Brauman J. I. Inorg. Chem. 2004, 43, 2672–2679. [DOI] [PubMed] [Google Scholar]
- Prokop K. A.; de Visser S. P.; Goldberg D. P. Angew. Chem., Int. Ed. 2010, 49, 5091–5095. [DOI] [PubMed] [Google Scholar]
- Neu H. M.; Quesne M. G.; Yang T.; Prokop-Prigge K. A.; Lancaster K. M.; Donohoe J.; DeBeer S.; de Visser S. P.; Goldberg D. P.. Chem. Eur. J., accepted; DOI: 10.1002/chem.201404349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansky D. E.; Mandimutsira B.; Ramdhanie B.; Clausen M.; Penner-Hahn J.; Zvyagin S. A.; Telser J.; Krzystek J.; Zhan R.; Ou Z.; Kadish K. M.; Zakharov L.; Rheingold A. L.; Goldberg D. P. Inorg. Chem. 2005, 44, 4485–4498. [DOI] [PubMed] [Google Scholar]
- Wright M. R.An Introduction to Chemical Kinetics; Wiley: Chichester, U.K., 2004. [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- Prokop K. A.; Neu H. M.; de Visser S. P.; Goldberg D. P. J. Am. Chem. Soc. 2011, 133, 15874–15877. [DOI] [PubMed] [Google Scholar]
- Lansky D. E.; Narducci Sarjeant A. A.; Goldberg D. P. Angew. Chem., Int. Ed. 2006, 45, 8214–8217. [DOI] [PubMed] [Google Scholar]
- Mandimutsira B. S.; Ramdhanie B.; Todd R. C.; Wang H.; Zareba A. A.; Czernuszewicz R. S.; Goldberg D. P. J. Am. Chem. Soc. 2002, 124, 15170–15171. [DOI] [PubMed] [Google Scholar]
- Jackson T. A.; Rohde J. U.; Seo M. S.; Sastri C. V.; DeHont R.; Stubna A.; Ohta T.; Kitagawa T.; Münck E.; Nam W.; Que L. Jr. J. Am. Chem. Soc. 2008, 130, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki K.; Nimri S.; Gross Z.; Proniewicz L. M.; Kincaid J. R. J. Am. Chem. Soc. 1996, 118, 2929–2935. [Google Scholar]
- Lansky D. E.; Goldberg D. P. Inorg. Chem. 2006, 45, 5119–5125. [DOI] [PubMed] [Google Scholar]
- Zdilla M. J.; Dexheimer J. L.; Abu-Omar M. M. J. Am. Chem. Soc. 2007, 129, 11505–11511. [DOI] [PubMed] [Google Scholar]
- Nakazawa J.; Ogiwara H.; Kashiwazaki Y.; Ishii A.; Imamura N.; Samejima Y.; Hikichi S. Inorg. Chem. 2011, 50, 9933–9935. [DOI] [PubMed] [Google Scholar]
- Leeladee P.; Baglia R. A.; Prokop K. A.; Latifi R.; de Visser S. P.; Goldberg D. P. J. Am. Chem. Soc. 2012, 134, 10397–10400. [DOI] [PubMed] [Google Scholar]
- Smeltz J. L.; Lilly C. P.; Boyle P. D.; Ison E. A. J. Am. Chem. Soc. 2013, 135, 9433–9441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.