

Abstract
The success of enantioselective olefin metathesis relies on the design of enantioenriched alkylidene complexes capable of transferring stereochemical information from the catalyst structure to the reactants. Cyclometalation of the NHC ligand has proven to be a successful strategy to incorporate stereogenic atoms into the catalyst structure. Enantioenriched complexes incorporating this design element catalyze highly Z- and enantioselective asymmetric ring opening/cross metathesis (AROCM) of norbornenes and cyclobutenes, and the difference in ring strain between these two substrates leads to different propagating species in the catalytic cycle. Asymmetric ring closing metathesis (ARCM) of a challenging class of prochiral trienes has also been achieved. The extent of reversibility and effect of reaction setup was also explored. Finally, promising levels of enantioselectivity in an unprecedented Z-selective asymmetric cross metathesis (ACM) of a prochiral 1,4-diene was demonstrated.
Introduction
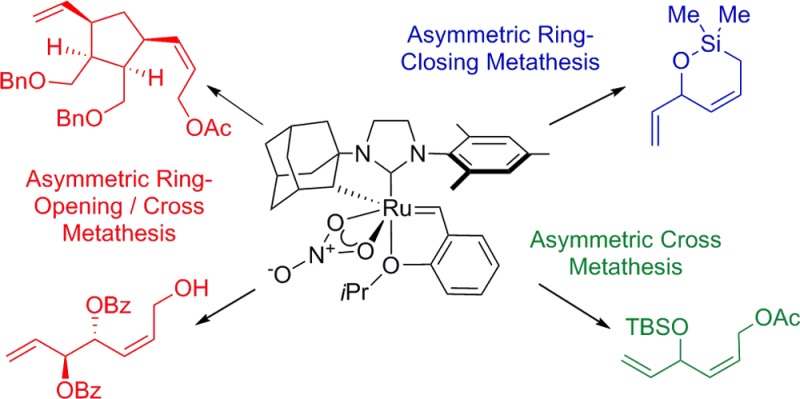
Olefin metathesis is a powerful method for the construction of C=C bonds in a large number of synthetic contexts, including target oriented synthesis,1 polymer chemistry,2 and renewable feedstock derivatization.3 Extensive efforts have been made to design tailored catalysts for each application.4 The development of asymmetric olefin metathesis catalysts has enabled the synthesis of enantioenriched compounds containing olefin functional groups, which are useful functional handles for further transformations. Generations of Mo- and Ru-based catalysts have been developed for asymmetric ring opening cross metathesis (AROCM), asymmetric ring closing metathesis (ARCM), asymmetric ring rearrangements (ARR) and asymmetric cross metathesis (ACM) (Figure 1). These methods have been applied in the synthesis of useful synthetic building blocks and natural products.5 Despite progress in catalyst design, however, significant challenges remain. Controlling olefin geometry in AROCM and ACM while maintaining high enantioselectivity is difficult. Furthermore, ARCM of unhindered trienes has so far been unsuccessful, resulting in extremely low enantioselectivities.
Figure 1.

Representative examples of the four manifolds of asymmetric olefin metathesis.
The first chiral Ru-based catalyst (1, Figure 2)6 possessed a C2-symmetric NHC ligand with chiral centers on the backbone of the NHC and unsymmetrical N-aryl substituents. This chiral information was relayed to the metal center through a gearing effect.7 Complex 1 catalyzed desymmetrizing ARCM to afford dihydropyrans in high ee. It was found that substitution of chloride for iodide ligands resulted in higher ee but lower yield. The highest levels of enantioinduction were obtained on substrates with E-trisubstituted enantiotopic olefins; Z-trisubstituted or 1,1-disubstituted enantiotopic olefins reacted with much lower selectivity. Subsequent modifications of the N-aryl substituents resulted in a more selective catalyst (2)8 for ARCM and AROCM, although the latter transformation took place with poor E/Z selectivity.9 C1-symmetric NHC ligands employing a geared arene substituent have also been developed by Collins10 and Blechert.11 For example, C1-symmetric catalyst 3 was capable of performing ARCM to generate tetrasubstituted olefins with good enantioselectivity.10e
Figure 2.

Selected enantioenriched Ru-based olefin metathesis catalysts.
Hoveyda has developed stereogenic-at-Ru complexes bearing a binaphthyl aryloxide NHC substituent.12 These complexes, which can be isolated as a single diastereomer, were used in E-selective AROCM and ARCM of trienes containing disubstituted enantiotopic olefins. Later, a modified complex 4 containing NHC backbone chirality and a biphenyl aryloxide substituent was reported to have improved activity in E-selective AROCM of terminal olefins,13 and to catalyze Z-selective AROCM with vinyl ethers and vinyl sulfides.14 Subsequent studies demonstrated that a higher energy diastereomer (differing in configuration at Ru) is accessible. These diastereomers can interconvert either through olefin metathesis, or a nonmetathesis based polytopal rearrangement (thermal or Brønsted acid catalyzed).15
Substantial progress has been made in the development of cyclometalated Ru complexes such as (rac)-5, which catalyze the Z-selective cross metathesis of terminal olefins.16 We anticipated that these complexes could also be highly enantioselective catalysts in asymmetric metathesis. A mechanistic proposal has been developed based on the preference of these complexes to react through side-bound metallacyclobutanes (syn to the NHC). This orientation forces all substituents in the forming metallacyclobutane to point away from the NHC N-aryl group, thus favoring the formation of the Z-olefin product.17 Recently, it has been shown that (rac)-5 can be resolved to generate enantioenriched 5. Complex 5 performs enantio- and Z-selective AROCM of norbornenes18 and cyclobutenes.19
Herein, we disclose a full account of our synthetic and mechanistic studies involving cyclometalated Ru-complexes 5 and 6 in Z-selective AROCM. Furthermore, these complexes are shown to provide promising levels of enantioinduction in two previously challenging transformations: ARCM of trienes composed of terminal olefins and ACM of prochiral 1,4-dienes. The impact of X-type ligand substitution on reactivity and selectivity is also analyzed leading to identification of nitrate 5 as the optimal catalyst for desymmetrizing transformations.
Results and Discussion
Catalyst Synthesis
Resolution of (rac)-5 was accomplished by ligand exchange of nitrate for iodide, which facilitated a second exchange with enantiopure silver carboxylate 8 (Scheme 1). Attempted exchange with several other carboxylates was unsuccessful, for example, reaction with the silver salts of α-methoxy-α-(trifluoromethyl)phenyl acetic acid, N-acetyl tert-leucine, or N-acetyl phenylglycine resulted in rapid decomposition. The mandelate-derived diastereomers 6a and 6b were chromatographically separable (under N2 atmosphere), to afford a 45% yield (90% of theoretical) of diastereomer 6a (>95:5 dr).18 A more rapid, but lower yielding, route was discovered wherein trituration of the mixture of 6a and 6b with 1:1 Et2O/pentane resulted in the isolation of pure 6a (77% of theoretical) due to the large difference in solubility between the diastereomers. The latter procedure is a marked improvement in the speed at which synthetically useful quantities of enantioenriched 5 can be produced. Waste is avoided by the easy recyclability of the washes containing the partially enriched diastereomeric mixture. Pure carboxylate 6a was then converted to the nitrate 5 by treatment with p-TsOH followed by NaNO3.20
Scheme 1. Resolution of (rac)-5.

(a) NaI, THF, 75% yield; (b) (S)-AgO2CCH(Ph)(OMe) (8) (2 equiv), THF, 23 °C, 1.5 h, 97%; (c) (1) 6a, pTsOH·H2O, THF, 5 min; (2) NaNO3, THF/MeOH, 15 min, 43%.
Asymmetric Ring Opening Cross Metathesis (AROCM)
AROCM of strained olefins is a powerful method for the construction of enantioenriched cyclic and acyclic dienes containing up to 5 stereocenters. The products contain two differentially substituted alkenes, which are poised for subsequent chemoselective transformations.9,11−15,18,19,21
We proposed that the cyclometalated NHC complexes 5 and 6, which contain stereogenic carbon and Ru atoms, would control the approach of the strained olefin reactant to the reactive metal center leading to a highly stereoselective AROCM reaction. The bulky adamantyl group limits approach of the reactant olefin solely toward the opposite face of the alkylidene. Strong preference for side-bound metallacyclic intermediates would result in higher fidelity communication of the stereochemical information stored in the NHC ligand. Finally, the pocket capped by the N-aryl substituent of the NHC is well suited for the Z-selective ring opening as it favors the formation of all-cis metallacyclobutanes, which had been previously observed in the context of ring opening metathesis polymerization (ROMP).22
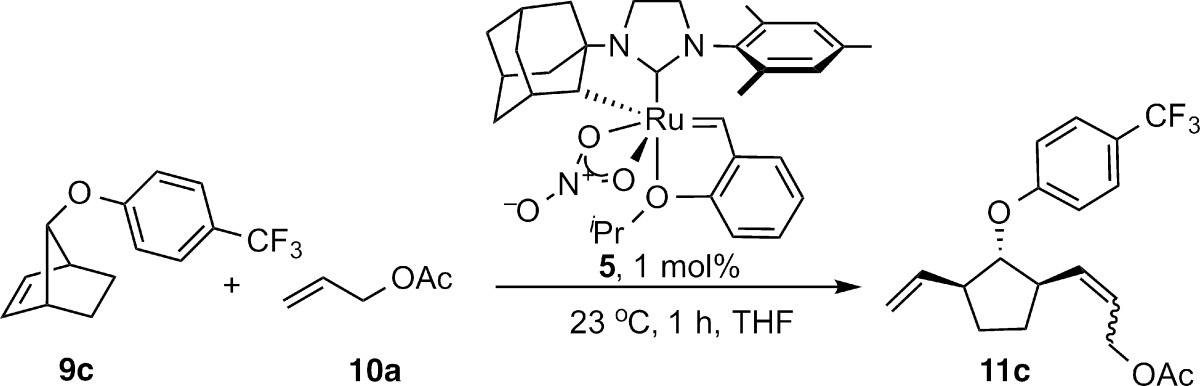
In accord with this proposal, enantioenriched cyclometalated complex 5 catalyzed the AROCM of norbornenes18 and cyclobutenes,19 resulting in the first ruthenium-catalyzed Z-selective and enantioselective ring opening of these strained rings with simple terminal olefins (Table 1). 2,3-Di-endo-substituted norbornenes afforded products in high Z-selectivity, and 2,3-dibenzyloxy substitution resulted in high enantioselectivity (93%, Table 1, entry 1). Substrates bearing 7-anti and 7-syn substitution (9c and 9d) were well tolerated, affording products in high ee (entries 3 and 4). However, the lack of 2,3 disubstitution appeared to have a strong influence on the diastereoselectivity of the reaction. The strongest effect arose from 7-syn substitution, which resulted in a preference for the E product.23 Benzonorbornadiene 9e, possessing sp2 carbons at the 2 and 3 positions, also resulted in reduced Z-selectivity, although the products were formed in excellent enantiomeric excess. Regardless of the E/Z selectivity, it was observed that in cases where both geometrical isomers could be isolated, the E and Z isomers were formed with identical enantioenrichment (entries 3–5).
Table 1. Selected Examples of Z-Selective AROCM Catalyzed by 5.
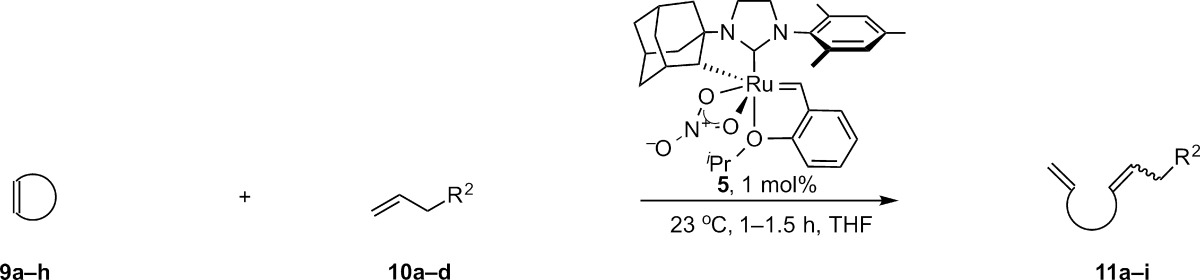
In contrast to the reactions employing norbornenes, the AROCM of cyclobutenes occurred with higher yield and comparable levels of Z-selectivity.19 AROCM of cyclobutenes tolerated commonly used oxygen protecting groups as well as free alcohols on both the cyclobutene (9f) and terminal olefin (10c) reactants. Notably, in stark contrast to the outcome for the norbornene AROCM, the ee’s of the Z and E products differed considerably. This difference was observed in all 7 cases where both isomers could be analyzed. The ee’s of Z and E products ranged from 91 and 67% ee, to 93 and 86% ee (for example, Table 1, entries 6–9).
To determine the stereochemical relationship between the double bond isomers, E-9e and Z-9e were hydrogenated to afford 12. Both reactions afforded the same major enantiomer of 12 as determined by chiral SFC (Scheme 2), demonstrating that the absolute configurations at the stereogenic carbons of E-9e and Z-9e were identical. The equal magnitude and sense of enantioenrichment suggests a common intermediate in the AROCM of norbornenes from which the E and Z products are ultimately generated.
Scheme 2. Determination of Relative Stereoinduction in E and Z Products.

A possible explanation for the identical enantioenrichment of the Z- and E-isomers is that a secondary metathesis process isomerizes some of the Z product to the more thermodynamically favored E product. However, this was ruled out by the observation that the E/Z ratio of AROCM products was constant throughout the course of the reaction and for several hours after complete conversion. Likewise in the cyclobutene AROCM, resubmission of product Z-11f to the reaction conditions in the presence of enantiopure catalyst 5 resulted in recovery of the pure Z product in identical enantioenrichment and Z/E ratio. These experiments strongly suggest that secondary metathesis proceeds at a negligible rate as compared to the productive AROCM reaction.
Our observations regarding AROCM of norbornenes and cyclobutenes suggest that the structure and strain energy of the cyclic olefin reactant dramatically alter the catalytic pathway responsible for the mono-cross products (Scheme 3). The identical enantioenrichment of the E and Z products formed in the AROCM of norbornenes suggests that a methylidene intermediate is involved in the enantiodetermining ring-opening step. The resultant alkylidene then reacts with an equivalent of terminal olefin to afford the monocross products. Since the enantiodetermining step precedes the olefin geometry determining step, the E and Z products must necessarily have identical enantioenrichment.
Scheme 3. Proposed Change in Mechanism for AROCM of Norbornenes and Cyclobutenes.

In contrast, the E and Z AROCM products derived from cyclobutenes are formed with different ee’s, suggesting that the initial ring-opening step occurs through the alkylidene derived from the terminal olefin. In the proposed pathway for the AROCM of cyclobutenes with 5, the initial ring opening of the strained olefin with an alkylidene derived from the terminal olefin is diastereo- and enantiodetermining, resulting in the difference in enantioenrichment for the E and Z products.24
The increased strain and lower steric demand of the cyclobutenes result in propagation through a Ru-alkylidene species, while the bulkier and less strained norbornenes result in propagation through a Ru-methylidene. The absolute configuration of the AROCM products requires that the methylidene intermediate of the cyclobutene AROCM possess the opposite configuration at ruthenium compared to the alkylidene in the norbornene AROCM. Nonproductive metathesis events could potentially be responsible for epimerization of the ruthenium center, providing access to the necessary active species.
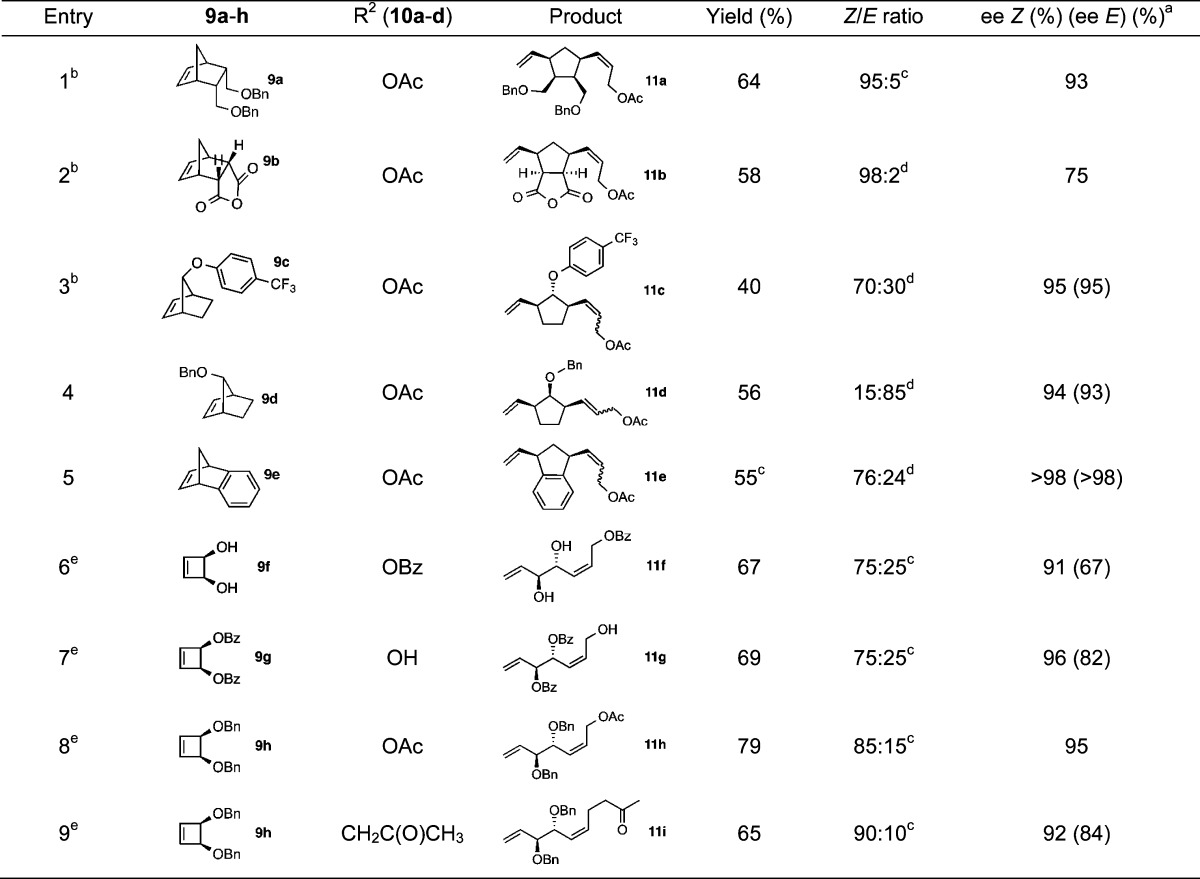
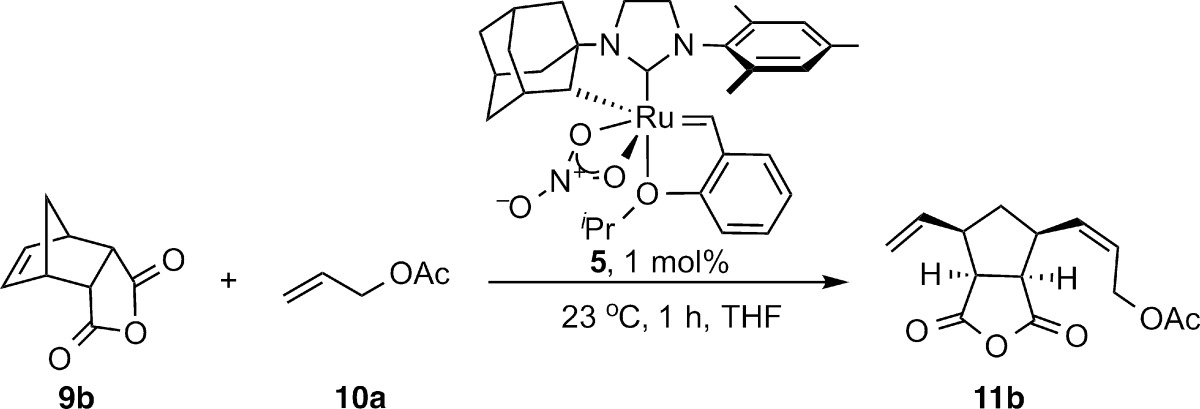
The effect of terminal olefin equivalents and concentration on stereoselectivity has previously been studied for certain catalysts.9,21a,21f In an attempt to better understand the impact of concentration and stoichiometry on the AROCM of norbornenes and cyclobutenes, the reactions of 9b and 9c with allyl acetate were studied in further detail. In the case of 2,3-di-endo substituted norbornene 9b, the Z/E ratio remained constant and ee was only slightly affected by concentration and equivalents of olefin (Table 2). A similar independence of diastereoselectivity and ee were observed in the AROCM of cyclobutenes.19
Table 2. Effect of Concentration and Equivalents of Terminal Olefin on the AROCM of Norbornene 9b.
| concentration (M) | equiv 10a | conversion (%)a | Z/E ratiob | Z ee (%)c |
|---|---|---|---|---|
| 0.05 | 7 | >95d | 97:3 | 75 |
| 0.1 | 7 | >95e | 97:3 | 74 |
| 0.5 | 7 | >95 | 98:2 | 75 |
| 0.5 | 3 | 40 | 97:3 | 72 |
Determined by 500 MHz 1H NMR.
Determined by GC.
Determined by chiral SFC on chromatographically purified products.
Full conversion achieved after 16 h.
Full conversion achieved after 4 h.
While the ee of Z-11c produced by the AROCM of 9c with allyl acetate was unaffected by concentration and equivalents of terminal olefin, the Z/E ratio was dependent on both variables with higher concentration and more terminal olefin favoring Z-11c (Table 3). Thus, in the absence of chelating substituents on the strained olefin component, the diastereodetermining cross metathesis with the terminal olefin is dependent on the concentration of the terminal olefin. A similar dependence of olefin geometry on concentration is observed in ROMP catalyzed by homogeneous alkylidene initiators.25 In ROMP, it has been proposed that the concentration dependence arises from a rate competition between first order rotation of the alkylidene and second order [2 + 2] cycloaddition with the incoming monomer. Higher concentrations have a greater influence on the second order process. In the current AROCMs, we propose that a similar effect is occurring with the dependence on concentration and the incoming reactant. This dependence suggests that cross metathesis to release the mono-cross product competes with rotation of the alkylidene.
Table 3. Effect of Concentration and Equivalents of Terminal Olefin on the AROCM of Norbornene 9c.
| concentration (M) | equiv 10a | conversion (%)a | Z/E ratiob | Z ee (%)c |
|---|---|---|---|---|
| 0.1 | 7 | >95 | 44:56 | 96 |
| 0.5 | 7 | >95 | 70:30 | 95 |
| 0.5 | 3 | >95 | 59:41 | 96 |
Determined by 500 MHz 1H NMR.
Determined by GC.
Determined by chiral SFC on chromatographically purified products
Asymmetric Ring Closing Metathesis (ARCM)
A considerable amount of work has been performed on catalyst development and applications for ARCM.6,8,10,11a,12b,26 The products of desymmetrizing ARCM are potentially useful in target-oriented synthesis since two differentiated olefins are present in the final enantioenriched product. These olefins provide an ideal platform for further functionalization. ARCM has been used as the key step in a number of natural product total syntheses.5b,26n,26p,27 Despite much progress, ARCM substrates have largely been limited to cases where the unique olefin is considerably less bulky than the enantiotopic olefins. Only isolated examples of all-terminal trienes lacking allylic quaternary substitution have proven successful.26h,26n,28 Furthermore, it has been noted in several reports that attempted ARCM of various all-terminal trienes have been unsuccessful, due either to low ee26f,26g,26o,26s or formation of oligomers.26i The ARCM of unhindered trienes is particularly challenging due to the difficulty in controlling the cyclization pathway, and the need to differentiate between relatively small enantiotopic groups.
The RCM of prochiral trienes can, in principle, proceed through two distinct pathways (Scheme 4). In pathway A, the initial alkylidene formation occurs on one of the two enantiotopic olefins, followed by cyclization with the unique olefin. If initial alkylidene formation is irreversible, then this step is enantiodiscriminating. In pathway B, initial alkylidene formation occurs with the unique olefin, and this alkylidene subsequently cyclizes with one of the two enantiotopic olefins in the enantiodiscriminating step. To a first approximation, an enantiodiscriminating cyclization step is more likely to be highly enantioselective than alkylidene formation, because of the ordered nature of a cyclic transition state. Furthermore, in order to achieve high enantioselectivity, it is desirable to ensure that only one pathway is operating. Competing pathways will lead to decreased enantioselectivity unless they are both highly enantioselective for the same product enantiomer (an unlikely scenario).
Scheme 4. Possible Pathways to ARCM Products.
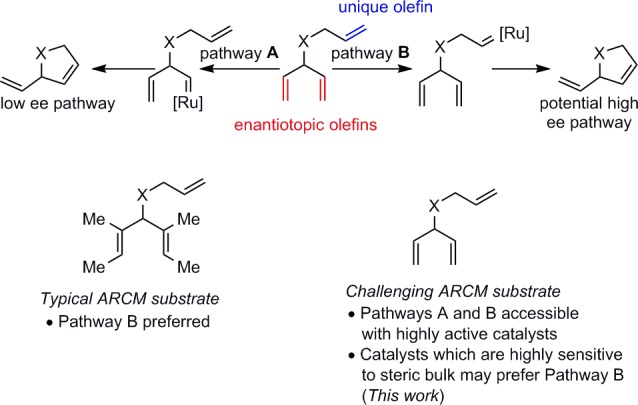
The substitution pattern of the enantiotopic olefins can also impact the relative energy of diastereomeric transition states. Cavallo has performed computational studies on the origin of stereoselectivity with geared NHC Ru complexes.7 It was found that the nonreacting olefin is oriented in pseudo-equatorial and pseudo-axial positions in the respective diastereomeric cyclization transition states. Higher selectivities are therefore expected when this substituent is large, leading to a large energy difference between pseudo-equatorial and pseudo-axial configurations.
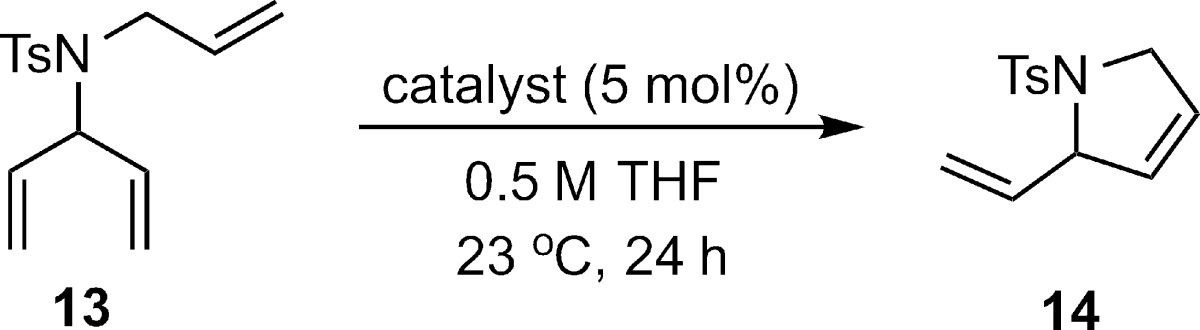
We felt that the necessity of employing highly substituted enantiotopic olefins has limited the potential utility of ARCM products. We have previously observed that chelated complexes such as (rac)-5 are sensitive to steric bulk at the allylic position.16e We hypothesized that resolved complex 5 would be an ideal candidate for ARCM of trienes such as 13, since this catalyst would likely disfavor initial alkylidene formation with the enantiotopic olefins and favor initial reaction with the allyl fragment. This preference would bias the system to undergo enantiodetermining ring closing metathesis, a pathway that is likely to lead to higher enantioinduction. Success of this strategy would improve the scope of the ARCM reaction by allowing the generation of cyclic products lacking the cumbersome substitution on the resultant product alkenes.
Furthermore, we wondered whether the addition of further steric bulk, through modification of the X-type ligand, could positively impact enantioselectivities. Complexes 6b–6i (Scheme 5) were prepared by ligand exchange from enantioenriched iodide ent-7.29 This reaction proceeded to full conversion based on 1H NMR and afforded products of sufficient purity after concentration, redissolution in benzene, and filtration through a short plug of Celite.
Scheme 5. Synthesis of Carboxylate Complexes.

Complexes containing achiral carboxylate ligands (6c–6e) and enantiopure carboxylates (6b, 6f–6i) were obtained. While the cyclometalated iodide complex ent-7 was inactive in RCM, all of the carboxylate complexes were found to be competent catalysts with varying levels of enantioselectivity (Table 4). Thus, while κ2 ligands are more active than monodentate ligands, the electronics and sterics of the carboxylate ligand also impact the ARCM reaction.
Table 4. Efficiency and Enantioselectivity of 1, 5, 6b–6i, and ent-7 in ARCM of 13.
| entry | catalyst | conversion (%)a | ee (%)b |
|---|---|---|---|
| 1 | ent-7 | <2 | ND |
| 2 | 6b | >98 | 42 |
| 3 | 6c | 35 | 36 |
| 4 | 6d | 35 | 53 |
| 5 | 6e | 76 | 46 |
| 6 | 6f | >98 | 18 |
| 7 | 6g | >98 | 42 |
| 8 | 6h | 48 | 43 |
| 9 | 6i | 72 | 40 |
| 10 | 5 | >98 | 54 |
| 11 | 1 | 20 | 0 |
Determined by 1H NMR spectroscopy;
Determined by chiral SFC analysis
More substituted aliphatic carboxylates, such as the pivalate 6e and N-acetyl amino carboxylates 6g–6i, were more competent catalysts than acetate 6c, forming the product in essentially full conversion (entries 5, 7–9). While the steric bulk of the amino acid side chain had little bearing on the enantioselectivity (no difference was observed between alanine and valine), the presence of an electron-withdrawing heteroatom in the α position of the carboxylate afforded a more enantioselective catalyst. For example, the 2-phenylbutyric acid-derived catalyst 6f generated the product in only 18% ee, while the relatively isosteric O-methyl mandelate 6b provided 13 in 42% ee (entries 6 and 2).30 The stereochemistry of the carboxylate has little influence on the stereochemical outcome of the ARCM as complexes 6g and 6h, containing either d- or l-alanine carboxylates, gave identical ee (entries 7 and 8). We concluded that the nitrate catalyst 5 (98% conversion, 54% ee, entry 10) was a significant improvement to the previous generation geared catalyst 1 (20% conversion, 0% ee, entry 11) and sought to study the reaction scope enabled by this advance.
We next probed the influence of substitution in the allylic position, nature of the heteroatom, and ring size on the efficiency and enantioselectivity of ARCM. Prochiral trienes composed of monosubstituted olefins were cyclized cleanly, resulting in generally high yields (Table 5). Moving from a dimethyl siloxy to the bulkier diphenyl siloxy tether resulted in a slower cyclization and required an increase in catalyst loading to achieve good conversion (entries 1 and 2). Triene 19, which contains trisubstituted enantiotopic olefins, did not undergo ring closure (entry 3). Saturated nitrogen-containing heterocycles were formed in high yield and moderate enantioselectivity, (entries 4 and 5).
Table 5. Scope of ARCM Reaction with 5a.
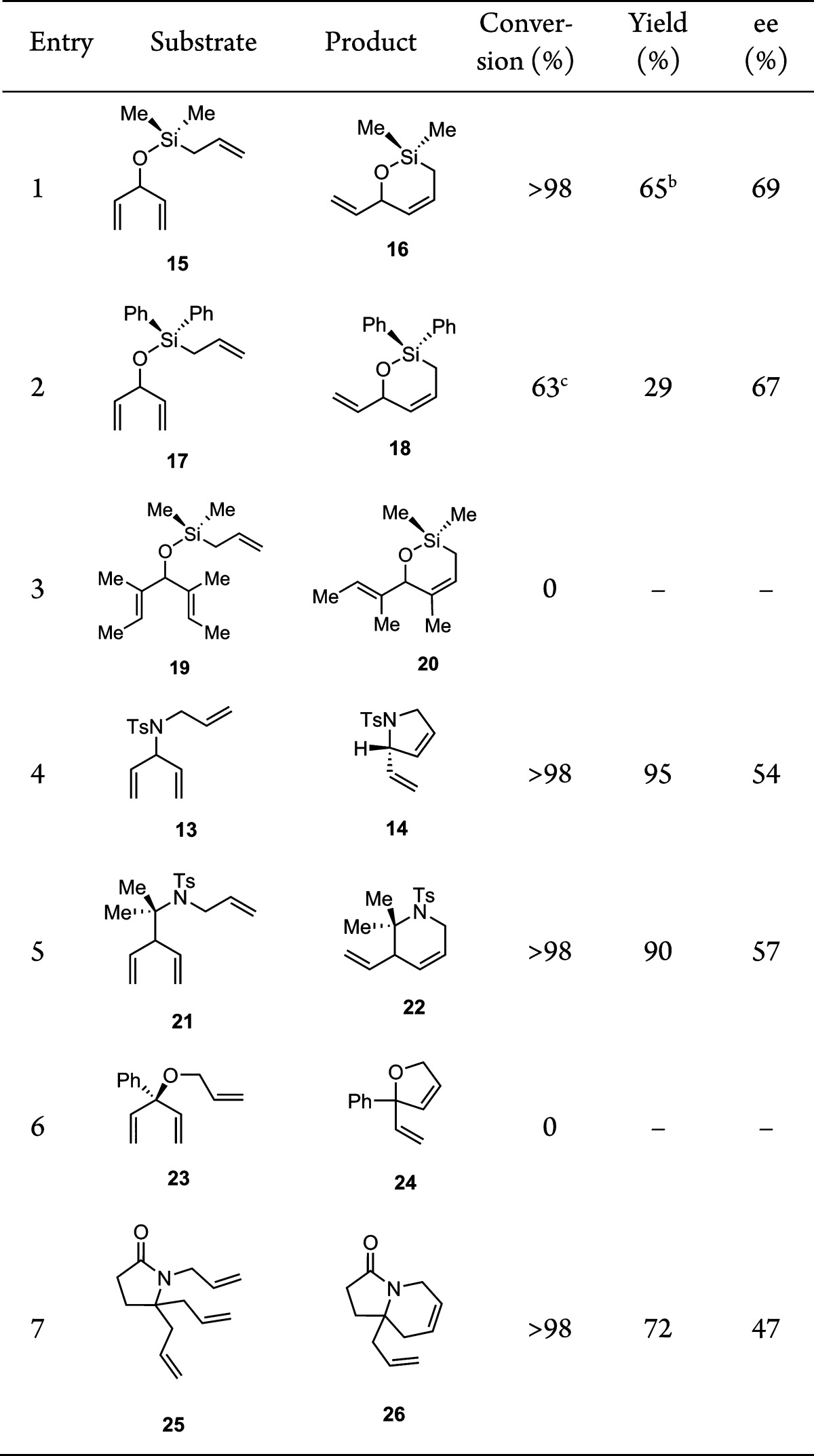
Reaction conditions: triene (0.5 M), 5 (5 mol %), THF, 23 °C, 24 h.
Determined by 1H NMR using mesitylene as an internal standard.
Using 10 mol % catalyst.
A particularly challenging substrate 23, bearing a fully substituted carbon in the allylic position of the 1,4-diene moiety, completely shut down the reaction (entry 6). On the other hand, the presence of a fully substituted carbon in the homoallylic position, as in 25, restored reactivity (entry 7). These results suggest that reducing the steric bulk of the catalyst, perhaps through the use of alternative cyclometalated NHC ligands, may expand the scope of the reaction to form synthetically challenging tertiary ether products.
Triene 27, containing a homoallyl diphenyl silyl group, was synthesized in order to test the efficiency of forming seven membered rings (Figure 4a). In contrast to triene 17, 27 underwent ring closure under the standard conditions in good yield, indicating that the additional methylene unit was sufficient to relieve the steric bulk of the diphenylsilyl unit. Surprisingly, the product was racemic. To probe whether enantioselectivity is lost due to reversibility, the reaction was performed in a sealed NMR tube and monitored by 1H NMR (Figure 3). After 4 h, 71% conversion had been achieved. However, the reaction eventually stalled at 78% conversion. Upon purging ethylene from the NMR tube, the reaction resumed and eventually reached 92% conversion. This result suggests that in a closed vessel, the RCM of 27 is reversible, and equilibrium can be reached prior to full conversion. The reversibility of the reaction erodes any enantioenrichment that is initially achieved. Therefore, efficient removal of ethylene is required to obtain enantioenrichment.
Figure 4.

(a) Effect of open vial on enantioselectivity in ARCM of triene 27; (b) Effect of open vial on enantioselectivity in ARCM of triene 13.
Figure 3.
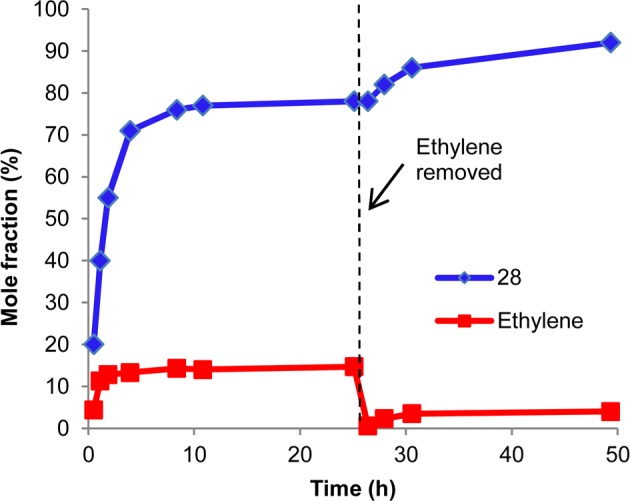
Time course for the ARCM of 27 (closed NMR tube).
To remove ethylene during the course of the ARCM reaction, the reaction was performed in an open vial in a nitrogen filled glovebox (Figure 4). After 24 h, full conversion of starting material was achieved, and the 7-membered product was generated in 37% ee. This result demonstrates the importance of assessing reversibility in ARCM reactions and suggests that removal of ethylene limits reversibility. Triene 13 was also subjected to open vial conditions (Figure 3b). Although reactivity was slightly diminished relative to closed vial conditions, the product was generated in an almost identical 58% ee, (compared to 54% ee for closed vial).31 Therefore, reversibility is not significant with triene 13. The reversible nature of the ARCM of 27, but not 13, is most likely due to the increased ring strain of 28.
The absolute configuration of diene 14 (Figure 5) produced by ARCM of 13 with 5 was determined by X-ray crystallographic analysis to be (2S). On the basis of the absolute configuration of 14, we propose that enantioinduction arises from the favorable conformational effect of placing the unreacted vinyl group of the 1,4-diene fragment in a pseudo-equatorial, as opposed to pseudo-axial, orientation (Figure 6).
Figure 5.
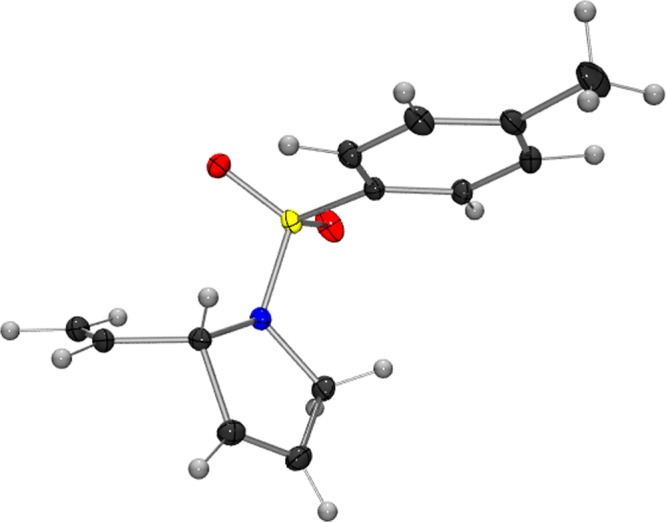
ORTEP drawing of 14.
Figure 6.

Tentative transition state model for the formation of (2S)-14.
Asymmetric Cross Metathesis (ACM)
Cross metathesis of prochiral 1,4, 1,5, or 1,6 dienes to afford desymmetrized metathesis products has remained an elusive method for the construction of allylic and homoallylic stereocenters. The lack of success is likely due to three factors: (1) difficulty in controlling the nature of the propagating species; (2) limiting secondary metathesis events resulting in symmetrical products; and (3) designing a chiral environment capable of high levels of enantioinduction. However, a previous example of ACM suggested that enantiopure Ru-based metathesis catalysts are capable of desymmetrizing cross metathesis, although in modest yields (17–54%) and ee’s (4–52%).9
After optimization of reaction conditions, we have observed that cyclometalated complex 5 catalyzes the ACM of 1,4 diene 29 with cis-1,4-diacetoxy-2-butene in 35% yield and with a promising ee of 50% (eq 1). In contrast to the previous report of E-selective ACM with C2-symmetric catalysts, this method provides the Z-isomer. While these results are preliminary, they suggest that further optimization of the ligand set and choice of the proper substitution on the pro-stereogenic carbon atom of the diene reactant may result in highly enantioenriched 1,4-diene products, which will be useful chiral building blocks in complex molecule synthesis.
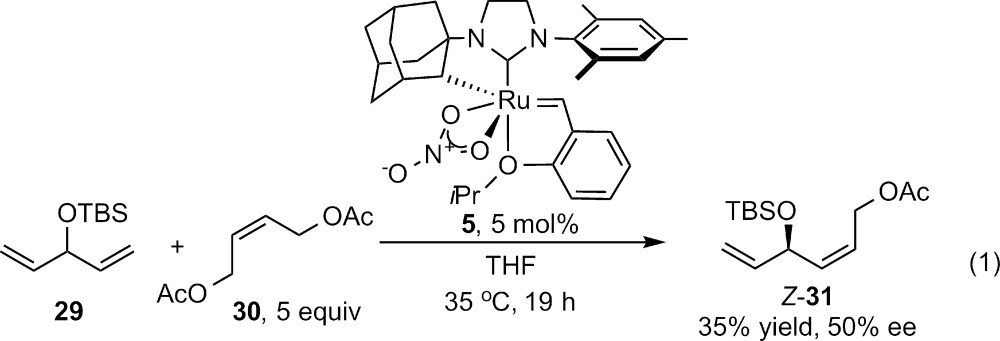 |
1 |
Conclusion
Cyclometalated ruthenium complexes, which are resolved by treatment with an enantiopure carboxylate and readily diversified by ligand exchange, have demonstrated high levels of enantioinduction in the reaction manifolds comprising enantioselective olefin metathesis. AROCM of cyclobutenes and norbornenes with 5 afforded, in many cases, highly Z and enantioenriched 1,4- and 1,6-dienes, respectively. In comparison to previous generations of C2 symmetric ruthenium alkylidenes, complexes 5 and 6 are capable of desymmetrizing prochiral trienes composed solely of monosubstituted olefins. Preliminary results suggest that 5 is capable of ACM with a level of enantioselectivity on par with the state of the art, and complementary in its ability to afford Z products.
Study of these reactions has led us to conclude that control of the active catalytic species through the manipulation of several experimental variables can greatly influence the outcome of the enantioselective olefin metathesis reactions. The influence of ring strain and steric bulk of the strained olefin on the mechanism of AROCM has led to a hypothesis for the active catalyst species in reactions catalyzed by 5. The efficiency and enantioselectivity of ARCM reactions catalyzed by cyclometalated catalysts is a function of both the X-type ligand and substitution pattern on the prochiral triene reactant. In cases where a medium-sized ring is formed, efficient removal of ethylene is required to prevent reversibility, which would otherwise erode enantioselectivity. The insights gained in this study will facilitate further developments in the field of asymmetric metathesis and will contribute more broadly to the development of new metathesis catalysts.
Acknowledgments
This work was financially supported by the NIH (5R01GM031332-27 to R.H.G.) and the NSF (CHE-1212767 to R.H.G.). Thanks to Mr. L. M. Henling for X-ray crystallography. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to the California Institute of Technology (CHE-0639094). NMR spectra were obtained by instruments supported by the NIH (RR027690). Materia, Inc. is thanked for its donation of metathesis catalysts. Dr. Daryl Allen of Materia, Inc., Dr. Jeffrey Cannon, Zachary Wickens and Dr. Scott Virgil of the Caltech Center for Catalysis and Chemical Synthesis are thanked for helpful advice.
Supporting Information Available
Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† J.H. and P.K.D. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Metathesis in Natural Product Synthesis: Strategies, Substrates, and Catalysts; 1st ed.; Cossy J., Arseniyadis S., Meyer C., Eds.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]; b Nicolaou K. C.; Bulger P. G.; Sarlah D. Angew. Chem., Int. Ed. 2005, 44, 4490–4527. [DOI] [PubMed] [Google Scholar]
- a Handbook of Metathesis; Grubbs R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]; b Sutthasupa S.; Shiotsuki M.; Sanda F. Polym. J. 2010, 42, 905–915. [Google Scholar]
- Nickel A.; Pedersen R. L. In Olefin Metathesis: Theory and Practice; Grela K., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp 335–348. [Google Scholar]
- Vougioukalakis G. C.; Grubbs R. H. Chem. Rev. 2010, 110, 1746–1787. [DOI] [PubMed] [Google Scholar]
- a Stenne B.; Collins S. K. In Olefin Metathesis: Theory and Practice; Grela K., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp 233–268. [Google Scholar]; b Hoveyda A. H.; Malcolmson S. J.; Meek S. J.; Zhugralin A. R. Angew. Chem., Int. Ed. 2010, 49, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kress S.; Blechert S. Chem. Soc. Rev. 2012, 41, 4389–4408. [DOI] [PubMed] [Google Scholar]; d Hoveyda A. H. J. Org. Chem. 2014, 79, 4763–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiders T. J.; Ward D. W.; Grubbs R. H. Org. Lett. 2001, 3, 3225–3228. [DOI] [PubMed] [Google Scholar]
- Costabile C.; Cavallo L. J. Am. Chem. Soc. 2004, 126, 9592–9600. [DOI] [PubMed] [Google Scholar]
- Funk T. W.; Berlin J. M.; Grubbs R. H. J. Am. Chem. Soc. 2006, 128, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin J. M.; Goldberg S. D.; Grubbs R. H. Angew. Chem., Int. Ed. 2006, 45, 7591–7595. [DOI] [PubMed] [Google Scholar]
- a Fournier P.-A.; Collins S. K. Organometallics 2007, 26, 2945–2949. [Google Scholar]; b Fournier P.-A.; Savoie J.; Stenne B.; Bédard M.; Grandbois A.; Collins S. K. Chem.—Eur. J. 2008, 14, 8690–8695. [DOI] [PubMed] [Google Scholar]; c Grandbois A.; Collins S. K. Chem.—Eur. J. 2008, 14, 9323–9329. [DOI] [PubMed] [Google Scholar]; d Savoie J.; Stenne B.; Collins S. K. Adv. Synth. Catal. 2009, 351, 1826–1832. [Google Scholar]; e Stenne B.; Timperio J.; Savoie J.; Dudding T.; Collins S. K. Org. Lett. 2010, 12, 2032–2035. [DOI] [PubMed] [Google Scholar]
- a Tiede S.; Berger A.; Schlesiger D.; Rost D.; Lühl A.; Blechert S. Angew. Chem., Int. Ed. 2010, 49, 3972–3975. [DOI] [PubMed] [Google Scholar]; b Kannenberg A.; Rost D.; Eibauer S.; Tiede S.; Blechert S. Angew. Chem., Int. Ed. 2011, 50, 3299–3302. [DOI] [PubMed] [Google Scholar]
- a Van Veldhuizen J. J.; Garber S. B.; Kingsbury J. S.; Hoveyda A. H. J. Am. Chem. Soc. 2002, 124, 4954–4955. [DOI] [PubMed] [Google Scholar]; b Van Veldhuizen J. J.; Gillingham D. G.; Garber S. B.; Kataoka O.; Hoveyda A. H. J. Am. Chem. Soc. 2003, 125, 12502–12508. [DOI] [PubMed] [Google Scholar]
- Van Veldhuizen J. J.; Campbell J. E.; Giudici R. E.; Hoveyda A. H. J. Am. Chem. Soc. 2005, 127, 6877–6882. [DOI] [PubMed] [Google Scholar]
- Khan R. K. M.; O’Brien R. V.; Torker S.; Li B.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 12774–12779. [DOI] [PubMed] [Google Scholar]
- a Khan R. K. M.; Zhugralin A. R.; Torker S.; O’Brien R. V.; Lombardi P. J.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 12438–12441. [DOI] [PubMed] [Google Scholar]; b Torker S.; Khan R. K. M.; Hoveyda A. H. J. Am. Chem. Soc. 2014, 136, 3439–3455. [DOI] [PubMed] [Google Scholar]
- a Endo K.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Keitz B. K.; Endo K.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 9686–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Herbert M. B.; Lan Y.; Keitz B. K.; Liu P.; Endo K.; Day M. W.; Houk K. N.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 7861–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Keitz B. K.; Endo K.; Patel P. R.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Quigley B. L.; Grubbs R. H. Chem. Sci. 2013, 5, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Rosebrugh L. E.; Herbert M. B.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 1464–1467. [DOI] [PubMed] [Google Scholar]
- Hartung J.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10183–10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung J.; Grubbs R. H. Angew. Chem., Int. Ed. 2014, 53, 3885–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The configurational stability of the adamantyl stereocenter throughout these X-ligand modifications was confirmed: Conversion of the enantioenriched nitrate complex 5 to the corresponding iodide followed by treatment with enantioenriched carboxylates resulted in the generation of only a single diastereomer (vide infra).
- a La D. S.; Ford J. G.; Sattely E. S.; Bonitatebus P. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 1999, 121, 11603–11604. [Google Scholar]; b La D. S.; Sattely E. S.; Ford J. G.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2001, 123, 7767–7778. [DOI] [PubMed] [Google Scholar]; c Tsang W. C. P.; Jernelius J. A.; Cortez G. A.; Weatherhead G. S.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2003, 125, 2591–2596. [DOI] [PubMed] [Google Scholar]; d Gillingham D. G.; Hoveyda A. H. Angew. Chem., Int. Ed. 2007, 46, 3860–3864. [DOI] [PubMed] [Google Scholar]; e Ibrahem I.; Yu M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 3844–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Yu M.; Ibrahem I.; Hasegawa M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 2788–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Keitz B. K.; Fedorov A.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 2040–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rosebrugh L. E.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10032–10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J. G.; Ivin K. J.; Rooney J. J. J. Mol. Catal. 1986, 36, 115–125. [Google Scholar]
- Since the E/Z selectivity is determined at different steps for AROCM of norbornenes and cyclobutenes, a direct comparison between the E/Z selectivity of these two substrate classes cannot be made. Increasing the steric demand of the NHC arene to improve the E/Z selectivity is a possible direction for further catalyst development.
- a Oskam J. H.; Schrock R. R. J. Am. Chem. Soc. 1993, 115, 11831–11845. [Google Scholar]; b Bishop J. P.; Register R. A. Polymer 2010, 51, 4121–4126. [Google Scholar]
- a Fujimura O.; Grubbs R. H. J. Am. Chem. Soc. 1996, 118, 2499–2500. [Google Scholar]; b Fujimura O.; Grubbs R. H. J. Org. Chem. 1998, 63, 824–832. [DOI] [PubMed] [Google Scholar]; c Alexander J. B.; La D. S.; Cefalo D. R.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 1998, 120, 4041–4042. [Google Scholar]; d La D. S.; Alexander J. B.; Cefalo D. R.; Graf D. D.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 1998, 120, 9720–9721. [Google Scholar]; e Zhu S. S.; Cefalo D. R.; La D. S.; Jamieson J. Y.; Davis W. M.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 1999, 121, 8251–8259. [Google Scholar]; f Dolman S. J.; Sattely E. S.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 2002, 124, 6991–6997. [DOI] [PubMed] [Google Scholar]; g Weatherhead G. S.; Houser J. H.; Ford J. G.; Jamieson J. Y.; Schrock R. R.; Hoveyda A. H. Tetrahedron Lett. 2000, 41, 9553–9559. [Google Scholar]; h Cefalo D. R.; Kiely A. F.; Wuchrer M.; Jamieson J. Y.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2001, 123, 3139–3140. [Google Scholar]; i Kiely A. F.; Jernelius J. A.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2002, 124, 2868–2869. [DOI] [PubMed] [Google Scholar]; j Dolman S. J.; Schrock R. R.; Hoveyda A. H. Org. Lett. 2003, 5, 4899–4902. [DOI] [PubMed] [Google Scholar]; k Tsang W. C. P.; Hultzsch K. C.; Alexander J. B.; Bonitate-bus P. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2003, 125, 2652–2666. [DOI] [PubMed] [Google Scholar]; l Dolman S. J.; Hultzsch K. C.; Pezet F.; Teng X.; Hoveyda A. H.; Schrock R. R. J. Am. Chem. Soc. 2004, 126, 10945–10953. [DOI] [PubMed] [Google Scholar]; m Jernelius J. A.; Schrock R. R.; Hoveyda A. H. Tetrahedron 2004, 60, 7345–7351. [Google Scholar]; n Sattely E. S.; Cortez G. A.; Moebius D. C.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2005, 127, 8526–8533. [DOI] [PubMed] [Google Scholar]; o Lee A.-L.; Malcolmson S. J.; Puglisi A.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2006, 128, 5153–5157. [DOI] [PubMed] [Google Scholar]; p Malcolmson S. J.; Meek S. J.; Sattely E. S.; Schrock R. R.; Hoveyda A. H. Nature 2008, 456, 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Harvey J. S.; Malcolmson S. J.; Dunne K. S.; Meek S. J.; Thompson A. L.; Schrock R. R.; Hoveyda A. H.; Gouverneur V. Angew. Chem., Int. Ed. 2009, 48, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Sattely E. S.; Meek S. J.; Malcolmson S. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]; s Ogasawara M.; Watanabe S.; Nakajima K.; Takahashi T. J. Am. Chem. Soc. 2010, 132, 2136–2137. [DOI] [PubMed] [Google Scholar]; t Grisi F.; Costabile C.; Gallo E.; Mariconda A.; Tedesco C.; Longo P. Organometallics 2008, 27, 4649–4656. [Google Scholar]; u Gawin R.; Pieczykolan M.; Malińska M.; Woźniak K.; Grela K. Synlett 2013, 24, 1250–1254. [Google Scholar]
- a Burke S. D.; Müller N.; Beaudry C. M. Org. Lett. 1999, 1, 1827–1829. [DOI] [PubMed] [Google Scholar]; b Funk T. W. Org. Lett. 2009, 11, 4998–5001. [DOI] [PubMed] [Google Scholar]
- It is unclear whether the mechanism involves a direct ARCM, or an RCM of the two enantiotopic olefins to generate an achiral cyclopentene, followed by ARCM/ring-opening metathesis isomerization.
- ent-7 was obtained from diastereomer 6b by treatment with NaI.
- Complexes 6b–6i possess opposite stereochemistry at ruthenium and the adamantyl stereocenter to that of 5 (nitrate), and all favored the opposite product enantiomer. This indicates that the ruthenium and adamantyl stereocenters are the primary determinants of the sense of enantioinduction.
- In open vial reactions, the solvent evaporated over the course of the reaction, and the reaction mixture became viscous. This change in reaction medium may explain the small increase in ee (from 54 to 58) and drop in yield with triene 13.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.