

Abstract
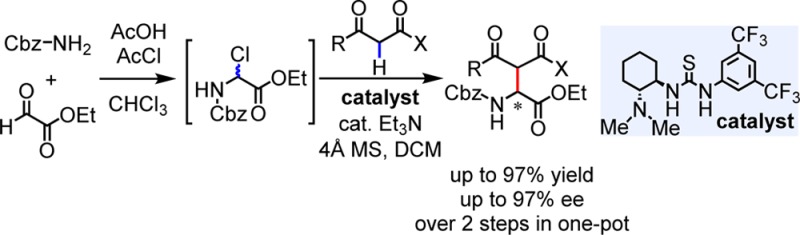
We report a scalable, one-pot Mannich route to enantioenriched α-amino esters by direct reaction of α-chloroglycine ester as a practical imino ester surrogate. The reaction is promoted by a chiral aminothiourea, which is proposed to operate cooperatively by generating an iminium ion by chloride abstraction and an enolate by deprotonation, followed by highly stereoselective C–C bond formation between both reactive intermediates associated non-covalently within the catalyst framework.
The Mannich reaction involves the enantioselective addition of enolate equivalents to aldimines or ketimines to produce β-amino esters.1 Practical, asymmetric methods have been enabled by the identification of various chiral metal and organic catalysts.1−3 Mannich reactions involving N-carbamoyl imino esters as electrophilic partners afford chiral carbamate-protected α-amino esters directly, and this represents a most attractive route to this important class of products.4 However, the instability of imino esters, which require cumbersome preparation and strictly controlled conditions in catalytic reactions, is an inherent limitation. To counter this obstacle, α-haloglycine esters5,6 and α-amido sulfones3f,3g,3i,7 have been exploited as imine surrogates in Mannich-type reactions (Scheme 1). These systems, however, require multi-pot operations and use of excess base for the generation of imine.8 To overcome these limitations, we envisioned the application of chiral bifunctional catalysts capable of both generating imine from its surrogate and inducing asymmetric addition of the enolate.
Scheme 1. Asymmetric Mannich Synthesis of α-Amino Esters.
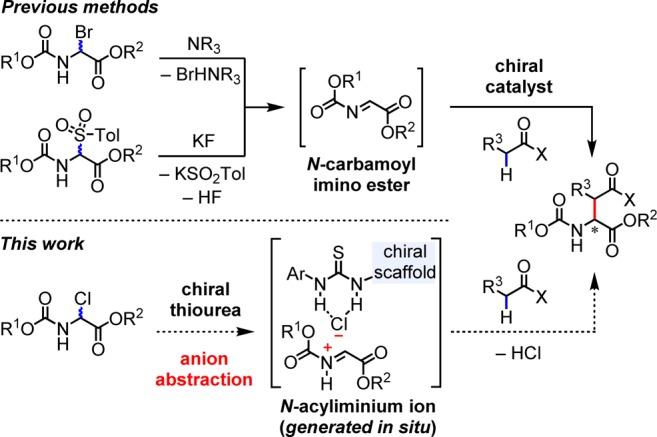
The use of chiral thioureas to promote anion abstraction from neutral organic electrophiles to generate highly reactive cationic intermediates has emerged as a powerful platform in asymmetric catalysis.9,10 Recently, the Roche group reported a practical route to α-chloroglycine esters by reaction of carbamates or amides with ethyl glyoxylate, acetyl chloride, and acetic acid in the context of a new route to racemic α-arylglycine esters.11 We considered whether α-chloroglycine esters prepared in this manner could be engaged directly in thiourea-catalyzed enantioselective Mannich reactions (Scheme 1).3 Specifically, thiourea-induced chloride abstraction could serve to generate a reactive N-acyliminium ion, while the basic amine functionality could generate and position an enolate for nucleophilic addition to give the desired enantioenriched α-amino esters. We describe here the application of such an anion-binding strategy in a practical Mannich synthesis of aspartic acid derivatives. This methodology circumvents the multi-pot, base-mediated preparation of N-carbamoyl imino esters from imine surrogates developed previously.3f,3g,3i,5,7
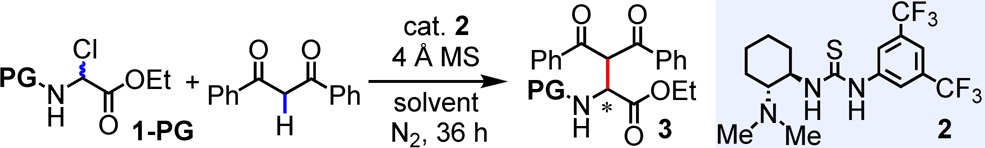
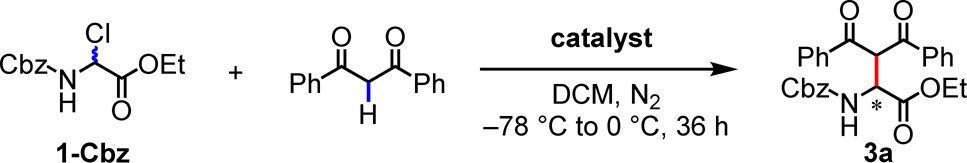
We examined the Mannich reactions using N-Cbz α-chloroglycine ethyl ester (1-Cbz) as the model substrate and bifunctional thioureas as potential catalysts. We discovered that Takemoto’s tertiary aminothiourea catalyst 2(3e,12) promoted the reaction between 1-Cbz and dibenzoylmethane in DCM at −30 °C in the presence of 4 Å molecular sieves (MS), affording the Mannich product in 90% yield and 93% ee (Table 1, entry 1).13−15 Thiourea catalysts lacking the tertiary amino group gave no desired product. Substrates bearing other carbamate or acyl N-protecting groups afforded significantly inferior results in comparison to Cbz with respect to reaction enantioselectivity (entries 2–8).16 Reaction temperature and solvent were also found to exert profound effects on enantioselectivity (entries 1, 9–16). Optimal results were obtained at −30 °C in DCM, with lower temperatures affording no advantage.
Table 1. Evaluation of Reaction Parametersa,b.
| entry | PG | solvent | temp (°C) | yield (%) | ee (%) | entry | PG | solvent | temp (°C) | yield (%) | ee (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cbz | DCM | –30 | 90 | 93 | 9 | Cbz | DCM | rt | 65 | 44 | |
| 2 | Fmoc | DCM | –30 | 71 | 66 | 10 | Cbz | DCM | 0 | 70 | 88 | |
| 3 | Troc | DCM | –30 | 83 | 58 | 11 | Cbz | DCM | –78 | 80 | 90 | |
| 4 | MeO2C– | DCM | –30 | 82 | 82 | 12 | Cbz | CHCl3 | –30 | 42 | 44 | |
| 5 | PhO2C– | DCM | –30 | 50 | 44 | 13 | Cbz | toluene | –30 | 61 | 80 | |
| 6 | Ac | DCM | –30 | 84 | 76 | 14 | Cbz | Et2O | –30 | 66 | 90 | |
| 7 | Bz | DCM | –30 | 78 | 43 | 15 | Cbz | TBME | –30 | 66 | 80 | |
| 8 | TFA | DCM | –30 | 40 | 25 | 16 | Cbz | THF | –30 | 71 | 81 |
Conditions: substrate (0.05 mmol), catalyst (10 mol%), diketone (0.1 mmol), 4 Å MS (20 mg), DCM (1 mL), under N2, initially cooled to −78 °C and stirred at the temperature denoted in the table for 36 h.
The yield was determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard.
We considered whether the stoichiometric HCl byproduct of the Mannich reaction might have a deleterious effect on catalyst performance by forming a salt with its tertiary amine moiety. Indeed, the HCl salt of 2 catalyzes formation of 3a in only 21% yield and 18% ee under the conditions of the model reaction (Table 2, entries 1 and 2). We explored whether added bases could serve to regenerate active catalyst 2 from the HCl salt. While inorganic bases conferred no improvement on reaction performance, introduction of Et3N (0.5 equiv) to the reaction with 2·HCl resulted in formation of 3a in 76% yield and 84% ee (entry 3). Addition of Et3N to the reaction with the free base had a striking, positive effect, with formation of 3a in 95% yield and 99% ee (entries 1 vs 4). Ultimately, it was found that addition of 25 mol% of Et3N was sufficient to obtain optimal results (entry 5). Reduction of the catalyst loading or omission of 4 Å MS13 had a deleterious effect on both yield and ee (entries 6 and 7).
Table 2. Effect of Et3Na,b.
| entry | catalyst (mol%) | additive (mol%) | yield (%) | ee (%) |
|---|---|---|---|---|
| 1 | 2 (10) | none | 70 | 88 |
| 2 | 2·HCl (10) | none | 21 | 18 |
| 3 | 2·HCl (10) | Et3N (50) | 76 | 84 |
| 4 | 2 (10) | Et3N (50) | 95 | 99 |
| 5 | 2 (10) | Et3N (25) | 96 | 99 |
| 6 | 2 (5) | Et3N (25) | 83 | 94 |
| 7c | 2 (10) | Et3N (50) | 70 | 93 |
Conditions: substrate (0.05 mmol), catalyst (10 mol%), diketone (0.1 mmol), DCM (1 mL), under N2, initially cooled to −78 °C and stirred at −0 °C, 36 h.
The yield was determined by the 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
Reaction carried out in the absence of added 4 Å MS.
A variety of 1,3-diketones were found to participate effectively in enantioselective Mannich reactions with 1-Cbz catalyzed by 2 (Table 3). Whereas some variability was observed in reactions carried out at 0.25 mmol scale on 0 °C, consistent and optimal results were obtained at −30 °C. Both symmetrical and unsymmetrical 1,3-diaryl-diketones afforded products in high ee (3a, 3e, 3f). α-Fluorinated β-diketones also underwent highly enantioselective reactions (3d, 3g). A nearly statistical mixture of diastereomers was obtained in the case of 3g, which bears a non-epimerizable β-dicarbonyl stereocenter. Unsymmetrical alkyl-aryl diketones were also found to be compatible substrates for the enantioselective reaction (3b); however, aliphatic 1,3-diketone underwent reaction with 1-Cbz with lower ee (3c).
Table 3. Asymmetric Mannich Reaction with 1,3-Diketonesa–d.
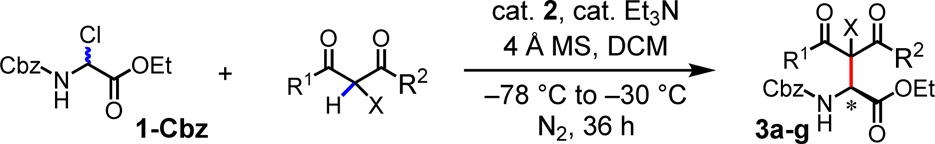
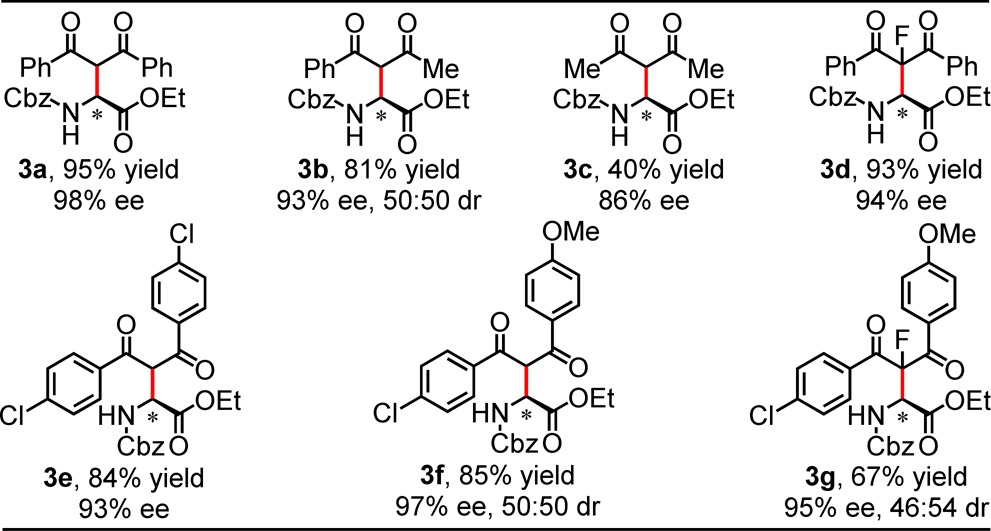
Conditions: substrate (0.25 mmol), catalyst (10 mol%), diketone (0.5 mmol), 4 Å MS (40 mg), Et3N (25 mol%), DCM (5 mL), under N2, initially cooled to −78 °C and stirred at −30 °C, 36 h.
Isolated yield.
dr was determined by 1H NMR and HPLC analyses of the crude product.
Absolute configuration assigned by analogy to product 4h (Table 4).
Reactions of 1-Cbz with β-ketoesters proceeded to afford Mannich products with moderate-to-high enantioselectivity (Table 4).17,18 The resulting ketodiesters may be subjected to decarboxylation to reveal valuable β-keto α-amino acid derivatives.19 In particular, products bearing electron-neutral or -deficient aryl ketone groups (4a, 4c–k) were obtained with generally good yields and ≥89% ee. Whereas variation of the size of the ester substituent had little impact on the reaction outcome (e.g., 4a vs 4g, 4i vs 4j), the use of benzhydryl esters had a significant positive effect on reaction enantioselectivity (e.g., 4a vs 4h). This latter effect made it possible to obtain alkyl ketone products in high ee and good yield (4k).
Table 4. Asymmetric Synthesis of Aspartic Acid Derivativesa–d.
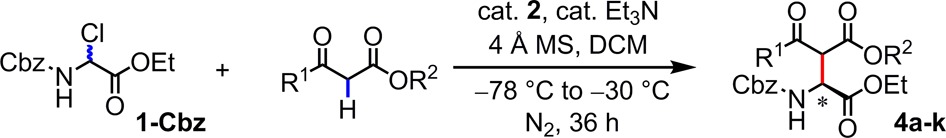
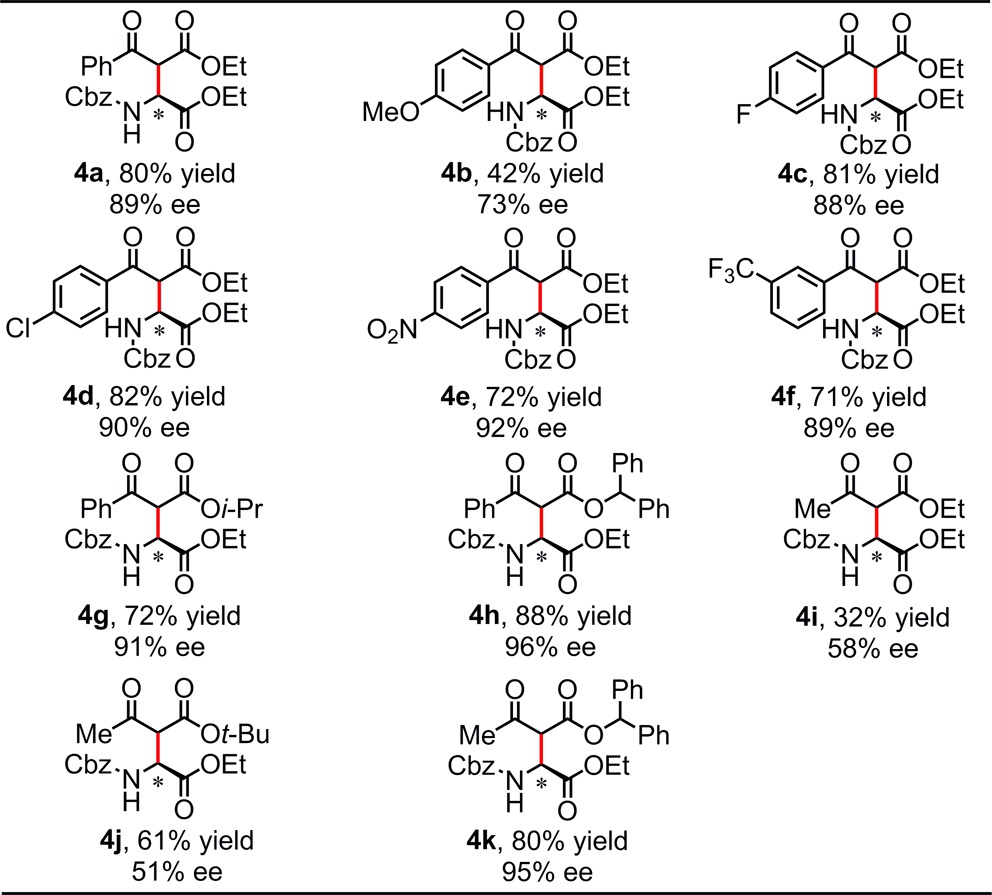
Conditions: substrate (0.25 mmol), catalyst (10 mol%), β-ketoester (0.5 mmol), 4 Å MS (40 mg), Et3N (25 mol%), DCM (5 mL), under N2, initially cooled to −78 °C and stirred at −30 °C, 36 h.
Isolated yield.
Products were isolated as the thermodynamic mixtures of diastereomers.
The structure and absolute configuration of 4h was established by X-ray crystallography, and the stereochemistry of all other products was assigned by analogy.
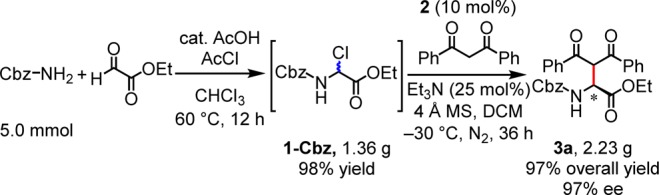
The practicality of this protocol was demonstrated in the sequential preparation of N-Cbz α-chloroglycine ester (1-Cbz) and enantioselective Mannich reaction in a one-pot protocol on a preparative scale (Scheme 2). As reported previously,111-Cbz could be prepared in nearly quantitative yield from commercial feedstock molecules. Removal of AcOH and AcCl from the product mixture of 1-Cbz under reduced pressure was found to be essential for the subsequent Mannich reaction. Reaction of 1-Cbz with 1,3-diphenyl-1,3-propanedione in the presence of 10 mol% catalyst afforded 3a in 97% overall yield and 97% ee.20 This two-step, one-pot synthesis of highly enantioenriched unnatural amino esters is accomplished using commercially available substrates and catalyst, and generates only AcOH, HCl, and Et3N·HCl as byproducts.
Scheme 2. Large-Scale, One-Pot Synthesis of 3a.
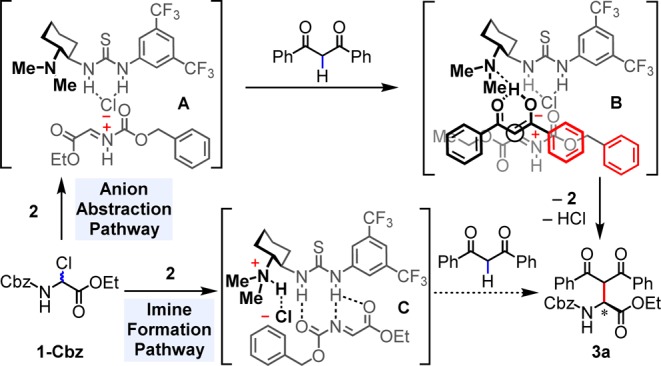
The potential mechanisms by which this enantioselective Mannich reaction of α-chloroglycine esters proceeds merit consideration. No measurable background reaction between 1-Cbz and dibenzoylmethane is observed in the absence of the thiourea catalyst, either with or without added Et3N. This observation suggests an essential role of the H-bond donor component of the aminothiourea catalyst 2 not only in enantiocontrol but also in the generation of the reactive electrophilic intermediate. Two possible mechanistic pathways involving thiourea activation of the α-chloroglycine are outlined in Scheme 3. In the anion abstraction pathway, the thiourea catalyst abstracts chloride from 1-Cbz to form a thiourea-bound acyliminium/chloride intermediate (A).10 Reaction of the tertiary amine moiety in A with the β-dicarbonyl compound (dibenzoylmethane in the scheme) generates the amine-bound enolate (B), which attacks the nearby iminium ion in an enantioselective manner to give the Mannich product 3a. The plausibility of this mechanism would require that the highly acidic iminium ion intermediate not interfere with the generation and addition of the enolate. An alternative reaction pathway involving the formation of a thiourea-bound N-Cbz iminoester intermediate (C) generated by the reaction between the catalyst’s tertiary amine moiety and 1-Cbz must therefore also be considered, although the mechanism of enolate generation is less evident in this pathway.
Scheme 3. Possible Mechanistic Pathways.
Regardless of the specific mechanism of catalysis, it is clear that interactions involving aromatic substituents on the substrates play a critical role in modulating both reactivity and enantioselectivity. Replacement of aryl groups for alkyl groups in the 1,3-diketone substrates leads to sizable decreases in enantioselectivity and product yield (3a → 3b → 3c, Table 3). In a similar manner, significantly improved results were obtained using benzhydryl esters (4h, 4k) relative to aliphatic esters (4a, 4g, 4i, 4j) in the addition of β-ketoesters to 1-Cbz. Finally, the electronic properties of the aryl substituents in aryl-β-ketoester nucleophiles correlate directly with product yield and ee (Table 4, 4c–4f), with electron-deficient substrates affording highest enantioselectivities. Further elucidation of the specific catalyst–substrate interactions at play in these reactions is a subject of ongoing interest in our laboratories.21
In summary, we have developed an efficient thiourea-catalyzed enantioselective Mannich reaction that provides access to a variety of N-carbamoyl α-amino esters. The products can be obtained using a one-pot protocol on a preparative scale from commercial substrates and catalyst. Further application of 1-Cbz and related substrates in asymmetric catalysis with H-bond donor catalysts is currently underway.
Acknowledgments
We gratefully acknowledge the National Institutes of Health (GM-43214) for financial support. We thank the Japan Society for the Promotion of Science (postdoctoral fellowship to M.W.), Herchel Smith-Harvard Research Fellowship (undergraduate fellowship to R.Y.L.), and Dr. Shao-Liang Zheng for help with the X-ray data collection and structure determination.
Supporting Information Available
Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For reviews of asymmetric Mannich reactions, see:; a Córdova A. Acc. Chem. Res. 2004, 37, 102. [DOI] [PubMed] [Google Scholar]; b Wenzel A. G.; Jacobsen E. N. In Enantioselective Synthesis of β-Amino Acids; Juaristi E., Soloshonok V., Eds.; Wiley-VCH: New York, 2005; Chapter 4. [Google Scholar]; c Ting A.; Schaus S. E. Eur. J. Org. Chem. 2007, 2007, 5797. [Google Scholar]; d Verkade J. M. M.; Hemert L. J. C. v.; Quaedflieg P. J. L. M.; Rutjes F. P. J. T. Chem. Soc. Rev. 2008, 37, 29. [DOI] [PubMed] [Google Scholar]; e Weiner B.; Szymanski W.; Janssen D. B.; Minnaard A. J.; Feringa B. L. Chem. Soc. Rev. 2010, 39, 1656. [DOI] [PubMed] [Google Scholar]; f Karimi B.; Enders D.; Jafari E. Synthesis 2013, 45, 2769. [Google Scholar]
- For pioneering examples of asymmetric Mannich synthesis of N-PMP α-amino esters with organocatalysts, see:; a List B. J. Am. Chem. Soc. 2000, 122, 9336. [Google Scholar]; b Notz W.; Sakthivel K.; Bui T.; Zhong G. F.; Barbas F. C. III. Tetrahedron Lett. 2001, 42, 199. [Google Scholar]; c Córdova A.; Watanabe S.-i.; Tanaka F.; Notz W.; Barbas C. F. J. Am. Chem. Soc. 2002, 124, 1866. [DOI] [PubMed] [Google Scholar]; d Cobb A. J. A.; Shaw D. M.; Ley S. V. Synlett 2004, 558. [Google Scholar]; e Ooi T.; Kameda M.; Fujii J.; Maruoka K. Org. Lett. 2004, 6, 2397. [DOI] [PubMed] [Google Scholar]
- Thiourea-catalyzed asymmetric Mannich reactions:; a Wenzel A. G.; Jacobsen E. N. J. Am. Chem. Soc. 2002, 124, 12964. [DOI] [PubMed] [Google Scholar]; b Taylor M. S.; Tokunaga N.; Jacobsen E. N. Angew. Chem., Int. Ed. 2005, 44, 6700. [DOI] [PubMed] [Google Scholar]; c Yoon T. P.; Jacobsen E. N. Angew. Chem., Int. Ed. 2005, 44, 466. [DOI] [PubMed] [Google Scholar]; d Bode C. M.; Ting A.; Schaus S. E. Tetrahedron 2006, 62, 11499. [Google Scholar]; e Yamaoka Y.; Miyabe H.; Yasui Y.; Takemoto Y. Synthesis 2007, 2571. [Google Scholar]; f Song J.; Shih H. W.; Deng L. Org. Lett. 2007, 9, 603. [DOI] [PubMed] [Google Scholar]; g Takada K.; Tanaka S.; Nagasawa K. Synlett 2009, 1643. [Google Scholar]; h Sohtome Y.; Tanaka S.; Takada K.; Yamaguchi T.; Nagasawa K. Angew. Chem., Int. Ed. 2010, 49, 9254. [DOI] [PubMed] [Google Scholar]; i Gao J.; Chuan Y.; Li J.; Xie F.; Peng Y. Org. Biomol. Chem. 2012, 10, 3730. [DOI] [PubMed] [Google Scholar]; j Bergonzini G.; Schindler C. S.; Wallentin C. J.; Jacobsen E. N.; Stephenson C. R. J. Chem. Sci. 2014, 5, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected early examples, see:; a Hagiwara E.; Fujii A.; Sodeoka M. J. Am. Chem. Soc. 1998, 120, 2474. [Google Scholar]; b Ferraris D.; Young B.; Dudding T.; Lectka T. J. Am. Chem. Soc. 1998, 120, 4548. [Google Scholar]; c Trost B. M.; Terrell L. R. J. Am. Chem. Soc. 2003, 125, 338. [DOI] [PubMed] [Google Scholar]; d Bernardi L.; Gothelf A. S.; Hazell R. G.; Jørgensen K. A. J. Org. Chem. 2003, 68, 2583. [DOI] [PubMed] [Google Scholar]; e Marigo M.; Kjaersgaard A.; Juhl K.; Gathergood N.; Jørgensen K. A. Chem.—Eur. J. 2003, 9, 2359. [DOI] [PubMed] [Google Scholar]
- For the use of N-acylimino ester generated from α-bromo glycine esters in metal-catalyzed Mannich reactions, see:; a Kobayashi S.; Kitagawa H.; Matsubara R. J. Comb. Chem. 2001, 3, 401. [DOI] [PubMed] [Google Scholar]; b Kobayashi S.; Matsubara R.; Kitagawa H. Org. Lett. 2002, 4, 143. [DOI] [PubMed] [Google Scholar]; c Kobayashi S.; Matsubara R.; Nakamura Y.; Kitagawa H.; Sugiura M. J. Am. Chem. Soc. 2003, 125, 2507. [DOI] [PubMed] [Google Scholar]; d Nakamura Y.; Matsubara R.; Kiyoara H.; Kobayashi S. Org. Lett. 2003, 5, 2481. [DOI] [PubMed] [Google Scholar]; e Matsubara R.; Nakamura Y.; Kobayashi S. Angew. Chem., Int. Ed. 2004, 43, 1679. [DOI] [PubMed] [Google Scholar]
- Poulsen T. B.; Alemparte C.; Saaby S.; Bella M.; Jørgensen K. A. Angew. Chem., Int. Ed. 2005, 44, 2896. [DOI] [PubMed] [Google Scholar]
- a Fini F.; Sgarzani V.; Pettersen D.; Herrera R. P.; Bernardi L.; Ricci A. Angew. Chem., Int. Ed. 2005, 44, 7975. [DOI] [PubMed] [Google Scholar]; b Palomo C.; Oiarbide M.; Laso A.; López R. J. Am. Chem. Soc. 2005, 127, 17622. [DOI] [PubMed] [Google Scholar]; c Gianelli C.; Sambri L.; Carlone A.; Bartoli G.; Melchiorre P. Angew. Chem., Int. Ed. 2008, 47, 8700. [DOI] [PubMed] [Google Scholar]; d Galzerano P.; Agostino D.; Bencivenni G.; Sambri L.; Bartoli G.; Melchiorre P. Chem.—Eur. J. 2010, 16, 6069. [DOI] [PubMed] [Google Scholar]
- Recently, a disulfonimide-catalyzed asymmetric synthesis of β-aryl β-amino esters via catalyst-mediated formation of iminoesters from amino sulfones was reported:Wang Q. G.; Leutzsch M.; van Gemmeren M.; List B. J. Am. Chem. Soc. 2013, 135, 15334. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Doyle A. G.; Jacobsen E. N. Chem. Rev. 2007, 107, 5713. [DOI] [PubMed] [Google Scholar]; b Zhang Z. G.; Schreiner P. R. Chem. Soc. Rev. 2009, 38, 1187. [DOI] [PubMed] [Google Scholar]; c Brak K.; Jacobsen E. N. Angew. Chem., Int. Ed. 2013, 52, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Taylor M. S.; Jacobsen E. N. J. Am. Chem. Soc. 2004, 126, 10558. [DOI] [PubMed] [Google Scholar]; b Raheem I. T.; Thiara P. S.; Peterson E. A.; Jacobsen E. N. J. Am. Chem. Soc. 2007, 129, 13404. [DOI] [PubMed] [Google Scholar]; c Raheem I. T.; Thiara P. S.; Jacobsen E. N. Org. Lett. 2008, 10, 1577. [DOI] [PubMed] [Google Scholar]; d Peterson E. A.; Jacobsen E. N. Angew. Chem., Int. Ed. 2009, 48, 6328. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Klausen R. S.; Jacobsen E. N. Org. Lett. 2009, 11, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lee Y.; Klausen R. S.; Jacobsen E. N. Org. Lett. 2011, 13, 5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S. P.; Samanta S. S.; Gosselin M. M. J. Chem. Commun. 2014, 50, 2632. [DOI] [PubMed] [Google Scholar]
- For reviews on the development of tertiary-aminothiourea catalysts by Takemoto, see:; a Takemoto Y. Org. Biomol. Chem. 2005, 3, 4299. [DOI] [PubMed] [Google Scholar]; b Miyabe H.; Takemoto Y. Bull. Chem. Soc. Jpn. 2008, 81, 785. [Google Scholar]; c Takemoto Y. Chem. Pharm. Bull. 2010, 58, 593. [DOI] [PubMed] [Google Scholar]
- Molecular sieves induce a measurable improvement in product yield by absorbing adventitious water and thereby suppressing formation of α-hydroxyglycine ester. Sieves may also help to sequester the HCl byproduct of the Mannich reaction, which also has a detrimental effect on reaction performance.
- See Supporting Information for catalyst screening data.
- Ethyl 2-cyano-2-phenylacetate underwent reaction with 1-Cbz in the presence of 2 to afford the Mannich product in >90% yield, but in only 37% ee. Similarly, silyl ketene acetals such as (1-methoxyvinyl)oxy)trimethylsilane participated in the catalyzed reaction, affording Mannich product in very low ee (<10%).
- The Boc-protected analogue of 1 could not be accessed, presumably due to its instability to the acidic conditions employed for its preparation.
- Erosion in dr of products 4a to 4k was observed due to slow epimerization of the stereocenter to yield the corresponding thermodynamically favored products.
- Methyl 1-oxo-1,2,3,4-tetrahydronaphthalene-2-carboxylate underwent reaction with 1-Cbz to afford the Mannich product in 72% yield but only 52% ee and 3:1 dr.
- a Marigo M.; Kjærsgaard A.; Juhl K.; Gathergood N.; Jørgensen K. A. Chem. −Eur. J. 2003, 9, 2359. [DOI] [PubMed] [Google Scholar]; b Yang C.-F.; Shen C.; Wang J.-Y.; Tian S.-K. Org. Lett. 2012, 14, 3092. [DOI] [PubMed] [Google Scholar]
- Product 3a could be obtained in 80% yield and 95% ee with 5 mol% catalyst.
- Knowles R. R.; Jacobsen E. N. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.