

Abstract
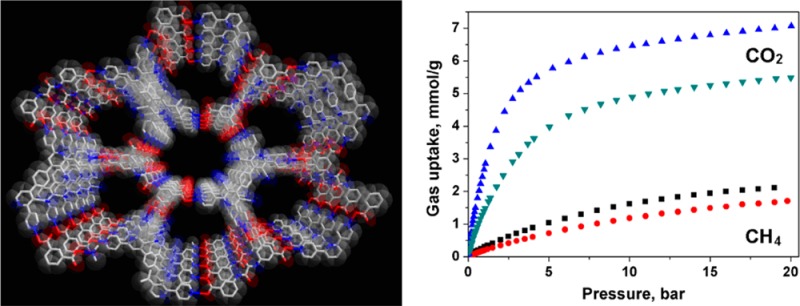
A robust binary hydrogen-bonded supramolecular organic framework (SOF-7) has been synthesized by solvothermal reaction of 1,4-bis-(4-(3,5-dicyano-2,6-dipyridyl)dihydropyridyl)benzene (1) and 5,5′-bis-(azanediyl)-oxalyl-diisophthalic acid (2). Single crystal X-ray diffraction analysis shows that SOF-7 comprises 2 and 1,4-bis-(4-(3,5-dicyano-2,6-dipyridyl)pyridyl)benzene (3); the latter formed in situ from the oxidative dehydrogenation of 1. SOF-7 shows a three-dimensional four-fold interpenetrated structure with complementary O–H···N hydrogen bonds to form channels that are decorated with cyano and amide groups. SOF-7 exhibits excellent thermal stability and solvent and moisture durability as well as permanent porosity. The activated desolvated material SOF-7a shows high CO2 adsorption capacity and selectivity compared with other porous organic materials assembled solely through hydrogen bonding.
Porous framework materials, such as porous carbon,1 zeolites,2 metal–organic frameworks,3 and porous organic frameworks,4−7 have attracted intensive research interest due to their potential applications in molecular storage and separation. Porous organic framework materials have become competitive materials because of their low framework density resulting from the use of light elements (typically H, C, N, O, B) and their low toxicity as well as their controllable assembly through organic synthesis and crystal engineering.4−7 For example, covalent organic frameworks (COFs)6 or porous organic polymers (POPs) and polymers of intrinsic microporosity (PIMs) represent7 a widely investigated family of porous organic materials that are typically prepared from organic coupling reactions of selected and/or designed precursors. However, the development of COFs/POPs/PIMs has been somewhat restricted by harsh reaction conditions, multistep syntheses, and the involvement of relatively expensive catalysts.
Supramolecular organic frameworks (SOFs) have recently been recognized as promising porous materials which are constructed from functional organic modules assembled via supramolecular interactions (e.g., hydrogen bonds, π–π stacking, CH···π, and van der Waals interactions).4,5 Special interest in SOF materials comes from the soft and flexible nature of their molecular interactions, the ease of manipulation of the modularity and functionality of the organic components, and the tunable guest selectivity achieved by decorating the pores with organic groups that can exploit specific interactions with different gas molecules. Moreover, SOF materials can be highly crystalline, which is an advantage not only for structural determination but also for investigation of structure–property relationships. However, upon guest removal many SOF materials undergo phase changes to give close-packed structures, lose porosity, and/or undergo structure collapse due to the relative weakness of the supramolecular interactions that underpin the framework structure.8 We have targeted organic modules with favorable molecular configurations that promote the formation of spatial voids and permanent cavities, noting that cooperative functional groups play a key role in stabilizing molecular assemblies via both intramolecular and intermolecular interactions.9
SOF materials have been reported in which a single type of organic molecule crystallizes into a porous phase via supramolecular hydrogen-bonding interactions5a−5f in which the resultant porous phase depends greatly on the solvents present. Specific intermolecular interactions such as hydrogen bonds can be optimized and balanced in two- and multicomponent materials via molecular recognition between functional organic modules.10 In this context, we have adopted a binary design strategy in which two different hydrogen-bonding tectons assemble to form a stable porous network.
In this work, 1,4-bis-(4-(3,5-dicyano-2,6-dipyridyl)dihydropyridyl)benzene (1) and 5,5′-bis-(azanediyl)-oxalyl-diisophthalic acid (2) (Scheme 1) have been chosen to build a binary SOF material for selective gas storage. Our approach is based upon the use of exo-pyridyl and carboxyl groups on two separate organic modules that give complementary directional hydrogen bonding and, at the same time, incorporate amide groups on 2 to give potential interactions with CO2.11
Scheme 1. Schematic View of the Organic Modules.

Reaction of 1 and 2 in a 1:1 molar ratio in dimethylformamide (DMF) at 90 °C resulted in the formation of orange prismatic crystals of SOF-7 after 72 h. This material is a solvated binary hydrogen-bonded cocrystal9a,9c,9g,9h with overall formula [(C18H12N2O10)·(C40H20N10)]·7DMF comprising a 1:1 combination of 2 and 1,4-bis-(4-(3,5-dicyano-2,6-dipyridyl)pyridyl)benzene (3), this latter species9i being formed in situ by oxidative dehydrogenation of 1. A single crystal X-ray structure determination (Table S1) reveals that SOF-7 crystallizes in the monoclinic space group C2/c and features a three-dimensional (3D), four-fold interpenetrating lattice containing channels decorated with cyano and amide groups (Figure 1a). The exo-carboxyl and pyridyl groups on 2 and 3 contribute to the O–H···N hydrogen bonds which direct the self-assembly process. Moreover, the lateral amide in 2 and cyano group in 3 may offer potential binding sites for guest molecules to enhance gas uptake. The network can be regarded as a cocrystal rather than an organic salt since complete proton transfer between the carboxyl and pyridyl groups is not observed with two different C to O distances (C–OH = ∼1.31 Å; C=O = ∼1.21 Å) observed, consistent with protonation of the carboxyl group.9g,9h
Figure 1.
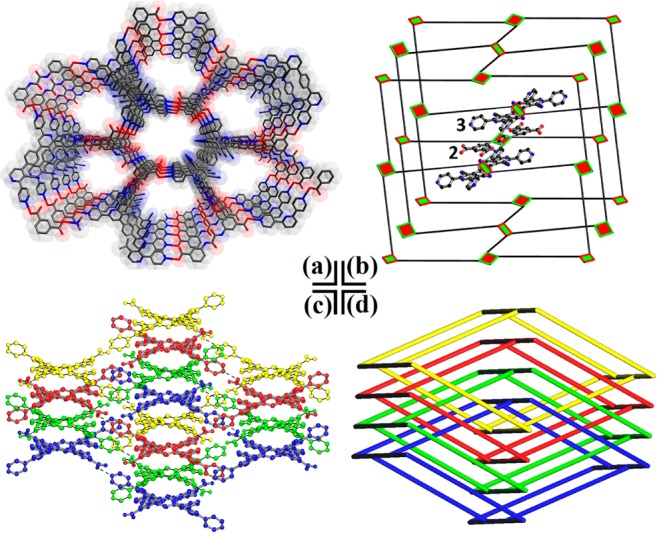
Views of SOF-7 (a) the 3D structure along a axis; (b) the simplified cds net (red node, 2; green node, 3); (c) the four-fold interpenetrating framework; (d) a simplified schematic view of the four-fold interpenetrating cds nets.
In SOF-7, each molecule of 2 interacts with four neighboring molecules of 3 through primary hydrogen bonds (Ocarboxyl–H···Npyridyl, 2.598(5), 2.599(4) Å) to form a 3D supramolecular organic network (Figure 1a). The guest DMF molecules reside within the channels of this material and interact with the internal amide groups in 2 via secondary hydrogen-bond interactions (Namide–H···Ocarbonyl, 2.890(5) Å). The network topology of SOF-7 was analyzed using TOPOS12 as a 65·8-cds (CdSO4) net, reflecting a square topology (Figure 1b). This topology has been identified as one of the most frequently observed nets to show framework interpenetration.13 This is also the case for SOF-7 in which four identical cds nets interpenetrate to give an overall four-fold interpenetrating framework (Figure 1c,d). Π–π interactions are observed between two exo-pyridyl groups in 3 (∼3.077 Å) from adjacent single nets as well as between a central pyridyl moiety in 3 and a phenyl group in 2 (∼3.421 Å) from adjacent single nets. Despite this network interpenetration, the total solvent-accessible volume of SOF-7 after removal of guest DMF molecules was estimated to be ∼48% as calculated using PLATON/VOID routine.14 The thermal stability of the SOF-7 framework was evaluated by thermogravimetric analysis (TGA), which showed a decomposition temperature of around 350 °C (Figure S1). The phase purity of the bulk sample of SOF-7 was confirmed by powder X-ray diffraction (PXRD, Figure S2). The DMF molecules within the pores of SOF-7 were exchanged with acetone, and the acetone-exchanged sample degassed under dynamic vacuum at 100 °C for 24 h to afford the activated, desolvated sample SOF-7a. SOF-7a retains its structural integrity and crystallinity upon solvent exchange as well as upon removal of guest molecules, as confirmed by PXRD patterns (Figure S2), which revealed a highly robust framework. Moreover, the desolvated sample SOF-7a exhibited excellent structural durability toward both common organic solvents and water (Figure S3).
The permanent porosity of SOF-7a was confirmed by gas adsorption studies. The results clearly show that SOF-7a exhibits selective adsorption for CO2 over N2, H2 and CH4 (Figures 2a,b, and S4). The N2 adsorption isotherm of SOF-7a at 77 K displays restricted adsorption behavior (Figure S4), with the Brunauer–Emmett–Teller (BET) surface area of SOF-7a calculated from N2 adsorption isotherm at 77 K being much lower than expected (21.03 m2·g–1). However, the CO2 isotherm recorded at 273 K reveals a reversible type-I adsorption behavior and gives an expected BET surface area based upon the crystal structure of 900.0 m2·g–1 (Figure S5). Furthermore, the pore volume estimated from the N2 adsorption isotherm (0.097 cm3·g–1) is significantly lower than the value estimated from the CO2 adsorption isotherm (0.233 cm3·g–1) using non-local density functional theory modeling. Interactions between N2 and the channel windows of SOF-7a at 77 K could hinder the diffusion of N2 into the material; restricted N2 uptake but higher type-I CO2 uptake has been observed previously in materials with pore sizes larger than the kinetic diameter of N2.5b,15 Thus, in this case, the BET surface area and the pore size distribution for SOF-7a calculated from the CO2 adsorption isotherm were 900.0 m2·g–1 and 13.6 Å (Figure S6), respectively, in good accordance with the channel window of ∼13.5 × 14 Å calculated from the single crystal X-ray data.
Figure 2.
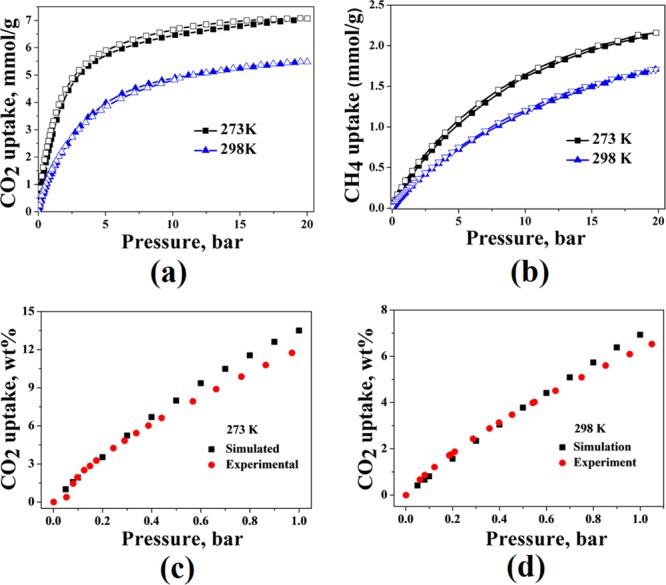
CO2 isotherms for SOF-7a at 273 K (black) and 298 K (blue) in the pressure range 0–20 bar (a); CH4 isotherms for SOF-7a at 273 K (black) and 298 K (blue) in the pressure range 0–20 bar (b); experimental (red) and simulated (black) CO2 isotherms up to 1 bar at 273 K (c); experimental (red) and simulated (black) CO2 isotherms up to 1 bar at 298 K (d).
SOF-7a was tested for CO2 adsorption at different temperatures. SOF-7a shows reversible CO2 adsorption with CO2 capacities of 12.54 wt % (2.85 mmol·g–1) and 6.53 wt % (1.49 mmol·g–1) at 273 and 293 K at 1 bar, respectively (Figure 2a). High-pressure (20 bar) CO2 adsorption of SOF-7a gives the total amount of 31.09 wt % (7.07 mmol·g–1) and 24.12 wt % (5.48 mmol·g–1) at 273 and 293 K (Figure 2a), respectively. The CO2 adsorption capacity of SOF-7a at 273 K and 1 bar is comparable to some of the best performing single component SOF materials;4a−4c,5b−5d for example, triptycene-tris(benzimidazolone) absorbs 15.9 wt % CO2 at 273 K and 1 bar5d (Table S2). The heat of adsorption for CO2 (Qst) was calculated via the Clausius–Clapeyron equation using CO2 isotherms at 273 and 298 K (Figure S7) and was found to be 21.6 kJ·mol–1 at zero loading, which is slightly lower than previously reported single component SOF materials.4a−4c,5b−5d
Uptake of methane by SOF-7a was tested at different pressures (up to 20 bar) and temperatures (273 and 298 K) (Table S3), the isotherms showing reversible CH4 uptake of 0.47 wt % (0.29 mmol·g–1) and 0.35 wt % (0.22 mmol·g–1) at 1 bar, and 3.38 wt % (2.11 mmol·g–1) and 2.74 wt % (1.71 mmol·g–1) at 20 bar (Figure 2b). The CH4 uptake of SOF-7a at 16 bar and 298 K (1.54 mmol·g–1) is comparable to that of SOF-1a (1.43 cm3·g–1). Strikingly, however, SOF-7a adsorbs ∼70% more of CO2 than SOF-1a at 16 bar and 298 K (5.30 vs 3.08 mmol·g–1). In comparison with the selectivity of CO2 over CH4 calculated for SOF-1a from Henry’s Law constant, SOF-7a shows significantly higher CO2/CH4 selectivity of 9.13 at 298 K and 1 bar, compared with 4.24 (at 298 K and 1 bar) for SOF-1a and 14.2 for SOF-7a and 5.60 for SOF-1a (at 273 K and 1 bar).
In order to analyze further the gas adsorption properties of SOF-7a, grand canonical Monte Carlo (GCMC) simulations of CO2 adsorption were performed (see ESI). The results of GCMC simulations of CO2 adsorption in SOF-7a are in good agreement with the experimental data at 273 and 298 K at up to 1 bar (Figure 2c,d). Moreover, in situ PXRD patterns of CO2-loaded SOF-7a were studied in order to monitor the possible dynamic structural changes related to CO2 adsorption. The in situ PXRD patterns remain essentially the same at 273 and 298 K up to 1 bar (Figure S8), suggesting that there are no significant structural changes or deterioration; this is consistent with the excellent match between simulated and experimental CO2 isotherms for SOF-7a at pressures of up to 1 bar.
GCMC simulations for CO2 adsorption at different pressures have also been conducted to analyze potential CO2 binding sites on the framework material of SOF-7a. Density functional calculations (DFT) have yielded binding energies (BE) and reveal the configurations corresponding to the strongest binding of CO2 in SOF-7a (Table S4). The three most preferred binding sites for CO2 in SOF-7a have been identified (Figure 3): the most stable configuration (BE = −35.19 kJ·mol–1) is characterized by strong N–H···O@CO2 hydrogen-bond interaction; the second most stable configuration (BE = −31.53 kJ·mol–1) is characterized by one N−H···O@CO2 hydrogen bond and two electrostatic attractions between the carbon of CO2 (q = 0.40 |e|) and the electronegative nitrogen atoms of the linker (q = −0.20 |e|); the third most stable configuration (BE = −29.75 kJ·mol–1) corresponds to a CO2 location near the amide group of the linker, stabilized by N–H···O@CO2 and C–H···O@CO2 hydrogen-bonding interactions and electrostatic interaction between C@CO2 and oxygen atoms in the linker. The calculation thus confirms that the amide and cyano groups in 2 and 3 contribute significantly to the highly selective binding of CO2 in SOF-7a.
Figure 3.
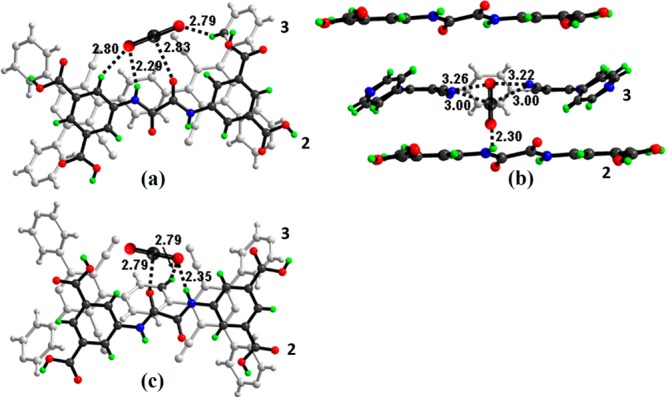
Binding of CO2 molecules to SOF-7a as determined by GCMC simulations and DFT calculations (labeled distances are in Å): (a) CO2 interacting with the amide group in 2 in a parallel position to also form N–H···O hydrogen bonds to 3; (b) CO2 interacting with the amide group in 2 in a perpendicular position to also form N–H···O hydrogen bonds to 3; (c) CO2 interacting with the cyano groups in 3 in a parallel position to also form N–H···O hydrogen bonds to 2.
In summary, we have demonstrated a new strategy using two different organic building blocks incorporating complementary hydrogen bonding donor–acceptor motifs to form new SOF materials via the formation of cocrystals. SOF-7 features a 3D four-fold interpenetrating structure incorporating channels decorated with cyano and amide groups. SOF-7 is crystalline, highly robust, and shows permanent porosity. Appropriate functionalization of the organic building blocks favors not only the successful isolation of SOF-7 but also excellent CO2 adsorption capacity and selectivity. GCMC simulation confirms the role of these functional groups as favorable binding sites for CO2 molecules, thereby enhancing CO2/CH4 selectivity. To our knowledge, SOF-7a represents the first binary hydrogen-bonded supramolecular organic framework material to exhibit gas adsorption. The design strategy described herein opens up new possibilities for the flexible synthesis of not only new modified and extended binary systems but may also be programmed and extended toward tertiary and higher component porous assemblies.
Acknowledgments
We thank the EPSRC and the University of Nottingham for funding, and the EPSRC National Crystallography Service and Diamond Light Source (Beamline I11) for data collection. We acknowledge the Royal Society and Sino-British Fellowship Trust for an Incoming Fellowship (to J.L.), the Royal Commission for Jubail and Yanbu, Jubail University College, Kingdom of Saudi Arabia, for a Ph.D. Fellowship (to N.H.A.), and Conacyt, Mexico (to C.P.K.) for funding. M.S. gratefully acknowledges receipt of an ERC Advanced Grant and J.L. and M.S. thank the NSFC-RS International Exchanges Scheme (2011 China Costshare project based on NSFC-21001105) for financial support.
Supporting Information Available
Experimental details and supporting cif files, figures, graphs, and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Raymundo-Piñero E.; Cazorla-Amorós D.; Salinas-Martinez de Lecea C.; Linares-Solano A. Carbon 2000, 38, 335. [Google Scholar]; b Zhao X.; Villar-Rodil S.; Fletcher A. J.; Thomas K. M. J. Phys. Chem. B 2006, 110, 9947. [DOI] [PubMed] [Google Scholar]; c Yang Z.; Xia Y.; Mokaya R. J. Am. Chem. Soc. 2007, 129, 1673. [DOI] [PubMed] [Google Scholar]; d Hao G.-P.; Li W.-C.; Qian D.; Wang G.-H.; Zhang W.-P.; Zhang T.; Wang A.-Q.; Schüth F.; Bongard H.-J.; Lu A.-H. J. Am. Chem. Soc. 2011, 133, 11378. [DOI] [PubMed] [Google Scholar]; e Hao G.-P.; Li W.-C.; Qian D.; Lu A.-H. Adv. Mater. 2010, 22, 853. [DOI] [PubMed] [Google Scholar]; f Sevilla M.; Valle-Vigón P.; Fuertes A. B. Adv. Funct. Mater. 2011, 21, 2781. [Google Scholar]; g Sevilla M.; Fuertes A. B. Energy Environ. Sci. 2011, 4, 1765. [Google Scholar]
- a Brändle M.; Sauer J. J. Am. Chem. Soc. 1998, 120, 1556. [Google Scholar]; b Hudson M. R.; Queen W. L.; Mason J. A.; Fickel D. W.; Lobo R. F.; Brown C. M. J. Am. Chem. Soc. 2012, 134, 1970. [DOI] [PubMed] [Google Scholar]; c Shang J.; Li G.; Singh R.; Gu Q.; Nairn K. M.; Bastow T. J.; Medhekar N.; Doherty C. M.; Hill A. J.; Liu J. Z.; Webley P. A. J. Am. Chem. Soc. 2012, 134, 19246. [DOI] [PubMed] [Google Scholar]
- a Zhang J. P.; Chen X. M. J. Am. Chem. Soc. 2009, 131, 5516. [DOI] [PubMed] [Google Scholar]; b Sumida K.; Rogow D. L.; Mason J. A.; McDonald T. M.; Bloch E. D.; Herm Z. R.; Bae T.-H.; Long J. R. Chem. Rev. 2012, 112, 724. [DOI] [PubMed] [Google Scholar]; c Millward A. R.; Yaghi O. M. J. Am. Chem. Soc. 2005, 127, 17998. [DOI] [PubMed] [Google Scholar]; d Yang S.; Lin X.; Lewis W.; Suyetin M.; Bichoutskaia E.; Parker J.; Tang C. C.; Allan D. R.; Rizkallah P. J.; Hubberstey P.; Champness N. R.; Thomas K. M.; Blake A. J.; Schröder M. Nat. Mater. 2012, 11, 710. [DOI] [PubMed] [Google Scholar]; e Yang S.; Sun J.; Ramirez-Cuesta A. J.; Callear S. K.; David W. I. F.; Anderson D.; Newby R.; Blake A. J.; Parker J. E.; Tang C. C.; Schröder M. Nat. Chem. 2012, 4, 887. [DOI] [PubMed] [Google Scholar]
- a Lim S.; Kim H.; Selvapalam N.; Kim K.-J.; Cho S. J.; Seo G.; Kim K. Angew. Chem., Int. Ed. 2008, 47, 3352. [DOI] [PubMed] [Google Scholar]; b Kim H.; Kim Y.; Yoon M.; Lim S.; Park S. M.; Seo G.; Kim K. J. Am. Chem. Soc. 2010, 132, 12200. [DOI] [PubMed] [Google Scholar]; c Tian J.; Ma S.; Thallapally P. K.; Fowler D.; McGrail B. P.; Atwood J. L. Chem. Commun. 2011, 47, 7626. [DOI] [PubMed] [Google Scholar]; d Hasell T.; Schmidtmann M.; Cooper A. I. J. Am. Chem. Soc. 2011, 133, 14920. [DOI] [PubMed] [Google Scholar]; e Bojdys M. J.; Briggs M. E.; Jones J. T. A.; Adams D. J.; Chong S. Y.; Schmidtmann M.; Cooper A. I. J. Am. Chem. Soc. 2011, 133, 16566. [DOI] [PubMed] [Google Scholar]; f Hasell T.; Chong S. Y.; Jelfs K. E.; Adams D. J.; Cooper A. I. J. Am. Chem. Soc. 2012, 134, 588. [DOI] [PubMed] [Google Scholar]
- a Sozzani P.; Bracco S.; Comotti A.; Ferretti L.; Simonutti R. Angew. Chem., Int. Ed. 2005, 44, 1816. [DOI] [PubMed] [Google Scholar]; b Yang W.; Greenaway A.; Lin X.; Matsuda R.; Blake A. J.; Wilson C.; Lewis W.; Hubberstey P.; Kitagawa S.; Champness N. R.; Schröder M. J. Am. Chem. Soc. 2010, 132, 14457. [DOI] [PubMed] [Google Scholar]; c He Y.; Xiang S.; Chen B. J. Am. Chem. Soc. 2011, 133, 14570. [DOI] [PubMed] [Google Scholar]; d Mastalerz M.; Oppel I. M. Angew. Chem., Int. Ed. 2012, 51, 5252. [DOI] [PubMed] [Google Scholar]; e Luo X.-Z.; Jia X.-J.; Deng J.-H.; Zhong D.-C. J. Am. Chem. Soc. 2013, 135, 11684. [DOI] [PubMed] [Google Scholar]; f Li P.; He Y.; Guang J.; Weng L.; Zhao J. C.-G.; Xiang S.; Chen B. J. Am. Chem. Soc. 2014, 136, 547. [DOI] [PubMed] [Google Scholar]; g Yamamoto A.; Hamada T.; Hisaki I.; Miyata M.; Tohnai N. Angew. Chem., Int. Ed. 2013, 52, 1709. [DOI] [PubMed] [Google Scholar]
- a Côté A. P.; Benin A. I.; Ockwig N. W.; O’Keeffe M.; Matzger A. J.; Yaghi O. M. Science 2005, 310, 1166. [DOI] [PubMed] [Google Scholar]; b El-Kaderi H. M.; Hunt J. R.; Mendoza-Cortés J. L.; Côté A. P.; Taylor R. E.; O’Keeffe M.; Yaghi O. M. Science 2007, 316, 268. [DOI] [PubMed] [Google Scholar]; c Ding X.; Chen L.; Honsho Y.; Feng X.; Saengsawang O.; Guo J.; Saeki A.; Seki S.; Irle S.; Nagase S.; Parasuk V.; Jiang D. J. Am. Chem. Soc. 2011, 133, 14510. [DOI] [PubMed] [Google Scholar]; d Ding S.-Y.; Wang W. Chem. Soc. Rev. 2013, 42, 548. [DOI] [PubMed] [Google Scholar]
- For example see:; a Ben T.; Ren H.; Ma S.; Cao D.; Lan J.; Jing X.; Wang W.; Xu J.; Deng F.; Simmons J. M.; Qiu S.; Zhu G. Angew. Chem., Int. Ed. 2009, 48, 9457. [DOI] [PubMed] [Google Scholar]; b Trewin A.; Cooper A. I. Angew. Chem., Int. Ed. 2010, 49, 1533. [DOI] [PubMed] [Google Scholar]; c Ben T.; Pei C.; Zhang D.; Xu J.; Deng F.; Jing X.; Qiu S. Energy Environ. Sci. 2011, 4, 3991. [Google Scholar]; d Dawson R.; Stöckel E.; Holst J. R.; Adams D. J.; Cooper A. I. Energy Environ. Sci. 2011, 4, 4239. [Google Scholar]; e Dawson R.; Cooper A. I.; Adams D. J. Prog. Polym. Sci. 2012, 37, 530. [Google Scholar]; f Msayib K. J.; Book D.; Budd P. M.; Harris K. D. M.; Helliwell M.; Tedds S.; Warren J. E.; Xu M. C.; McKeown N. B. Angew. Chem., Int. Ed. 2009, 48, 3273. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Bezzu C. G.; Carta M.; Tonkins A.; Jansen J. C.; Bernardo P.; Bazzarelli F.; McKeown N. B. Adv. Mater. 2012, 24, 5930. [DOI] [PubMed] [Google Scholar]
- a Dalgarno S. J.; Thallapally P. K.; Barbour L. J.; Atwood J. L. Chem. Soc. Rev. 2007, 36, 236. [DOI] [PubMed] [Google Scholar]; b Maspoch D.; Ruiz-Molina D.; Veciana J. Chem. Soc. Rev. 2007, 36, 770. [DOI] [PubMed] [Google Scholar]
- a Kuduva S. S.; Craig D. C.; Nangia A.; Desiraju G. R. J. Am. Chem. Soc. 1999, 121, 1936. [Google Scholar]; b Vishweshwar P.; McMahon J. A.; Peterson M. L.; Hickey M. B.; Shattock T. R.; Zaworotko M. J. Chem. Commun. 2005, 41, 4601. [DOI] [PubMed] [Google Scholar]; c Braga D.; Grepioni F. Chem. Commun. 2005, 41, 3635. [DOI] [PubMed] [Google Scholar]; d Gilli P.; Pretto L.; Bertolasi V.; Gilli T. Acc. Chem. Res. 2009, 42, 33. [DOI] [PubMed] [Google Scholar]; e Khan M.; Enkelmann V.; Brunklaus G. J. Am. Chem. Soc. 2010, 132, 5254. [DOI] [PubMed] [Google Scholar]; f Berná J.; Alajarín M.; Orenes R.-A. J. Am. Chem. Soc. 2010, 132, 10741. [DOI] [PubMed] [Google Scholar]; g Cheney M. L.; Weyna D. R.; Shan N.; Hanna M.; Wojtas L.; Zawarotko M. J. Cryst. Growth Des. 2010, 10, 4401. [Google Scholar]; h Schultheiss N.; Lorimer K.; Wolfe S.; Desper J. CrystEngComm 2010, 12, 742. [Google Scholar]; i Ghozlan S. A. S.; Hassanien A. Z. A. Tetrahedron 2002, 58, 9423. [Google Scholar]
- a Aakeröy C. B.; Beatty A. M.; Helfrich B. A. Angew. Chem., Int. Ed. 2001, 40, 3240. [DOI] [PubMed] [Google Scholar]; b Bhogala B. R.; Basavoju S.; Nangia A. Cryst. Growth Des. 2005, 5, 1683. [Google Scholar]; c Martí-Rujas J.; Colombo L.; Lü J.; Dey A.; Terraneo G.; Metrangolo P.; Pilati T.; Resnati G. Chem. Commun. 2012, 48, 8207. [DOI] [PubMed] [Google Scholar]
- a Dincă M.; Long J. R. Angew. Chem., Int. Ed. 2008, 47, 6766. [DOI] [PubMed] [Google Scholar]; b Dietzel P. D. C.; Besikiotis V.; Blom R. J. Mater. Chem. 2009, 19, 7362. [Google Scholar]; c Zheng B.; Bai J.; Duan J.; Wojtas L.; Zaworotko M. J. J. Am. Chem. Soc. 2011, 133, 748. [DOI] [PubMed] [Google Scholar]; d Duan J.; Yang Z.; Bai J.; Zheng B.; Li Y.; Li S. Chem. Commun. 2012, 48, 3058. [DOI] [PubMed] [Google Scholar]
- Blatov V. A.IUCr Computing Commission Newsletter; International Union of Crystallography: Chester, England, 2006; Vol. 7, p 4; see also http://www.topos.ssu.samara.ru
- Alexandrov E. V.; Blatov V. A.; Kochetkov A. V.; Proserpio D. M. CrystEngComm 2011, 13, 3947. [Google Scholar]
- Spek A. L. PLATON, Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Maji T. K.; Matsuda R.; Kitagawa S. Nat. Mater. 2007, 6, 142. [DOI] [PubMed] [Google Scholar]; b Ok K. M.; Sung J.; Hu G.; Jacobs R. M. J.; O’Hare D. J. Am. Chem. Soc. 2008, 130, 3762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.