

Abstract
The synthesis of a pentadentate ligand with strategically designed fluorinated arene groups in the second coordination sphere of a nonheme iron center is reported. The oxidatively resistant fluorine substituents allow for the trapping and characterization of an FeIV(O) complex at −20 °C. Upon warming of the FeIV(O) complex, an unprecedented arene C–F hydroxylation reaction occurs. Computational studies support the finding that substrate orientation is a critical factor in the observed reactivity. This work not only gives rare direct evidence for the participation of an FeIV(O) species in arene hydroxylation but also provides the first example of a high-valent iron–oxo complex that mediates aromatic C–F hydroxylation.
Determining the first- and second-coordination sphere elements that control the reactivity of nonheme iron complexes has been the focus of intense efforts, in part because of the potential to identify the key factors that allow nonheme Fe enzymes to mediate an impressive array of oxidative transformations.1,2 Learning how to rationally tune the reactivity of nonheme iron complexes also has direct implications for the development of earth-abundant, environmentally compatible catalysts. One subclass of nonheme Fe enzymes are pterin-dependent amino acid (AA) hydroxylases, which perform arene hydroxylations with mononuclear iron centers. A high-valent, electrophilic iron(IV)-oxo intermediate has been spectroscopically characterized as the active oxidant in two of these systems: Tyr and Phe hydroxylases.3 Synthetic nonheme Fe complexes that promote arene hydroxylation have been known for over 20 years. Much speculation has centered on the nature of the active intermediate(s) in this chemistry, with proposals suggesting that FeIV(O) or FeV(O) intermediates may be the key players. However, direct evidence in these reactions for such high-valent intermediates is mostly unknown.1a,4 Identification of the iron-based intermediate(s) that mediate arene hydroxylation remains a challenging goal.
Well-characterized FeIV(O) species are strikingly inert in the oxidation of arene substrates.1a,4e,4g,4h However, the few documented cases of intramolecular arene hydroxylation have been proposed to operate through putative FeIV(O) intermediates.1a,4a−4d We recently tested this dichotomy by constructing a nonheme iron(II) complex with two arene groups built into the second coordination sphere of [FeII(N4Py2Ph)(CH3CN)]2+.1a Rapid intramolecular arene hydroxylation was observed, while, in contrast, no intermolecular arene hydroxylation chemistry could be seen for the FeIV(O) complex of the untethered N4Py analog. Although indirect evidence suggested that an FeIV(O) species was the active oxidant in the intramolecular case, we were unable to provide any direct observation of this elusive species.
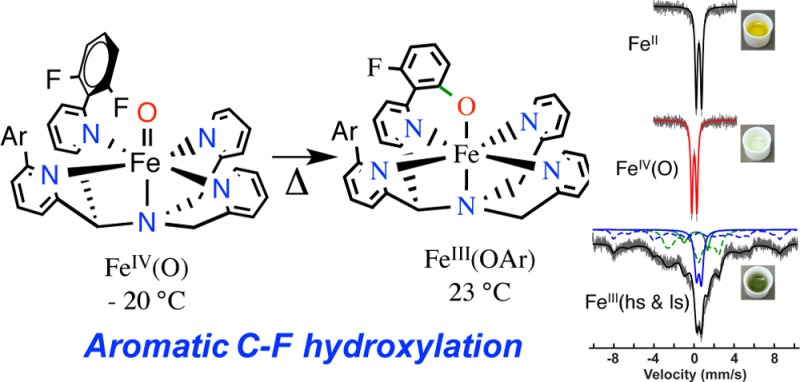
Herein we have designed and synthesized a new ligand related to N4Py2Ph, in which fluorine groups were incorporated to allow for trapping of a putative FeIV(O) intermediate. This ligand, N4Py2PhF2 (1), was used to prepare the new FeII complex, [FeII(N4Py2PhF2)(NCMe)]2+ (2), which then leads to trapping of an FeIV(O) complex [FeIV(O)(N4Py2PhF2)]2+ at low temperature as designed. However, upon controlled warming of the FeIV(O) complex, an unprecedented arene C–F hydroxylation reaction occurs, resulting in formation of [FeIII(N4PyArO,PhF2)]2+ (3). This controlled hydroxylation provided us with the first direct, time-dependent spectroscopic observations of a nonheme FeIV(O) complex mediating arene hydroxylation. Density functional theory (DFT) studies support a mechanism involving electrophilic attack of FeIV(O) on the arene substrate and indicate that proper orientation of the substrate is essential for reactivity. Although C–F cleavage of aromatic substrates has been observed for iron enzymes,5 to our knowledge this report presents the first example of C–F hydroxylation mediated by a nonheme iron complex.
A polyfluorinated ligand, N4Py2PhF2 (1), was synthesized as shown in Scheme S1, and reaction of 1 with FeII(BF4)2 led to dark red crystals of [FeII(N4Py2PhF2)(CH3CN)](BF4)2 (2a) (Figure 1) in 70% yield. The X-ray structure revealed a six-coordinate FeII complex with CH3CN bound in the open site. The Fe–N distances are typical for low-spin (ls) iron(II) complexes,6 which is consistent with Mössbauer spectroscopy (5.8 K, δ = 0.49 mm s–1; ΔEQ = 0.54 mm s–1). The BF4– counterion in 2a can be replaced by ClO4–, giving FeII(N4Py2PhF2)(CH3CN)](ClO4)2 (2b) (X-ray structure, Figure S2). Both the ClO4– and BF4– complexes were employed in further reactivity studies (vide infra).
Figure 1.
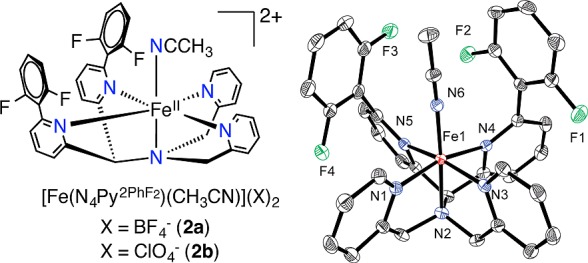
FeII complexes of 2a–b (left), and displacement ellipsoid plot (50% probability) for the cation of 2a (right). H-atoms were omitted for clarity.
The iron(II) complexes 2a–b react rapidly with a slight excess of the soluble O-atom transfer reagent isopropyl-2-iodoxybenzoate (IBX-ester) (1 equiv) in CH3CN at 23 °C to immediately form a pale yellow intermediate with a weak absorption band at 750 nm. This yellow species then converts over 50 min into a dark green species with a more intense, red-shifted maximum at 785 nm (Figure 2). The weak band seen at 750 nm is diagnostic of nonheme FeIV(O) complexes.7 However, the final spectrum with the more intense peak centered at 785 nm is suggestive of an FeIII(OAr) LMCT band.1a Analysis by LDI-MS of the final reaction mixture revealed a parent ion at m/z 643.87 corresponding to [Fe(N4Py2PhF2)+O–F]+ (calcd m/z 644.14), suggesting that, remarkably, one of the C–F bonds had been cleaved and replaced by a C–O group. An isotope labeling experiment with PhI(18O) shifts the peak at m/z 643.8 by +2 units, confirming that the oxygen atom in the product comes from the oxidant (70% incorporation) (Figure S11). The monodefluorinated, monohydroxylated free ligand was isolated by demetalation and characterized by accurate mass determination by FAB-MS.
Figure 2.
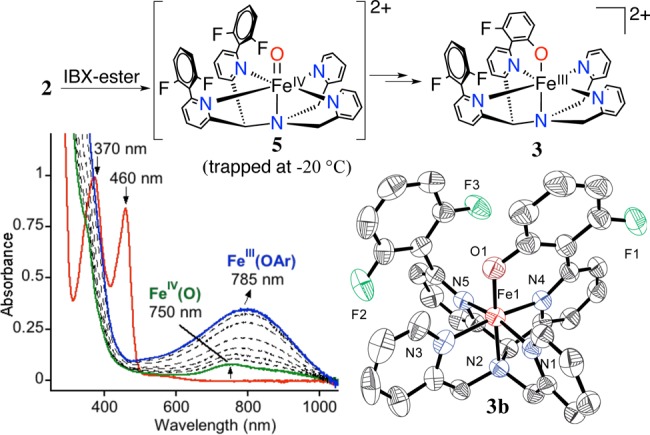
Overall scheme for aromatic hydroxylation (top), UV–vis spectral changes for 2a + IBX-ester over 50 min in CH3CN at 23 °C (bottom left), displacement ellipsoid plot (50% probability) for the dication of 3b (bottom right). H-atoms were omitted for clarity.
Definitive evidence for the occurrence of C–F hydroxylation was obtained by using the perchlorate starting material 2b. Reaction of 2b with 1 equiv of IBX-ester in CH3CN led to the isolation of green crystals from Et2O/CH3CN suitable for X-ray structure determination, which revealed the structure for [FeIII(N4PyArO,PhF2)](ClO4)2 (3b) (Figure 2). The Fe–N (1.951(4)–2.040(4) Å) and Fe–O (1.787(3) Å) bond lengths in 3b are consistent with a related ls (S = 1/2) FeIII(OAr) complex.1a A very small amount of yellow-green crystals precipitated together with 3b, and X-ray diffraction revealed that these crystals are the iron(III)–fluoride complex [FeIII(N4Py2PhF2)(F)](ClO4)2 (4b) (Figure S4). Bond lengths for 4b are consistent with a high-spin (hs) (S = 5/2) FeIII ion.
Further characterization of reaction mixtures by EPR spectroscopy revealed a rhombic, ls-FeIII signal with g 2.42, 2.13, 1.90, as well as peaks for hs-FeIII at g 6.35, 4.24 (Figure S12). The ls-FeIII spectrum is a close match to that seen for [FeIII(N4PyPhO,Ph)](BF4)21a and can be assigned to the phenolato–iron(III) complex 3a. The hs-FeIII signals may arise at least in part from the minor side-product 4a. A yield of 75–80% for 3b was measured by UV–vis spectroscopy on final reaction mixtures following hydroxylation.
Low-temperature methods were employed to trap the metastable 750 nm species seen in Figure 2. Addition of IBX-ester to 2a in CH3CN at −20 °C leads to the isosbestic conversion seen in Figure S17, with a final, weak band appearing at 750 nm (ε = 250 M–1 cm–1). This spectrum is stable for at least 4 h at −20 °C. Cold-spray ionization mass spectrometry (CSIMS) was performed on the reaction run at −20 °C and revealed a parent ion at m/z 331.5679 which matches [FeIV(O)(N4Py2PhF2)]2+ (calcd m/z 331.5667). The Mössbauer spectrum for the 57Fe-labeled FeII starting material is shown together with the spectrum for [57FeIV(O)(N4Py2PhF2)]2+ in Figure 3. The sharp doublet seen for the FeII complex clearly shifts to a lower isomer shift upon oxidation, giving a new, sharp doublet characterized by δ = 0.03 mm/s, ΔEQ = 0.54 mm/s. The low isomer shift is a hallmark of an FeIV(O) complex (5a). The quadrupole splitting parameter for this species is smaller than the ΔEQ values observed for most other ls-FeIV(O) complexes7 and falls closer to those for high-spin (S = 2) FeIV(O) complexes. However, density functional theory (DFT) calculations suggest that 5a has a low-spin (S = 1) ground state (vide infra).
Figure 3.
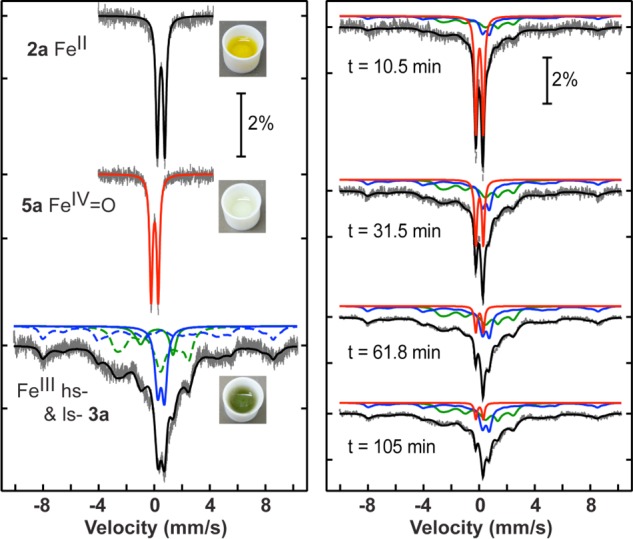
Mössbauer spectra (data collected at 5.8 K) for 2a, 5a and the final solution after warming 5a (left) and time-dependent conversion of 5a to 3a (right).
Gradual warming of the FeIV(O) complex 5a from −20 to 23 °C triggers the direct conversion of the weak band at 750 nm (yellow) to the relatively intense band at 780 nm (green) for [FeIII(N4PyArO,PhF2)]2+. These spectral changes are similar to those seen at room temperature. This conversion was also monitored by CSIMS. The mass spectrum for 5a disappears upon warming, and a new spectrum is observed with a parent ion at m/z 322.0679, which matches the expected peak for 3a ([FeIII(N4PyArO,PhF2)]2+ (calcd: m/z 322.0675). The EPR spectrum for 5a generated at −20 °C is featureless as expected, except for a residual high-spin FeIII spectrum (g = 4.22). Upon warming, the EPR spectrum revealed peaks at g 2.42, 2.13, 1.90, indicative of the ls-FeIII(OAr) complex 3a. The kinetics of conversion to the C–F hydroxylation product could also be directly monitored by UV–vis spectroscopy at rt, where the 750 nm band was shown to convert directly into the 785 nm species. A plot of absorbance at 785 nm versus time fit well to a first-order kinetic model up to 5 half-lives (Figure S24). Fitting of the data yielded a first-order rate constant of k = 0.9 × 10–3 s–1. The rate constant is independent of the concentration of the iron complex, supporting an intramolecular mechanism for the C–F hydroxylation.
Time-dependent Mössbauer spectroscopy provided conclusive evidence for the intermediacy of the FeIV(O) species in C–F hydroxylation (Figure 3). The FeIV(O) complex 5a was first generated at −20 °C and then was slowly warmed and allowed to react at 15–19 °C for varying reaction times. The doublet for 5a smoothly converts to a new spectrum over 105 min. This new spectrum consists of a mixture of hs (blue) and ls (green) FeIII signals that are both slow (dashed lines) and fast (solid lines) relaxing. The isomer shift and quadrupole splittings of both FeIII species were determined by measuring the sample at 170 K, at which temperature the spectrum almost fully relaxes into a set of quadrupole doublets (hs-FeIII: δ 0.49 mm/s, ΔEQ = 0.49 mm/s; ls-FeIII: δ 0.18 mm/s, ΔEQ = 2.33 mm/s). The full hyperfine constants are given in Tables S1–S2. The relative area of each species was determined at different time points, and the data are plotted in Figure S23. The FeIV(O) complex undergoes a single exponential decay with a rate constant of k = 1.2 × 10–3 s–1, which is in excellent agreement with the rate constant determined by UV–vis spectroscopy. Upon warming the FeIV(O) complex trapped at −20 °C, equal proportions of hs- and ls-FeIII species are produced. Although the ls-FeIII species can be assigned to 3a, the hs-FeIII component may partly be composed of the FeIII(F) side-product 4a as noted earlier. However, a hydroxylation reaction carried out at rt and then subsequently frozen for Mössbauer spectroscopy showed higher proportions of ls-FeIII, more consistent with the higher yields of 3a obtained by UV–vis.
Taken together, all of the data indicate that the FeIV(O) species is the key intermediate that initiates the arene hydroxylation reaction. Electrophilic attack of FeIV(O) on the deactivated, difluorophenyl ring in [FeIV(O)(N4Py2PhF2)]2+ is the most reasonable next step, with electron transfer from the arene ring to the Fe center to give an arene radical concomitant with C–O bond formation. The release of a fluorine radical is unfavorable, and therefore it is more likely that the radical intermediate is reduced by one electron from an as yet unidentified reductant, allowing for the facile elimination of F– and formation of 3. A recent example of a C−F hydroxylation reaction mediated by a high-valent copper species is also promoted by a sacrificial reductant.100
DFT calculations were performed to analyze the initial electrophilic attack of the FeIV(O) species (Figure 4). The calculations indicate a triplet spin ground state for 5, with a low-lying quintet excited state. The O-atom attacks the aromatic ring via transition states 3,5TSC–O, leading to stable radical intermediates 3,5IntIIIC–O. The triplet spin pathway is the lowest in energy, proceeding via 3TSC–O,π (18.1 kcal/mol), which is unusual in nonheme iron reactivity because the high-spin quintet barrier is normally lower in energy.4g,8 The triplet spin state is calculated to react by accepting an electron into a π*xz/yz orbital (π-pathway), which requires the substrate to orient with an angle of Fe–O–C ≈ 120° for good overlap. In contrast, the quintet spin state can, in principle, react through an additional channel involving one electron transfer to a σ*z2 orbital (σ-pathway, Fe–O–C ≈ 180°), because this acceptor orbital comes down in energy for the quintet state. An orientation with Fe–O–C = 116.8° (116.0°) is obtained for 3TSC–O (5TSC–O) indicating that the aromatic attack takes place via a π-pathway for both spin states. Group spin densities also confirm π-pathways for both states. Attempts to swap orbitals and create σ-pathway intermediates and TSs in the quintet spin state failed, converging back to the lower lying solutions described in Figure 4. Typically, the triplet π-pathway, which requires an approximate perpendicular substrate approach, is prohibitively high in energy because of the steric clash with the equatorial ligands, preventing intermolecular arene hydroxylation.4g,9 Thus, orientation of the substrate in the second coordination sphere of [FeIV(O)(N4Py2PhF2)]2+ has caused a dramatic lowering of the triplet π-pathway. The availability of this pathway is likely responsible for the observed intramolecular arene hydroxylation, as proposed previously.1a
Figure 4.
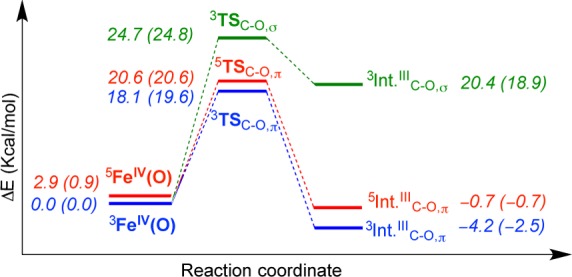
Potential energy diagram for the initial electrophilic attack of 5. The numbers in parentheses are ΔG at 298 K.
Conclusive evidence has been provided to show that aromatic C−F hydroxylation is mediated by a metastable FeIV(O) intermediate. A key aspect of the inherent reactivity of the FeIV(O) complex comes from the proper orientation of the arene substrate in the second coordination sphere. In an earlier study, we showed that positioning of a phenyl ring in the second-coordination sphere of [FeII(N4Py2Ph)]2+ facilitated arene hydroxylation and postulated that this reaction proceeded via a key, yet unidentified FeIV(O) intermediate. In the current work we successfully arrested the arene hydroxylation chemistry through fluorination of the second-coordination sphere Ph groups, allowing for the FeIV(O) intermediate to be trapped and characterized. However, this intermediate was still capable of intramolecular arene hydroxylation at higher temperatures and its direct conversion into hydroxylated product could be monitored in real time. These results together with DFT calculations help to clarify long-standing, yet puzzling observations in the literature1a,4c,4e,4g,4h that indicate that FeIV(O) species can mediate intramolecular, but not intermolecular, arene hydroxylations. These studies suggest that the inherent reactivity of ls-FeIV(O) complexes could be unleashed through further ligand design to facilitate challenging reactions. These results also support the importance of substrate orientation in tuning the reactivity at the metal center of nonheme Fe enzymes.
Acknowledgments
The NIH (D.P.G., GM62309) is acknowledged for financial support. S.S. is grateful for a Martin and Mary Kilpatrick Fellowship. S.d.V. thanks the NSCCS for CPU time, and M.G.Q. thanks the BBSRC for a studentship. G.N.L.J. thanks The International Mobility Fund administered by Royal Society of New Zealand. I.I.-B. and M.D. acknowledge the “Solar Technologies Go Hybrid” initiative of the State of Bavaria.
Supporting Information Available
Experimental and DFT details, Scheme S1, Tables S1–S10, and Figures S1–S28. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Sahu S.; Widger L. R.; Quesne M. G.; de Visser S. P.; Matsumura H.; Moënne-Loccoz P.; Siegler M. A.; Goldberg D. P. J. Am. Chem. Soc. 2013, 135, 10590–10593. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Widger L. R.; Davies C. G.; Yang T.; Siegler M. A.; Troeppner O.; Jameson G. N. L.; Ivanović-Burmazović I.; Goldberg D. P. J. Am. Chem. Soc. 2014, 136, 2699–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Krebs C.; Fujimori D. G.; Walsh C. T.; Bollinger J. M. Jr. Acc. Chem. Res. 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bruijnincx P. C. A.; van Koten G.; Klein Gebbink R. J. M. Chem. Soc. Rev. 2008, 37, 2716–2744. [DOI] [PubMed] [Google Scholar]
- a Eser B. E.; Barr E. W.; Frantom P. A.; Saleh L.; Bollinger J. M. Jr.; Krebs C.; Fitzpatrick P. F. J. Am. Chem. Soc. 2007, 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Panay A. J.; Lee M.; Krebs C.; Bollinger J. M. Jr.; Fitzpatrick P. F. Biochemistry 2011, 50, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hegg E. L.; Ho R. Y. N.; Que L. Jr. J. Am. Chem. Soc. 1999, 121, 1972–1973. [Google Scholar]; b Mekmouche Y.; Ménage S.; Toia-Duboc C.; Fontecave M.; Galey J.-B.; Lebrun C.; Pécaut J. Angew. Chem., Int. Ed. 2001, 40, 949–952. [PubMed] [Google Scholar]; c Jensen M. P.; Lange S. J.; Mehn M. P.; Que E. L.; Que L. Jr. J. Am. Chem. Soc. 2003, 125, 2113–2128. [DOI] [PubMed] [Google Scholar]; d Mehn M. P.; Fujisawa K.; Hegg E. L.; Que L. Jr. J. Am. Chem. Soc. 2003, 125, 7828–7842. [DOI] [PubMed] [Google Scholar]; e Oh N. Y.; Seo M. S.; Lim M. H.; Consugar M. B.; Park M. J.; Rohde J.-U.; Han J.; Kim K. M.; Kim J.; Que L. Jr.; Nam W. Chem. Commun. 2005, 5644–5646. [DOI] [PubMed] [Google Scholar]; f Nielsen A.; Larsen F. B.; Bond A. D.; McKenzie C. J. Angew. Chem., Int. Ed. 2006, 45, 1602–1606. [DOI] [PubMed] [Google Scholar]; g de Visser S. P.; Oh K.; Han A.-R.; Nam W. Inorg. Chem. 2007, 46, 4632–4641. [DOI] [PubMed] [Google Scholar]; h Makhlynets O. V.; Rybak-Akimova E. V. Chem.—Eur. J. 2010, 16, 13995–14006. [DOI] [PubMed] [Google Scholar]; i Thibon A.; Jollet V.; Ribal C.; Sénéchal-David K.; Billon L.; Sorokin A. B.; Banse F. Chem.—Eur. J. 2012, 18, 2715–2724. [DOI] [PubMed] [Google Scholar]; j Bigi J. P.; Harman W. H.; Lassalle-Kaiser B.; Robles D. M.; Stich T. A.; Yano J.; Britt R. D.; Chang C. J. J. Am. Chem. Soc. 2012, 134, 1536–1542. [DOI] [PubMed] [Google Scholar]; k Ansari A.; Kaushik A.; Rajaraman G. J. Am. Chem. Soc. 2013, 135, 4235–4249. [DOI] [PubMed] [Google Scholar]
- a Murphy C. D. Biotechnol. Lett. 2010, 32, 351–359. [DOI] [PubMed] [Google Scholar]; b Fox B. G.; Borneman J. G.; Wackett L. P.; Lipscomb J. D. Biochemistry 1990, 29, 5300–5306. [DOI] [PubMed] [Google Scholar]
- Prat I.; Company A.; Corona T.; Parella T.; Ribas X.; Costas M. Inorg. Chem. 2013, 52, 9229–9244. [DOI] [PubMed] [Google Scholar]
- McDonald A. R.; Que L. Jr. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar]
- Serrano-Plana J.; Garcia-Bosch I.; Miyake R.; Costas M.; Company A. Agnew. Chem. Int. Ed. 2014, 53, 9608–9612. [DOI] [PubMed] [Google Scholar]
- a de Visser S. P. J. Am. Chem. Soc. 2006, 128, 9813–9824. [DOI] [PubMed] [Google Scholar]; b Hirao H.; Kumar D.; Que L. Jr.; Shaik S. J. Am. Chem. Soc. 2006, 128, 8590–8606. [DOI] [PubMed] [Google Scholar]; c Geng C.; Ye S.; Neese F. Angew. Chem., Int. Ed. 2010, 49, 5717–5720. [DOI] [PubMed] [Google Scholar]; d Wilson S. A.; Chen J.; Hong S.; Lee Y.-M.; Clémancey M.; Garcia-Serres R.; Nomura T.; Ogura T.; Latour J.-M.; Hedman B.; Hodgson K. O.; Nam W.; Solomon E. I. J. Am. Chem. Soc. 2012, 134, 11791–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Decker A.; Clay M. D.; Solomon E. I. J. Inorg. Biochem. 2006, 100, 697–706. [DOI] [PubMed] [Google Scholar]; b Neidig M. L.; Decker A.; Choroba O. W.; Huang F.; Kavana M.; Moran G. R.; Spencer J. B.; Solomon E. I. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12966–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shaik S.; Chen H.; Janardanan D. Nat. Chem. 2011, 3, 19–27. [DOI] [PubMed] [Google Scholar]; d Wong S. D.; Bell C. B. III; Liu L. V.; Kwak Y.; England J.; Alp E. E.; Zhao J.; Que L. Jr.; Solomon E. I. Angew. Chem., Int. Ed. 2011, 50, 3215–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ye S.; Neese F. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Srnec M.; Wong S. D.; England J.; Que L. Jr.; Solomon E. I. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.