

Abstract
Described here are mechanistic details of the chemical reactivities of two modified/saturated pyrimidine residues that represent naturally occurring forms of DNA damage: 5-thyminyl-5,6-dihydrothymine, commonly referred to as the “spore photoproduct” (SP), and 5,6-dihydro-2′-deoxyuridine (dHdU), formed via ionizing radiation damage to cytosine under anoxic conditions and also serving as a general model of saturated pyrimidine residues. It is shown that due to the loss of the pyrimidine C5–C6 double bond and consequent loss of ring aromaticity, the C4 position of both these saturated pyrimidines is prone to the formation of a hemiaminal intermediate via water addition. Water addition is facilitated by basic conditions; however, it also occurs at physiological pH at a slower rate. The hemiaminal species so-formed subsequently converts to a ring-opened hydrolysis product through cleavage of the pyrimidine N3–C4 bond. Further decomposition of this ring-opened product above physiological pH leads to DNA strand break formation. Taken together, these results suggest that once the aromaticity of a pyrimidine residue is lost, the C4 position becomes a “hot spot” for the formation of a tetrahedral intermediate, the decay of which triggers a cascade of elimination reactions that can under certain conditions convert a simple nucleobase modification into a DNA strand break.
Introduction
Pyrimidine nucleobases account for half of the nucleotide composition of any given genomic DNA and possess chemical and physical reactivities that can lead to unique modifications of their native structures.1 One of the most common DNA damage events involving pyrimidines is the formation of photodimers upon UV irradiation.2 Of the three naturally occurring thymine dimers identified to date (Figure 1): the cyclobutane pyrimidine dimer (CPD),3 the pyrimidine (6-4) pyrimidone photoproduct (6-4PP),4 and 5-thyminyl-5,6-dihydrothymine (spore photoproduct, “SP”),5 SP is formed as a result of the unique environment found within endospores, where a low hydration level and the presence of DNA binding proteins named small acid soluble proteins (SASPs) alter the genomic DNA from a B-form to an A-form conformation.6 UV irradiation of this A-form DNA produces SP as the dominant (>95%) photolesion found in endospores.5a,7 Toward understanding the chemistry of this DNA lesion, the synthesis of dinucleotide SP has been accomplished by Kim et al.8 and can also be prepared via dinucleotide TpT photoreaction in dry films.9 Recently, our group prepared the SP TpT phosphoramidite, which has enabled the incorporation of SP into synthetic oligonucleotides with high efficiency and high purity.10 Therefore, the chemical reactivity of SP in vitro and its impact on the stability of genomic DNA can be studied readily using dinucleotide SP and/or SP-containing oligonucleotides.
Figure 1.

Naturally occurring thymine dimers and dHdU that possess a saturated pyrimidine ring.
To investigate details of the fundamental chemical reactivity of SP, the behavior of SP was examined under alkaline conditions as well as at physiological pH. Results from these studies indicated that the loss of 5′-thymine aromaticity upon formation of SP facilitates water addition and formation of a hemiaminal intermediate at the C4 position of this nucleobase at or above neutral pH. The decay of this hemiaminal species under basic conditions leads to rupture of the N3–C4 bond, producing an SP hydrolysis product which readily triggers a cascade of elimination reactions that can lead, ultimately, to DNA strand cleavage. Moreover, a similar reaction was observed with another form of damaged pyrimidine that results from ionizing radiation damage to cytosine under anoxic conditions: 5,6-dihydro-2′-deoxyuridine (dHdU, Figure 1).11 For our purposes this particular lesion also serves as a general example representing saturated pyrimidine lesions. In light of the observed behaviors of both these pyrimidine-based systems, formation of a hemiaminal intermediate is likely a general property of a saturated pyrimidine residue at or above physiological pH.
Understanding the chemical reactivities of the altered nucleobases noted above provides important knowledge pertaining to the behavior of 5,6-saturated pyrimidines that are produced in DNA by various forms of damaging agents and events; as noted, saturated pyrimidine nucleobases can occur as a result of both photodamage and through ionizing radiation. Such knowledge is important toward ongoing synthetic, analytical, and biological studies, i.e., handling and generating DNA containing these lesions, the development of detection assays, elucidation of repair routes, and may provide fundamental insight into their biological consequences.
Methods
Materials and General Methods
All solvents and chemicals were of analytical grade and purchased from Sigma, Fisher or VWR and used without further purification. 1H NMR spectra were obtained using a Bruker 500 MHz NMR Fourier transform spectrometer using deuterium oxide as a solvent and with residual water acting as an internal standard. Mass spectrometric (MS) analyses were obtained via electrospray ionization (ESI) employing an ion-trap mass analyzer. HR-MS and MS/MS analyses were performed using a Q-TOF LC/MS spectrometer; data were acquired via “Agilent MassHunter Workstation Data Acquisition (B.03.00)” software and analyzed via “Qualitative Analysis of MassHunter Acquisition Data (B.03.00)” software. Oligonucleotides were prepared via automated DNA synthesis procedures using an ABI 394 DNA/RNA synthesizer.
Synthesis of Dinucleotide SP TpT and SP-Containing Oligonucleotides
The dinucleotide SP TpT was synthesized using a procedure originally developed by Kim et al.8 and later modified by our group.9 The SP-containing oligonucleotide 5′-TT(SP)T was synthesized via the SP phosphoramidite prepared in house10 and standard automated solid phase DNA synthesis procedures.
Synthesis of dHdU-Containing Oligonucleotides
A solution of 2′-deoxyuridine (2.05 g) and rhodium on alumina (5%, 200 mg) in MeOH/H2O (30 mL/30 mL) was stirred under 1 atm hydrogen gas for 2 days. After filtration to remove the catalyst and evaporation of solvent under vacuum, dHdU was collected as a colorless solid in quantitative yield and subsequently protected as its 5′-O-dimethoxytrityl derivative. The phosphoramidite of this nucleoside was prepared by first dissolving 5′-O-dimethoxytrityl-dHdU (1.09 g, 2.05 mmol) in dichloromethane (10 mL) to which DiPEA (1.40 mL, 8.20 mmol) was then added dropwise followed by addition of chloro-2-cyanoethyl-N,N-diisopropylphosphoramidite (0.60 g, 2.5 mmol). The reaction solution was stirred at room temperature for 30 min. The mixture was concentrated via rotary evaporation and then purified by flash chromatography using 1:1 hexane/ethyl acetate as an eluent to afford the dHdU phosphoramidite as a white solid (1.13 g, 75%). Using this purified phosphoramidite, the dHdU containing oligonucleotide 5′-TT(dHdU)TT was synthesized using standard automated solid phase DNA synthesis procedures.
Formation of SP Hydrolysis Product (1) in 0.2 M KOH
Dinucleotide SP TpT was dissolved in 0.2 M KOH to a final concentration of 0.75 mM. The resulting solution was maintained at room temperature until reaction equilibrium was attained (∼2 days) as assessed by monitoring 1 μL aliquots of the reaction mixture by HPLC. The maximum yield of 1 was ∼70% under conditions of 0.2 M KOH upon attainment of equilibrium.
Formation of SP TpT Hydrolysis Product (1) at Various pH Values
Different buffering systems were employed to achieve the basic pH solutions used in this study: 0.8 M KOH (pH 13.8); 0.2 M KOH (pH 13.3); 50 mM KOH (pH 12.7); and 100 mM K2HPO4 buffer (pH 11.0). SP TpT was dissolved in these different buffers to a final concentration of 0.75 mM. The resulting solutions were maintained at ambient temperature for 48–96 h to allow each reaction to achieve equilibrium, as confirmed by HPLC analyses of 1 μL aliquots of each solution (extracted); longer incubation times did not result in increased product formation.
Formation of SP Hydrolysis Product (1) in 18O Water
Different buffering systems were employed to achieve the basic solutions used in this study: 0.2 M KOH (pH 13.3); 100 mM K2HPO4 (pH 12.0 and 10.5); 100 mM Tris buffer (pH 8.7 and 7.4 at 37 °C). The buffers were first prepared in Milli-Q water and subsequently lyophilized overnight prior to redissolution in 100 μL of 97% 18O labeled water. SP TpT was subsequently dissolved in 100 μL of these individual 18O buffers to final concentrations of 0.75 mM. The resulting solutions were maintained at ambient temperature or at 37 °C for various time intervals. One μL of each of the resulting solutions was extracted and analyzed by direct injection into HPLC.
Formation of dHdU Hydrolysis Product (9) in 0.2 M KOH
dHdU was dissolved in 0.2 M KOH to a final concentration of 0.75 mM. The resulting solution was maintained at room temperature for 0.5 h. One μL of the resulting mixture was analyzed by HPLC; analyses indicated that all dHdU was converted to 9.
Hydrolysis of dHdU at Various pH Values in 18O Water
KOH (0.2 M, pH 13.3) and 100 mM K2HPO4 (pH 11) were used in this study. The buffers were first prepared using non-18O water, and the resulting solutions were lyophilized overnight before being redissolved in 100 μL of 97% 18O labeled water. dHdU was subsequently dissolved in 100 μL of each of these 18O buffers to final concentrations of 0.75 mM, and the resulting solutions were maintained at ambient temperature for various times. At different time points, 1 μL of each reaction solution was extracted and analyzed by HPLC.
HPLC Product Analyses
HPLC analyses were performed at room temperature using a Waters (Milford, MA) HPLC system coupled to a 2489 UV–vis detector at 268 nm. An Agilent ZORBAX Bonus-RP column (5 μm particle size, 250 × 4.6 mm i.d.) was equilibrated in solvent A (10 mM ammonium acetate in 99% water and 1% acetonitrile, pH 6.5), and compounds were eluted with an ascending gradient (1% ∼ 10%) of acetonitrile in 20 min at a flow rate of 1 mL/min. Semipreparative HPLC analyses were performed at room temperature with the same Waters HPLC setup. An XBridge OST C18 column (2.5 μm particle size, 50 × 10 mm i.d.) was equilibrated in solvent A (10 mM ammonium acetate in 99% water and 1% acetonitrile, pH 6.5), and compounds were eluted with an ascending gradient (1–10%) of acetonitrile in 20 min at a flow rate of 4.73 mL/min. Products were confirmed by LC/MS spectrometry and NMR spectroscopy.
LC/MS Product Analyses
LC/MS-based assays of 18O incorporation were conducted via an Agilent 6520 Accurate Mass Q-TOF LC/MS spectrometer using an Agilent ZORBAX Bonus-RP column (5 μm particle size, 250 × 4.6 mm i.d.). The column was equilibrated in solvent A (5 mM ammonium acetate in 99% water and 1% acetonitrile, pH 6.5), and compounds were eluted with an ascending gradient (1–10%) of acetonitrile (solvent B) in 20 min at a flow rate of 0.5 mL/min. The mass signals were monitored using both positive and negative ion modes. The LC/MS analyses of alkali treated 5′-TT(SP)T were conducted via the same Agilent LC/MS setup using an Agilent Eclipse Plus C18 column (3.5 μm particle size, 100 × 4.6 mm i.d.). The column was equilibrated in solvent A (5 mM ammonium acetate in 99% water and 1% acetonitrile, pH 6.5), and compounds were eluted with an ascending gradient (2–10%) of acetonitrile (solvent B) in 20 min at a flow rate of 0.5 mL/min. The mass signals were monitored using negative ion mode.
Product Analyses via Tandem Mass Spectrometry (MS/MS)
The MS/MS analyses of 1 were conducted using an Agilent 6520 Accurate Mass Q-TOF LC/MS spectrometer. The column was equilibrated in solvent A (5 mM ammonium acetate in 99% water and 1% acetonitrile, pH 6.5), and samples were eluted with an ascending gradient of acetonitrile (1–7% in the first 4 min, then 7% in the next 16 min) at a flow rate of 0.5 mL/min. The mass signals were monitored using both positive and negative ion modes.
Decomposition of the SP Hydrolysis Product (1) at pH 7.4
To a freshly isolated SP hydrolysis product 1 solution (0.5 mM, 10 μL) in 5 mM ammonium acetate solution (pH 6.5) was added pH 7.4 Na2HPO4/NaH2PO4 buffer (100 mM, 10 μL). The pH value of the resulting solution was determined to be 7.4. The solution was then heated on a heating mantle at 90 °C for various time periods. The reaction was analyzed by immediate injection of 1 μL of the resulting solution via HPLC as described above for a given time point.
SP Hydrolysis Product (1) Formation and Decomposition within Oligonucleotides
The SP containing oligonucleotide 5′-TT(SP)T was dissolved in 0.2 M KOH. The resulting solutions were transferred to 0.5 mL Eppendoff tubes and heated to 90 °C for 0.5 h on a heating mantle. The reaction products were analyzed immediately by LC/MS spectroscopy.
dHdU Hydrolysis Product (9) Formation and Decomposition within Oligonucleotides
The dHdU-containing oligonucleotide 5′-TT(dHdU)TT was dissolved in 0.2 M KOH for 2 h at ambient temperature. The resulting dHdU-H2O adduct (9) contained within 5′-TT(9)TT was isolated by HPLC, dissolved in pH 7.4 phosphate buffer, and heated to 90 °C on a heating mantle for 0.5 h. The products were then analyzed by LC-MS using the procedures noted above.
Results
Formation of an SP-Water Adduct under Alkaline Conditions
Incubation of dinucleotide SP TpT in 0.2 M KOH for 24 h at ambient temperature resulted in the formation of a new product (1) in approximately 65% yield (Figure 2A). ESI-MS analyses of this product (negative ion mode) revealed that it possessed an m/z value of 563.1, corresponding to the [M – H]− signal with M possessing a molecular mass of SP + 18 amu. This gain of mass suggests that 1 is likely a water adduct of SP formed at one of the four available carbonyl moieties. To shed light on the exact water addition site, MS/MS analyses were performed on both the SP TpT control and the putative water adduct 1. As shown in Figure 2B, the fragmentation patterns for these two compounds differed substantially: the fragments of SP TpT mainly resulted from loss of 2′-deoxyriboses; in contrast, the most abundant fragment of 1 was observed at an m/z value of 520.1 (negative ion mode), corresponding to the loss of NH=C=O. Fragmentation of 1 due to 2′-deoxyribose loss was also observed if the collision energy used for the MS/MS analyses was increased.
Figure 2.

(A) HPLC chromatograph (monitored at 260 nm) of the SP TpT hydrolysis reaction in the presence of 0.2 M KOH at ambient temperature for 24 h. (B) MS/MS analyses (negative ion mode, [M – H]−) of dinucleotide SP TpT (lower) and the SP hydrolysis product 1 (upper) as well as the structures of other major fragments formed.
Additional control analyses indicated that similar water adducts were not observed when undamaged, intact thymine residues were subjected to the same conditions. Thus, it is highly unlikely that the 3′-thymine of SP, which maintains its aromaticity (Figure 1), is involved in the reaction, leaving two possible positions for water addition within SP, both contained within the now saturated 5′-T ring (at C2 and C4). Of these two positions, if water addition occurs at C2, the hydrolysis product is likely to rupture the C2–N3 bond, yielding 2 (Scheme 1, path B). In contrast, reaction at C4 would lead to the hydrolytic cleavage of the N3–C4 bond and the formation of 1 (Scheme 1, path A). In support of C4 as the most likely site of water addition, we note that the C2 carbonyl is stabilized by resonance provided by the two lone pairs on N1 and N3, while the C4 carbonyl is stabilized only by N3. Thus, the C4 position should be more reactive toward an addition reaction. This rationale is supported directly by the observed MS/MS fragmentation pattern: The loss of NH=C=O from 1 is reasonable, as the N1–C2 bond can be hydrolyzed readily whereas it should be considerably more difficult to eliminate NH=C=O from 2 as it would require breakage of a strong C–C bond (Scheme 1, path B). Moreover, formation of 1 is consistent with findings reported upon the hydrolysis of 6-4PP, where the N3–C4 bond of the 5′-thymine ruptures.12 We thus conclude that the new product observed in the chromatogram shown in Figure 2A is most likely a water adduct possessing the structure shown for 1 (Scheme 1).
Scheme 1.

SP Treatment in 18O Water
To confirm that 1 is an adduct with water, the reactions described above were performed in 18O labeled water containing 0.2 M KOH for 48 h. Subsequent ESI-MS analyses indicated that 18O was indeed incorporated into 1. However, to our surprise, the incorporation of two18O atoms was observed. Analysis of unreacted SP TpT remaining in these reaction mixtures revealed a mass of unlabeled SP + 2 amu, suggesting that intact SPs exchange one 18O atom as well,13 which likely results from an oxygen exchange between the C4=O on the saturated 5′-thymine of SP and water.
To understand the oxygen exchange process noted above, the hydrolysis of SP in 18O water/0.2 M KOH was re-examined by ESI-MS. As shown in Figure 3, a linear incorporation of 18O into SP was observed within the first 1.5 h (the rate of 18O incorporation is reported in Table 1). After 4 h the reaction rate slowed considerably and reached completion after 2 days, as indicated by the fact that 96% of the SP present contained one 18O atom, equal to the 18O abundance in the 18O water employed. In comparison, analysis of 1 revealed that within the first 30 min ∼92% of 1 contained a single 18O label; the double-18O labeled 1 became obvious after 1 h of the reaction and reached completion in 2 days (Figure 3C). These results clearly indicate that the oxygen at the C4=O moiety exchanged with water and, further, that a tetrahedral hemiaminal intermediate must exist (Scheme 2) which can either eliminate the OH– to restore the thymine ring, resulting in the 18O incorporation into SP TpT or break the N3–C4 bond, generating the ring-open SP hydrolysis product 1. After the 16O at the C4=O of SP has been exchanged by 18O, the subsequent hydrolysis produces the double-18O labeled 1. The formation of double-18O labeled 1 is slower than the 18O incorporation into SP, in agreement with this rationale.
Figure 3.

ESI-MS spectra (negative ion mode) of the SP hydrolysis reaction in 0.2 M KOH at ambient temperature in 97% 18O water. (A) The gradual incorporation of 18O into SP. The peaks indicated by blue arrows exhibit an m/z of 545.1, corresponding to the [M – H]− signal of SP without any 18O incorporation. The peaks indicated by red arrows exhibit an m/z of 547.1, corresponding to the [M – H]− signal of SP with one 18O incorporated. (B) The gradual incorporation of 18O into the SP hydrolysis product 1. The peaks indicated by blue arrows exhibit an m/z of 563.1, corresponding to the [M – H]− signal of 1 without any 18O incorporation. The peaks indicated by orange arrows exhibit an m/z of 565.1, which correspond to the [M – H]− signal of 1 with one 18O incorporated and gradually decrease during the course of the reaction. The peaks indicated by green arrows exhibit an m/z of 567.1, which correspond to the [M – H]− signal of 1 with two 18O incorporated. (C) Time course indicating the incorporation of 18O into SP and SP hydrolysis product 1. The incorporation of the second 18O into 1 is slower than the 18O incorporation into SP, suggesting that the second 18O incorporation occurs by reacting with the 18O labeled SP.
Table 1. Rates of Formation of 1 and 18O Incorporation into SP TpT.
| reaction pH | formation of 1 (μM/h)a | 18O incorporation into SP TpT (μM/h)b |
|---|---|---|
| 13.9 | 427.3 ± 20.0 | |
| 13.3 | 118.4 ± 7.1 | 107.0 ± 5.8 |
| 12 | 8.3 ± 0.7 | 79.1 ± 3.5 |
| 10.5 | 4.0 ± 0.25 | |
| 8.7 | 1.05 ± 0.09 | |
| 7.4 | 0.14 ± 0.02 |
HPLC analysis.
ESI-MS analysis.
Scheme 2.

Formation and Stability of 1 at Various pH Values in 18O Water
As indicated by the reaction rates listed in Table 1, the formation of 1 is clearly driven by the presence of hydroxide anion. While the yield of 1 was too low to be observed by our HPLC assay when the pH was <11, we sought to determine whether the hemiaminal precursor to 1 was nonetheless formed. Presence of the hemiaminal species is reflected by 18O incorporation into SP; the compositions of SP in 18O water at different pH values were thus investigated via ESI-MS spectroscopy. ESI-MS results indicated that 18O incorporation indeed occurs at lower pH values, albeit at slower rates. Even at physiological pH, 18O incorporation was obvious after 6 days at 37 °C.13 By integrating the mass spectroscopic signals, the amount of 18O labeled SPs can be determined, which allowed a measurement of 18O incorporation rates (Table 1). The rate of 18O incorporation was determined to be 140 ± 20 nM/h at pH 7.4, a rate that clearly indicates the existence of an SP hemiaminal species under physiological conditions. Decay of this intermediate would produce 1, although the formation rate of 1 is beyond the detection limit of our assay.
It is known that the 6-4PP hydrolysis product leads to DNA strand cleavage upon hot alkaline treatment,12c we therefore investigated whether 1 could induce DNA strand scission. To reveal possible reaction intermediates, we chose pH 7.4 for this analysis instead of the strong basic conditions used in previous 6-4PP studies. The solution of 1 was heated to 90 °C and analyzed by HPLC. As shown in Figure 4A, ∼ 80% of 1 was consumed after 30 min, and all of 1 was reacted after 90 min. The reaction resulted in six major products: 3, 4, 5, SP TpT, and two SP isomers (as judged by the ESI-MS analysis and their identical MS/MS fragmentation pattern; formation of these SP isomers is currently under investigation and will be reported elsewhere). MS/MS analyses indicate that 3, 4, and 5 all resulted from elimination reactions of 1 (Figure 4B). This observation confirms that 1 decomposes via two possible pathways: β-elimination to remove the attached 2′-deoxyribose, or reversion to SP through loss of the added water.
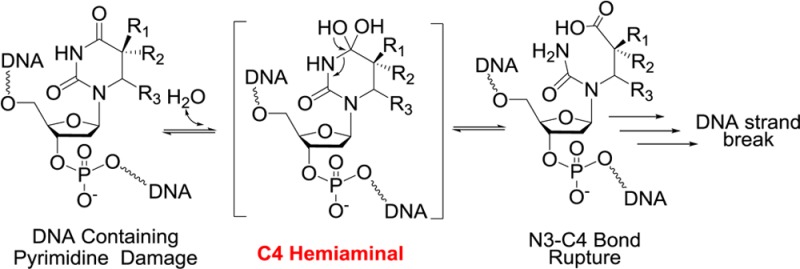
Figure 4.
(A) HPLC chromatograph (260 nm) of the decomposition reaction of 1 at 90 °C for 30, 90, and 240 min, respectively, at pH 7.4. The red dotted line represents the HPLC chromatograph of compound 1 immediately after HPLC purification; no obvious decomposition can be observed at this point. The majority of 1 has reacted after 0.5 h of heating; the unreacted 1 is indicated by the shoulder at the black arrow. All of 1 was consumed after 90 min. About 55% of decomposed 1 was converted to compounds 3, 4, and 5 after a 4 h reaction. 3 and 4 are possible reaction intermediates, and 5 is the final product, as suggested by the reaction kinetics. The remainder of 1 that decomposed reverted to SP and its isomers by loss of the added water. The two peaks marked by * are SP isomers, (see main text). (B) MS/MS analyses of the decomposition products 3, 4, and 5 (negative ion mode, [M – H]−). The chemical structures of these compounds and the fragmentation sites are indicated. The possible structures of the fragments are shown in Supporting Information. The structures confirm that 3 and 4 are two key intermediates during the β-elimination reaction and that 5 is the final product.
Given that the relative peak intensities of SP and its isomers remain constant during the course of the reaction described above, it is likely that they formed simultaneously. In contrast, in the elimination pathway, the major products are 3 and 4 during the first 30 min of the reaction, and a 90 min incubation results in 5 as the major product at the expense of 3 and 4. After 4 h, 5 became the dominant species. These observations indicated that 5 was the final decomposition product, while 3 and 4 are likely reaction intermediates en route to the formation of 5. This conclusion is further supported by the MS/MS analysis (Figure 4B). These analyses strongly suggest that 1 is prone to deglycosylation to yield 3 and 4; formation of 5 is achieved via a 1 → 3 → 4 → 5 pathway (Scheme 3). The abasic site in 4 can be easily converted to the keto form bearing acidic hydrogens at C2′.1b Loss of a proton at C2′ subsequently triggers a β-elimination reaction to remove the 5′-deoxyribose resulting in 5. If such a reaction were to occur in an oligonucleotide, strand cleavage should be the outcome.
Scheme 3.

It is worth mentioning that at 90 °C, the decomposition product 5 was also observed in 0.2 M KOH as a minor product, although little 3 and 4 could be detected, suggesting that these intermediates are unstable under heated basic conditions.13 This result contrasts to the accelerated decay of 1 at 90 °C and pH 7.4 described above, suggesting that under basic conditions, the major decomposition route of 1 is to revert back to SP. At neutral pH, 1 readily undergoes elimination. Such an elimination process is slightly more competitive than the reverse reaction, as indicated by the ∼55% collective yield of 3, 4, and 5, making strand scission a major decay pathway.
SP Hydrolysis Product Formation and Decomposition in a Model Oligonucleotide
To confirm that the species observed in the dinucleotide SP TpT reaction can lead to SP-mediated DNA strand cleavage, an SP-containing oligonucleotide, 5′-TT(SP)T, was treated with 0.2 M KOH at 90 °C for 30 min. HPLC and ESI-MS analyses revealed that SP hydrolysis occurred as expected, generating 8, an oligonucleotide containing the “ring-opened” SP water adduct 1 (Figure 5). The formation of 8 precedes oligonucleotide fragmentation, yielding, upon breakdown, oligonucleotide fragments 6 and 7, which correspond to the predicted 3′- and 5′-portions of the cleaved 5′-TT(SP)T due to the formation of 5.13 This result further supports our conclusion that the SP hydrolysis product 1, formed via an initial hemiaminal species, is the key intermediate that can lead to oligonucleotide strand scission.
Figure 5.

HPLC chromatograph (260 nm) of the SP-induced strand cleavage reaction of the oligonucleotide 5′-TT(SP)T in 0.2 M KOH at 90 °C for 0.5 h.
Formation of dHdU Hemiaminal, Water Adduct, and Oligonucleotide Strand Fragmentation under Alkaline Conditions
Given that the saturated 5′-thymine of SP supports hydrolysis, while the aromatic 3′-thymine does not, suggests that this reactivity results from an intrinsic property of a saturated pyrimidine. To test this hypothesis, the reactivity of dHdU was examined as a general model of saturated pyrimidines. Treatment of dHdU with 0.2 M KOH for 30 min resulted in its complete conversion to the predicted hydrolysis product dHdU-H2O, 9 (Scheme 4, confirmed by MS/MS and NMR analyses),13 which contrasts to the slower and incomplete conversion of SP to 1 under the same reaction conditions (Figure 2). This observation suggests that relative to SP, the N3–C4 bond in dHdU is more activated toward hydrolysis.
Scheme 4.

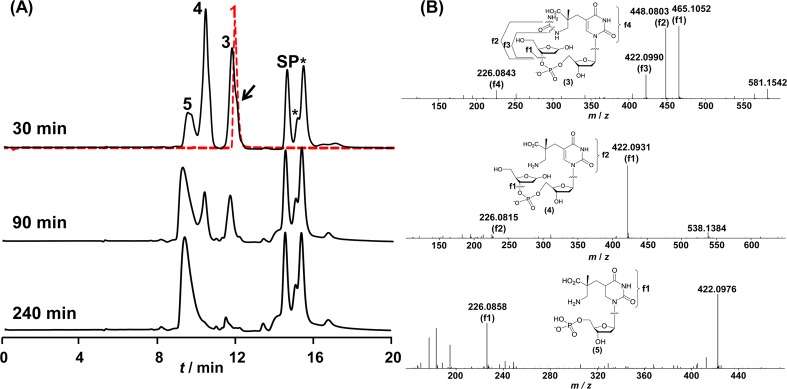
To prove the presence of a hemiaminal intermediate corresponding to that observed with SP during the formation of 9, 18O incorporation experiments were again conducted (Figure 6). Under conditions employed in the study of 1, the ratio between the single- and double-18O labeled 9 formed from dHdU was found to be 8.5:1 after 30 min, which remained constant after 48 h of incubation. The presence of double-18O labeled 9 indicated that the 18O-labeled dHdU must be formed, most likely through a hemiaminal intermediate similar to that formed from SP. However, the unchanged ratio between single- and double-labeled 9 during prolonged incubation suggested that the formation of 9 from dHdU was not prone to reversal, in contrast to the reversible formation of 1 from SP. To examine whether a similar hemiaminal intermediate is involved, the 18O labeling experiment was repeated at pH 11 where the conversion of dHdU to 9 was much slower. Indeed, after 4 h > 90% of dHdU remained; MS analyses indicated that >90% of the dHdU present was labeled by one 18O atom (Figure 6C). Also, as expected, both single- and double-18O labeled 9 were detected. While the double-labeled species continued to increase, its increase correlated well with the increase in the overall yield of 9. This result is consistent with the observation made in 0.2 M KOH, indicating that although the formation of the hemiaminal intermediate is reversible, the formation of 9 from the decomposition of the hemiaminal species is not (Scheme 4).
Figure 6.
ESI-MS spectra describing the dHdU hydrolysis reaction at pH 11 and ambient temperature in 97% 18O water. (A) The gradual incorporation of 18O into dHdU. The peaks indicated by blue and red arrows exhibit an m/z of 229.1 and 231.1, corresponding to the [M – H]− signal of dHdU with zero and one 18O incorporated, respectively. (B) The gradual incorporation of 18O into the dHdU hydrolysis product 9. The peaks indicated by blue, orange, and green arrows correspond to the [M – H]− signal of 9 with zero, one and two 18O incorporated, respectively. (C) Time course indicating the incorporation of 18O into dHdU and its hydrolysis product 9. The incorporation of the second 18O into 9 is slower than the 18O incorporation into dHdU, suggesting that the second 18O incorporation is due to the reaction with the single-18O labeled dHdU. However, the increase of double-18O labeled 9 correlates well with the increase of overall yield of 9; the ratio between single- and double 18O-labeled 9 remain constant if no new 9 is formed. This observation is in contrast to the reversible SP hydrolysis reaction, suggesting that formation of 9 from dHdU is irreversible, as indicated by Scheme 4
To determine the stability of 9 in the context of an oligonucleotide, dHdU was incorporated into 5′-TT(dHdU)TT via solid phase DNA synthesis followed by treatment with 0.2 M KOH to yield 5′-TT(9)TT in a stoichiometric yield (Figure 7B). After heating the solution at 90 °C/pH 7.4 for 30 min, the peak corresponding to 5′-TT(9)TT disappeared, concomitant with the formation of three new products (Figure 7C). As assessed by LC/MS, the major product formed resulted from deglycosylation at dHdU, generating an abasic site (10). Formation of 10 was accompanied by the appearance of two fragments (11 and 12) resulting from β-eliminations at the abasic site. This observation is consistent with the decay pattern observed upon decomposition of the SP hydrolysis product (Scheme 3); however, the lack of a methylene bridge within dHdU (as in SP) makes it impossible to trap intermediates formed during the decomposition of 9 via HPLC. Despite this, the decay of 9 at pH 7.4 was observed to form the predicted 2,5-dihydrofuran-2-ol intermediate, 12, in a sufficiently stable form to be observed. In contrast, such an intermediate is readily decomposed via another β-elimination in 0.2 M KOH, resulting in a complete removal of the ribose as reflected by the formation of 7 in the SP induced strand cleavage reaction (Figure 7). Together, these observations suggest that hydrolysis product formation via a hemiaminal intermediate is a common reactivity of saturated thymine residues, decay of which can lead to strand cleavage of oligomeric DNA under appropriate conditions.
Figure 7.

HPLC chromatograph (260 nm) of (A) 5′-TT(dHdU)TT, (B) formation of 5′-TT(9)TT after treatment of 5′-TT(dHdU)TT with 0.2 M KOH for 1 h at ambient temperature, (C) strand cleavage products resulting from thermal decay of 9 accelerated by heat treatment of 5′-TT(9)TT at 90 °C for 0.5 h at pH 7, and (D) likely structures of the thermal decay products of 9.
Discussion
Understanding details of the chemical reactivities of damaged DNA nucleobases is important toward supporting efforts aimed at characterizing these lesions, their successful total chemical syntheses, the development of bioanalytical methods for their detection, the elucidation of possible biological repair routes, or the consequences of their persistence in a genome. Among the nucleobases present in DNA, pyrimidines are prone to photodamage, oxidation, and the impact of ionizing radiation. The examples emphasized here, SP and dHdU, represent lesions resulting from photodamage to adjacent T residues and ionizing radiation damage to C nucleobases, respectively. Common to each of these instances of damage is the loss of pyrimidine residue aromaticity. In the case of SP, the crystal structure of this lesion revealed clearly that the 5′-thymine ring is distorted from a planar structure, with the C6 and the methyl moiety located ∼0.5 Å above the plane defined by the other five atoms.14 With dHdU, loss of aromaticity leads to a similar outcome as revealed by an NMR spectroscopic study.11a Although little structural alteration was observed at the C4 amide moiety due to the remaining resonance interactions among the carbonyl moieties and the lone pairs of the two N atoms, in both these instances, loss of aromaticity and ring distortion likely activate the C4 position of these saturated nucleobases, promoting the generation of hemiaminal intermediates as revealed by our studies here. The formation of a hemiaminal also indicates the presence of a labile oxygen at C4 that is exchangeable with the aqueous solution. Although similar hemiaminal intermediates were indicated to mediate the deamination reactions of saturated cytidine residues14 and of cytidine or adenosine catalyzed by cytidine15 and adenosine deaminases,16 respectively, to the best of our knowledge the reactivity of hemiaminals derived from saturated thymine residues has been largely overlooked in the past.
The formation of hemiaminal intermediates in SP and dHdU occurs at pH 7.4, but this process is also facilitated by basic conditions. Alkaline treatment has long been used to reveal DNA modifications as some modifications lead to strand cleavage under these conditions.1b For instance, strand scission induced by hot alkaline treatment was utilized to reveal the presence of 6-4PP in UV-irradiated genomic DNA.12b,17 Indeed, rupture of the N3–C4 bond at the 5′-thymine is the first step in 6-4PP hydrolysis en route to DNA strand cleavage upon base treatment.12c This is consistent with what we have observed with SP. However, subsequent strand cleavage in 6-4PP containing DNA was suggested to occur through deglycosylation at the 3′-thymine,12a,12b in contrast to the 5′-thymine deglycosylation observed in our SP studies. The molecular basis for the reactivity difference between these two thymine dimers is currently unclear.
These same hemiaminal intermediates appear also to be responsible for SP-induced DNA strand cleavage. As revealed by the rates shown in Table 1, a high OH– concentration such as that found in a pH 13 buffer forces the vast majority of the hemiaminal intermediate to decompose through rupture of the N3–C4 bond, resulting in the SP hydrolysis product 1 or its equivalent within an oligonucleotide (8). In contrast, decreased hydroxide concentration alters the fate of the hemiaminal, favoring OH– elimination to reform SP. Once formed, however, the SP hydrolysis product 1 or its equivalent in an oligonucleotide (8) is unstable at neutral pH leading to a cascade of elimination reactions and DNA strand scission.
While 1 is unstable under neutral conditions, under strong basic conditions (pH > 12) the major decay pathway of 1 is to eliminate water and revert back to SP via the hemiaminal intermediate. Such a reverse process is indicated by the observation that the double-18O labeled 1 is formed at the expense of the single-18O labeled 1 during prolonged SP hydrolysis in basic 18O-labeled water (Figure 3); the reaction kinetics suggest that this conversion occurs through a reverse reaction that reforms SP. This observation is surprising as 1 contains a carboxylate in the basic solution and is generally considered unreactive. We tentatively ascribe the occurrence of this reverse reaction to the stability of the restored six-member ring in SP.
Although SP and dHdU share the same general pathways to DNA strand cleavage, we show that the reactivities of the respective hemiaminal intermediates are clearly different. The vast majority of the SP hemiaminal intermediate decays back to SP, as indicated by the negligible yield of 1 upon treatment of SP at pH 11 and the mere ∼70% yield of 1 under conditions of concentrated KOH.13 In contrast, the major decay pathway for the dHdU hemiaminal intermediate is to break the N3–C4 bond, as indicated by the 100% conversion to 9 observed during the treatment of dHdU with 0.2 M KOH. This observation suggests that although formation of a hemiaminal may be a common property of a saturated pyrimidine residue, its decay route appears to be influenced by the chemical environment of the ring.
Although the work reported here is conducted using saturated thymidine/2′-deoxyuridine as working models, the chemistry is likely applicable to cytosine as well. The C4-NH2 moieties in damaged cytosine (C) and 5-methylcytosine (5mC) residues are known to be prone to deamination reactions at neutral pH,3a,18 which are also indicated to be mediated by a hemiaminal intermediate.14 Collectively, it appears that in a saturated pyrimidine residue, the C4 position becomes a “hot spot” for subsequent water addition–elimination reactions via a tetrahedral intermediate in living cells. The C4 position may be the weakest link for pyrimidine bases with a reduced electron density at the ring.
DNA strand cleavage upon alkaline treatment is generally considered a common property of damaged DNA in a basic environment with little correlation to behavior at physiological pH.1b Here we show that the hemiaminal intermediate can be formed at neutral pH, the vast majority of which reverts back to SP. Therefore, despite the constant formation of hemiaminals at SP, the genomic DNA is reasonably stable in endospores and a normal genomic DNA structure is thus maintained. However, given that the C–O bond associated with the hemiaminal intermediate can be ruptured at pH 7.4 (as indicated by the 18O incorporation experiment), we speculate that in a relatively rare event, the hemiaminal may decompose via rupture of the N3–C4 bond to form 1. Once 1 is formed, its low stability at physiological pH, as we have shown, may trigger a cascade of decomposition reactions, leading to the formation of an abasic site and eventually DNA strand cleavage.
Acknowledgments
We thank the National Institute of Environmental Health Sciences (R00ES017177) as well as IUPUI startup funds for financial support. The NMR and MS facilities at IUPUI are supported by National Science Foundation MRI grants CHE-0619254 and DBI-0821661, respectively.
Supporting Information Available
Characterizations of the SP and dHdU decomposition products, ESI-MS analyses of the reaction intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lukin M.; de los Santos C. Chem. Rev. 2006, 106, 607. [DOI] [PubMed] [Google Scholar]; b Burrows C. J.; Muller J. G. Chem. Rev. 1998, 98, 1109. [DOI] [PubMed] [Google Scholar]
- a Cadet J.; Vigny P. In Bioorganic Photochemistry; Morrison H., Ed.; Wiley: New York, 1990; Vol. 1, p ix. [Google Scholar]; b Cadet J.; Wagner J. R.. Cold Spring Harbor Perspect. Biol., 2013, 5, a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cadet J.; Mouret S.; Ravanat J. L.; Douki T. Photochem. Photobiol. 2012, 88, 1048. [DOI] [PubMed] [Google Scholar]
- a Cannistraro V. J.; Taylor J.-S. J. Mol. Biol. 2009, 392, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Park H.; Zhang K.; Ren Y.; Nadji S.; Sinha N.; Taylor J.-S.; Kang C. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Taylor J. S.; Brockie I. R.; O’Day C. L. J. Am. Chem. Soc. 1987, 109, 6735. [Google Scholar]
- a Mizukoshi T.; Hitomi K.; Todo T.; Iwai S. J. Am. Chem. Soc. 1998, 120, 10634. [Google Scholar]; b Iwai S.; Shimizu M.; Kamiya H.; Ohtsuka E. J. Am. Chem. Soc. 1996, 118, 7642. [Google Scholar]
- a Donnellan J. E. Jr.; Setlow R. B. Science 1965, 149, 308. [DOI] [PubMed] [Google Scholar]; b Desnous C. L.; Guillaume D.; Clivio P. Chem. Rev. 2010, 110, 1213. [DOI] [PubMed] [Google Scholar]
- a Fairhead H.; Setlow P. J. Bacteriol. 1992, 174, 2874. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Setlow B.; Sun D.; Setlow P. J. Bacteriol. 1992, 174, 2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller R.; Douki T.; Cadet J.; Stackebrandt E.; Nicholson W. L.; Rettberg P.; Reitz G.; Horneck G. Int. Microbiol. 2007, 10, 39. [PubMed] [Google Scholar]
- Kim S. J.; Lester C.; Begley T. P. J. Org. Chem. 1995, 60, 6256. [Google Scholar]
- Lin G.; Li L. Angew. Chem., Int. Ed. 2010, 49, 9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian Y.; Li L. J. Org. Chem. 2013, 78, 3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Villanueva J. M.; Pohl J.; Doetsch P. W.; Marzilli L. G. J. Am. Chem. Soc. 1999, 121, 10652. [Google Scholar]; b Geacintov N. E.; Broyde S.; In The Chemical Biology of DNA Damage; Wiley-VCH: Weinheim, 2010; p xxii. [Google Scholar]
- a Arichi N.; Yamamoto J.; Takahata C.; Sano E.; Masuda Y.; Kuraoka I.; Iwai S. Org. Biomol. Chem. 2013, 11, 3526. [DOI] [PubMed] [Google Scholar]; b Arichi N.; Inase A.; Eto S.; Mizukoshi T.; Yamamoto J.; Iwai S. Org. Biomol. Chem. 2012, 10, 2318. [DOI] [PubMed] [Google Scholar]; c Higurashi M.; Ohtsuki T.; Inase A.; Kusumoto R.; Masutani C.; Hanaoka F.; Iwai S. J. Biol. Chem. 2003, 278, 51968. [DOI] [PubMed] [Google Scholar]
- See Supporting Information.
- a Singh I.; Jian Y.; Li L.; Georgiadis M. M. Acta Cryst. D 2014, 70, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lin G.; Chen C.-H.; Pink M.; Pu J.; Li L. Chem.—Eur. J. 2011, 17, 9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludek O. R.; Schroeder G. K.; Liao C.; Russ P. L.; Wolfenden R.; Marquez V. E. J. Org. Chem. 2009, 74, 6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick L.; Wolfenden R.; Smal E.; Baker D. C. Biochemistry 1986, 25, 1616. [DOI] [PubMed] [Google Scholar]
- a Lippke J. A.; Gordon L. K.; Brash D. E.; Haseltine W. A. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 3388. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pfeifer G. P.; Drouin R.; Riggs A. D.; Holmquist G. P. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 1374. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yoon J.-H.; Lee C.-S.; O’Connor T. R.; Yasui A.; Pfeifer G. P. J. Mol. Biol. 2000, 299, 681. [DOI] [PubMed] [Google Scholar]
- Lemaire D. G. E.; Ruzsicska B. P. Biochemistry 1993, 32, 2525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.