

Abstract
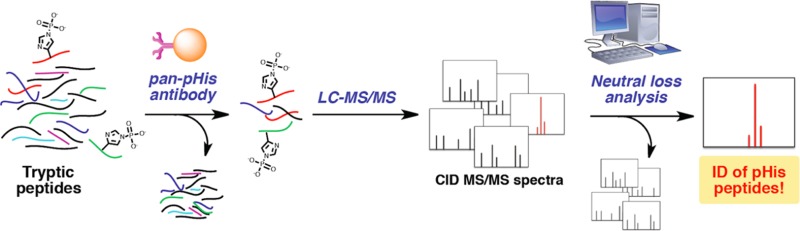
Protein histidine phosphorylation is increasingly recognized as a critical posttranslational modification (PTM) in central metabolism and cell signaling. Still, the detection of phosphohistidine (pHis) in the proteome has remained difficult due to the scarcity of tools to enrich and identify this labile PTM. To address this, we report the first global proteomic analysis of pHis proteins, combining selective immunoenrichment of pHis peptides and a bioinformatic strategy based on mechanistic insight into pHis peptide gas-phase fragmentation during LC–MS/MS. We show that collision-induced dissociation (CID) of pHis peptides produces prominent characteristic neutral losses of 98, 80, and 116 Da. Using isotopic labeling studies, we also demonstrate that the 98 Da neutral loss occurs via gas-phase phosphoryl transfer from pHis to the peptide C-terminal α-carboxylate or to Glu/Asp side chain residues if present. To exploit this property, we developed a software tool that screens LC–MS/MS spectra for potential matches to pHis-containing peptides based on their neutral loss pattern. This tool was integrated into a proteomics workflow for the identification of endogenous pHis-containing proteins in cellular lysates. As an illustration of this strategy, we analyzed pHis peptides from glycerol-fed and mannitol-fed Escherichia coli cells. We identified known and a number of previously speculative pHis sites inferred by homology, predominantly in the phosphoenolpyruvate:sugar transferase system (PTS). Furthermore, we identified two new sites of histidine phosphorylation on aldehyde-alcohol dehydrogenase (AdhE) and pyruvate kinase (PykF) enzymes, previously not known to bear this modification. This study lays the groundwork for future pHis proteomics studies in bacteria and other organisms.
Introduction
Protein phosphorylation plays an essential role in cell signaling events, and its dysregulation can have pathologic consequences.1,2 Much effort has been focused on the global analysis of protein phosphorylation sites and on the enzymes that control phosphosite occupancy.3 While the most well-studied phosphorylated amino acid residues are serine, threonine, and tyrosine, phosphorylation of other amino acids has been observed and in some cases has been known for many decades.4−6 In particular, phosphohistidine (pHis) was first discovered over 50 years ago in bovine liver mitochondria,7 and has since been detected in other eukaryotic and prokaryotic systems. In prokaryotes, pHis plays important roles in two-/multicomponent signaling systems and in facilitating sugar uptake through the phosphoenolpyruvate phosphotransferase system (PTS).8,9 In eukaryotes, pHis has been observed as a dynamic regulatory modification or a direct enzymatic participant in the context of chromatin, central carbon metabolism, and ion channel activity.4 Nonetheless, in contrast to phosphoserine (pSer), phosphothreonine (pThr), and phosphotyrosine (pTyr), still relatively little is known about the pHis modification. Progress in the study of pHis has been significantly hindered by the lack of available research tools4 due in large part to the labile nature of the pHis phosphoramidate moiety. Hydrolysis of the phosphoramidate in pHis releases roughly twice the energy of the phosphoester of pSer, pThr, or pTyr (ΔG° of hydrolysis −12 to −13 kcal/mol vs −6.5 to −9.5 kcal/mol). Accordingly, under acidic conditions, pHis is rapidly hydrolyzed to phosphoric acid and histidine, with a half-life of <30 s in 1 M HCl. Thus, the facile dephosphorylation of pHis has made studies of pHis biology extremely difficult.4
To overcome these limitations, our lab has recently developed antibodies that can recognize pHis. Initially, we developed a sequence-specific rabbit polyclonal antibody to phosphorylated histone H4, employing a synthetic histone H4 peptide with the stable analogue, phosphoryltriazolyl alanine (pTza).10 More recently, we generated a sequence-independent (pan) pHis antibody using the small molecule, phosphoryltriazolyl ethylamine (pTze), as a hapten.11 Using this pan-pHis antibody, together with mass spectrometric (MS) analyses, we characterized histidine phosphorylation of Escherichia coli PEP synthase (PpsA) and the changes in the levels of this modification as a function of cell state.11 During the course of that study, we noticed that pHis peptide ions consistently displayed a set of distinct neutral losses upon fragmentation by collision-induced dissociation (CID). Specifically, we observed a prominent neutral loss of 98 Da by CID, consistent with previous observations by others,12−15 with ancillary losses of 80 and 116 Da. Indeed these neutral losses often dominated the ion current in the MS/MS spectra, leading to reduced efficiency of peptide backbone fragmentation and downstream identification by database search engines.
The neutral loss of phosphoric acid (Δ98 Da) by CID has long been recognized as a hallmark of peptides bearing pSer and pThr.16 Loss of phosphoric acid occurs at the pSer or pThr residue site by β-elimination, or a charge-directed mechanism, which has been exploited to determine the site of phosphorylation on the phosphopeptide.17,18 For pHis peptides, the CID-induced neutral loss of 80 Da is not surprising, as this constitutes loss of HPO3 upon fragmentation of the labile P–N bond in the pHis residue. However, the observation of Δ98 Da as the most prominent CID-induced neutral loss for pHis peptides is puzzling. It suggests that, in addition to losing HPO3 (Δ80 Da) by fragmentation at the labile P–N bond, an additional water moiety (Δ18 Da) is somehow concomitantly lost from the peptide at a location other than the histidine residue.
To resolve this enigma, here we explored the gas-phase reaction mechanism of the pHis peptide Δ98 Da neutral loss, using isotopic labeling and peptide fragmentation under CID conditions. Furthermore, we have exploited the pattern of pHis peptide neutral losses that we observe by CID (dominant loss of 98 Da and accompanying losses of 80 and 116 Da) to enhance our ability to detect and characterize pHis peptides from proteomic samples. For this purpose, we developed a software tool, which we call TRIPLET, to identify MS/MS spectra that exhibit the characteristic CID neutral loss triplet pattern (Δ98, Δ80 and Δ116 Da) of pHis peptides. We also designed an MS-based workflow that incorporates this software tool in two ways: (1) to aid in the database search assignment of CID MS/MS spectra to pHis peptides, and (2) to flag potential pHis peptides for subsequent reanalysis by LC–MS using alternative fragmentation techniques. Finally, we employed this workflow in conjunction with the first reported peptide-level enrichment of pHis by immunoprecipitation using a pan-pHis antibody to achieve a global MS-based pHis proteomic analysis of E. coli cells.
Results
Prominent Neutral Loss of Phosphoric Acid (Δ98 Da) from a Known pHis-Containing Peptide by CID
As an example of the pHis neutral loss phenomenon, we show in Figure 1 the CID MS/MS spectrum of a tryptic peptide containing a pHis site from the protein PtsI, a known pHis-containing protein in the E. coli PTS pathway.11 The spectrum displays a dominant base peak derived from the neutral loss of 98 Da from the parent ion, along with ions of lesser intensity derived from losses of 80 and 116 Da. These neutral loss species dominate the MS/MS ion current, and the observed backbone fragment intensity is low to minimal. This MS/MS analysis was performed by CID in the linear ion trap (LIT) of the Orbi Elite instrument used in this study (see Supporting Information). We could also observe similar neutral loss species for the peptide by higher-energy collisional dissociation (HCD) on the same instrument, though their production was subject to large variation by small alterations in HCD parameter settings (data not shown). Therefore, we expect that these pHis peptide neutral losses would be readily observed on any instrument platform, particularly those with trap-based CID, albeit to a greater or lesser extent depending on collision cell parameters.
Figure 1.
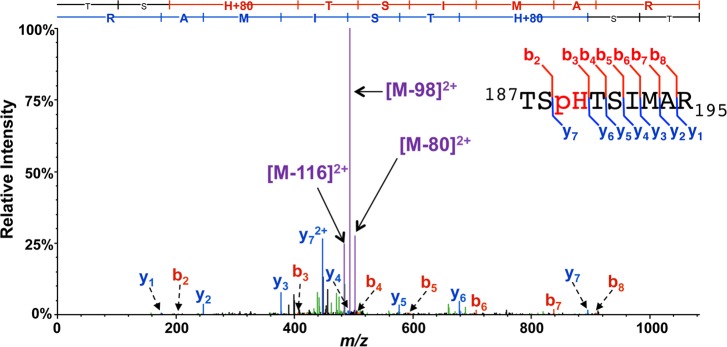
MS/MS spectrum of a pHis tryptic peptide, TSpHTSIMAR, from the endogenously phosphorylated E. coli protein PtsI. MS/MS was performed by linear ion trap CID, during which prominent species derived from the neutral loss of 98, 80, and 116 Da dominate the ion current. Major b- and y-ion backbone fragments and species derived from neutral losses off the precursor (M) are indicated in red, blue, and purple, respectively. Inset into the spectrum is a summary sequence, flagged to indicate the detected b- and y-ions, also shown in the sequence ladder above the spectrum.
Observation of Δ98, Δ80, and Δ116 Neutral Losses during CID of Diverse pHis-Containing Peptides
To determine whether the observed pattern of neutral losses in PtsI might be a general phenomenon of all pHis tryptic peptides, we conducted LC–MS/MS analysis using CID on a diverse array of pHis peptides from multiple sources. These include: (1) a known pHis protein (DhaM) overexpressed and endogenously phosphorylated in E. coli,11 (2) a known pHis protein (PpsA) enzymatically histidine-phosphorylated in vitro,11 (3) synthetic pHis peptides (AAASpHYAAR, TSpHYSIMAR, TSpHTSIMAR), and (4) bovine serum albumin (BSA) that had been chemically phosphorylated on histidine residues and then digested with trypsin. Each of these pHis peptides displayed the same triplet neutral loss of 98, 80, and 116 Da as the PtsI pHis peptide (Figure 2). Interestingly, this neutral loss pattern did not appear to depend on the position of the histidine residue in the peptide sequence and was observed both for 2+ and for 3+ pHis peptide ions (Figure 2b).
Figure 2.

Neutral losses of 98, 80, and 116 Da in CID MS/MS spectra of diverse pHis peptides. Prominent neutral loss triplets (Δ98, Δ80, and Δ116 Da) were observed in the CID MS/MS spectra for (a) tryptic peptides from known pHis proteins (DhaM, PpsA) and chemically phosphorylated synthetic peptides, respectively, and (b) pHis peptides derived from chemically phosphorylated BSA. The Δ116, Δ98, and Δ80 Da product ion species are indicated in purple with each peak labeled with a green, red, or blue diamond, respectively. Precursor peptide sequences, charge state, and m/z values are shown to the right of each spectrum. MS/MS spectra showing the full recorded m/z range for these CID experiments are shown in Figure S1.
Role of the Peptide C-Terminal Carboxylate in Gas-Phase Loss of Phosphoric Acid from pHis Peptides by CID
We next sought to understand the gas-phase ion chemistry involved in the CID-induced neutral loss of 98 Da from pHis peptides. This phenomenon involves the loss of water (18 Da) in addition to the loss of HPO3 (80 Da) expected from the breakage of the labile P–N bond in pHis. This additional loss of water clearly must occur at a location different from the pHis residue itself. The sequence diversity of pHis peptides showing this 98 Da loss (Figure 2a and b) implied that a sequence-independent peptide functional group was likely the source of the additional water loss. This reasoning led us to consider the carboxylate moiety of either the peptide C-terminus (α-carboxyl group) or aspartate/glutamate residue side chains, if present, as the potential source of the water loss. By analogy to bacterial two-component signaling systems, in which the phosphoryl group from pHis in a histidine kinase is transferred to an aspartate side chain,8 we hypothesized that a similar intramolecular phosphoryl transfer from pHis to the α-carboxyl group or a carboxylate side chain within the peptide, to give an acyl-phosphate, may be occurring in the gas phase CID reaction (Figure 3a and 3c). To test this idea, we subjected a synthetic pHis PtsI peptide that is free of carboxylate side chains, TSpHTSIMAR, to C-terminal 18O labeling using trypsin in the presence of 18O-water.19 CID of the doubly 18O-labeled species (TSpHTSIMAR-C18O18OH, +Δ4 Da) resulted in the prominent neutral loss of 100 Da (Figure 3b), without observable loss of 98 Da. An identical trend was observed for two other 18O-labeled pHis peptides that do not contain aspartate/glutamate residues (MGpHAGAIIAGGK-C18O18OH, LIpHGQVATR-C18O18OH), where prominent neutral loss of 100 Da, and not 98 Da, occurred upon CID fragmentation (Figure S2 and S3). This loss of 100 Da (HP16O3 + H218O) from the C-terminally labeled peptide provides direct mechanistic evidence for participation of the α-carboxyl in the gas-phase phosphoric acid loss of pHis peptides. In contrast, CID MS/MS analysis of the doubly 18O-labeled pSer-version of the PtsI peptide (TpSHTSIMAR-C18O18OH, +Δ4 Da) resulted in detection of only Δ98 and Δ116 Da ion species (Figure S4). This implies that phosphoric acid loss from pSer peptides does not involve water-loss from the peptide C-terminus.
Figure 3.

Neutral loss of 98 Da from pHis peptides occurs predominantly through the C-terminal or a side chain carboxylate. (a) Proposed model showing loss of phosphoric acid from pHis peptides via the C-terminal carboxylate during CID. (b) CID MS/MS of the [M + 2H]2+ pHis peptide TSpHTSIMAR-C18O18OH ion showing the prominent neutral loss of 100 Da. Inset into the spectrum is a high resolution MS spectrum of the precursor species at m/z 544.23763 (left panel) and a high resolution MS/MS spectrum showing species corresponding to the primary loss of 100 Da and less prominent losses of 80, 118, and 116 Da (right panel). (c) Proposed model showing loss of phosphoric acid from pHis peptides via a side chain carboxylate during CID. (d) CID MS/MS of the [M + 2H]2+ pHis peptide SpHEFMNK-C18O18OH ion showing the prominent neutral loss of 98 Da. Inset into the spectrum is a high resolution MS spectrum of the precursor species at m/z 488.69006 (left panel) and a high resolution MS/MS spectrum showing species corresponding to the primary loss of 98 Da and less prominent losses of 80, 118, and 116 Da (right panel).
To explore whether a side chain carboxylate group can also participate in CID-induced 98 Da neutral losses for pHis peptides, we performed a similar experiment as above, wherein aspartate/glutamate-containing pHis peptides were C-terminally 18O-labeled labeled and subjected to CID fragmentation. We found that CID of a phosphopeptide from the sensor histidine kinase DcuS (SpHEFMNK-C18O18OH) resulted in prominent neutral loss of 98 Da, and not the 100 Da that we observed in peptides lacking Asp/Glu residues (Figure 3d and vide supra). An identical trend was observed for two other 18O-labeled pHis peptides containing Asp/Glu residues (TTLTDLTpHSLK, pHGESQWNK), where prominent neutral loss of 98 Da, and not 100 Da, occurred upon CID fragmentation (Figure S5 and S6). This prominent loss of 98 Da (HP16O3 + H216O) in the presence of an 18O-labeled labeled C-terminus indicates that a side chain carboxylate group can take priority over the α-carboxyl group in the gas-phase phosphoric acid loss of pHis peptides.
In considering the mechanism of phosphoric acid loss from pHis peptide ions, we wondered whether the acyl phosphate moiety generated upon phosphoryl transfer might undergo a β-elimination process to afford a ketene species (Figure S7). To probe this hypothesis, we substituted the C-terminal α-hydrogen atom of a pHis peptide with deuterium. Elimination in this case would lead to a loss of 99 Da (H2DPO4) during CID. Note that it has been previously shown that α-deuteration of phosphoserine leads to a loss of 99 Da through β-elimination during CID, albeit at low levels presumably due to the isotope effect.18 Accordingly, we prepared a synthetic pHis peptide with a C-terminal deuterated alanine, TSpHTSIMA(d4-A), and then subjected it to CID. The CID MS/MS spectrum of this peptide revealed the formation of only the Δ98 Da neutral loss species with no evidence for a loss of 99 Da (Figure S7). This argues against a β-elimination process in this context, although we cannot rule out suppression of this pathway due to a strong isotope effect. Nonetheless, there appears to be an alternate pathway for phosphoric acid loss from the C-terminal acyl phosphate intermediate, which we speculate involves nucleophilic attack on the acyl phosphate by a side chain or backbone group, leading to loss of phosphoric acid and generation of the corresponding cyclic peptide fragment species (Figure S7).
Exploitation of the pHis Neutral Loss Pattern to Screen for MS/MS Spectra of pHis Peptides
We next investigated whether this triplet pattern could be used to distinguish pHis peptides from other common phosphopeptide types by MS/MS. Serine, threonine, and tyrosine phosphopeptide ions are well-known to undergo facile neutral loss upon CID-type fragmentation, resulting in major neutral loss species of Δ116 and Δ98 Da for phosphoserine/threonine, and Δ80 Da for phosphotyrosine.16 To ascertain whether the observed triplet of neutral losses could act as a distinguishing feature of pHis peptides, we prepared an initial series of isobaric phosphopeptides based on the sequence TSHYSIMAR, where the phosphorylation site was either on Ser2, His3, or Tyr4 (TpSHYSIMAR, TSpHYSIMAR, TSHpYSIMAR). All three phosphopeptides displayed neutral losses upon CID; however, the pattern of these losses was distinctly different (Figure 4). While the pHis variant showed the expected triplet losses of 98, 80, and 116 Da, the pSer version displayed only loss of 98 and 116 Da, and the pTyr variant displayed only a minor loss species at Δ80 Da. We confirmed these observations through an additional set of LC–MS experiments using model phosphopeptides AAApSHYAAR, AAASpHYAAR, and AAASHpYAAR, which showed neutral loss patterns identical to their TSHYSIMAR counterparts (Figure S8). These two examples suggest that the neutral loss triplet (Δ98, Δ80 Da, and Δ116) and their relative abundance pattern may serve as a distinguishing feature of pHis peptides, at least under the conventional LIT CID conditions employed in this study.
Figure 4.

CID Fragmentation of pHis, pTyr, and pSer peptides display distinct neutral loss patterns. CID MS/MS spectra of a family of phosphopeptides containing the same underlying peptide sequence, TSHYSIMAR. The pHis peptide TSpHYSIMAR exhibited the distinct CID-induced neutral loss pattern of Δ98, Δ80 and Δ116 Da. This triplet neutral loss pattern was not observed for the isobaric pSer (TpSHYSIMAR) and pTyr (TSHpYSIMAR) peptides. b- and y-ions and neutral loss species are indicated in the spectra, as in Figure 1. Precursor peptide sequences are as labeled.
To further explore whether this triplet neutral loss pattern is a selective feature of pHis peptides, we prepared a 45-member phosphopeptide library derived from nine previously described pHis sites in E. coli. In this library, nine tryptic peptide sequences were synthesized to contain pHis at the endogenous pHis site. Alternatively, the pHis site was replaced with His, pSer, pThr, or pTyr, resulting in 45 peptides in total (Figure 5a). CID MS/MS analysis of the pHis peptides from this library resulted in the identical triplet neutral loss pattern as reported for other pHis peptides above (Figure S9–S11). To investigate whether the CID-induced triplet neutral loss pattern could be detected for other members of the peptide library, and to evaluate their comparative prevalence, we calculated the percentage of triplet neutral loss-positive MS/MS spectra with respect to the total number of MS/MS spectra for each phosphopeptide type. To tally the triplet neutral loss-positive MS/MS spectra, we developed a software tool in house, which we call TRIPLET (Tool for Retrieving IDs of Phosphopeptides by their neutral Loss fingErprinTs), in which MS/MS spectra from LC–MS experiments can be searched and filtered for those that possess the pHis neutral loss fingerprint (Figure S12). This tool builds on the concept of previous software for detection of neutral loss species;20 however, we have added the capability to filter for multiple co-occurring neutral losses with independent abundance thresholds, so as to capture unique patterns of losses that may be characteristic of particular classes of peptides. This software is instrument vendor neutral, as the input for it consists of processed MS/MS peaklist files from LC–MS runs. Additionally, TRIPLET possesses an MS/MS spectrum viewer, so that candidate spectra that meet the pattern filtering criteria may be visually inspected (see Supporting Information for more details on TRIPLET).
Figure 5.

Specificity of a second-generation pHis antibody and its use for pHis peptide immunoprecipitation in combination with triplet neutral loss filtering for highly selective discrimination of pHis from pSer/pThr/pTyr peptide analogues. (a) Phosphopeptide library sequences, consisting of tryptic peptides containing the known pHis sites of 9 E. coli proteins. The library was diversified by replacing the pHis site with pSer, pThr, pTyr, or His residues. (b) Library peptides were analyzed by CID MS/MS, and the percentage of individual MS/MS spectra displaying the Δ98, Δ80, and Δ116 Da neutral loss pattern (black bars) or the Δ98 neutral loss (gray bars) was calculated for each phosphotype. Note that these neutral losses were not observed for any of the nonphosphorylated peptides (not shown). (c) Dot blot analysis of the library of phosphopeptides in (a), pooled by phosphotype, using a newly developed α-pHis antibody and an α-pTyr antibody (4G10, Millipore) (see Figure S14 for loading control). (d) Library peptides from (a) were analyzed by CID MS/MS before and after immunoprecipitation with the α-pHis antibody. Tallies of MS/MS spectra displaying the triplet neutral loss pattern were generated for each of the library constituents by the TRIPLET software, and the results were grouped according to pHis, pSer, pThr, or pTyr phosphotype.
Using the TRIPLET software filter, we found that, as expected, pHis peptides displayed a much higher percentage of triplet neutral loss production than for pSer, pThr, and pTyr peptides (Figure 5b). Notably, the latter group had low, but nonzero levels of triplet neutral loss formation, although precise levels were subject to slight variation based on chosen cutoff threshold parameters in TRIPLET for m/z error tolerances and Δ116/ Δ80 Da peak intensities (Figure S13). Thus, while the triplet neutral losses can be detected at low frequencies in the MS/MS spectra of pSer, pThr, pTyr peptides, their pattern can be largely distinguished from the pHis peptide neutral loss pattern under the appropriate, user-defined settings, demonstrating the potential of TRIPLET as a bioinformatic filter for pHis peptide mass spectra. This discrimination of pHis peptides was not possible when we employed only the Δ98 neutral loss as an MS/MS spectra filtering criterion (Figure 5b).
We next investigated whether the triplet neutral loss pattern could be used in combination with other pHis peptide enrichment methods to further enhance the specificity of global pHis proteomic analysis. Given the power of phosphopeptide enrichment in proteomic analyses and the impact that it has had on the field,21−23 we focused our efforts on developing a peptide-based pHis immunoprecipitation methodology for integration into a workflow for pHis proteomic analyses. We used the previously described pTze hapten11 to prepare a second-generation pan-pHis antibody capable of specifically immunoprecipitating pHis-containing peptides, a property that the previous antibody lacked. The specificity of this antibody for the pHis epitope was analyzed by dot blot and was found to be highly selective for pHis peptides over pSer, pThr, and pTyr species (Figure 5c and Figure S14). The antibody was also shown to be effective at immunoprecipitation, and its sensitivity threshold for recovering authentic pHis signal from a complex E. coli lysate peptide matrix was measured through spike-in titration experiments to be ∼1 pmol pHis peptide (Figure S15). Moreover, we demonstrated that the use of pHis peptide immunoprecipitation in combination with the TRIPLET software, led to significant enrichment of pHis peptides from the phosphopeptide library (Figure 5d). This suggests that the pHis antibody and TRIPLET software can be used in tandem to improve the specificity of detection of pHis peptides from a complex mixture such as a lysate. It is important to note that while CID fragmentation in our workflow provides a means to generate the pHis triplet neutral loss pattern, it does not alone provide optimal pHis peptide MS/MS spectra for identification purposes, due to reduced levels of peptide backbone fragmentation. To overcome this, we incorporated follow-up MS/MS analyses by alternate fragmentation methods for improved peptide ion identification (see next section) and potentially to aide in phosphosite localization13 (see Figure S16 for an exploration of the effect of different fragmentation methodologies on phosphosite localization scoring).
Use of the TRIPLET Software to Enhance MS/MS-Based Peptide Identification
We envisioned the TRIPLET analysis could play an important role in global pHis proteomic analysis in two ways; (i) to validate CID peptide spectral matches to pHis peptides from standard database searches, (ii) to screen CID LC–MS/MS runs for spectra indicative of potential pHis peptides so as to place these peptides onto inclusion lists (parent ion lists) for subsequent targeted reanalysis by LC–MS/MS using alternative fragmentation methodologies, such as multistage activation (MSA) or electron transfer dissociation (ETD). These fragmentation methods, despite their additional costs in terms of sensitivity and instrument duty-cycle time, can improve MS/MS analysis of phosphopeptides by generating more informative backbone fragments for peptide identification.24,25 Importantly, this follow-up targeted reanalysis of potential pHis peptides allows for the dedication of valuable instrument resources to pHis peptides which are likely to be sparse, present at low levels, or poorly matched in the database search of results from the first round of analysis.
To demonstrate the feasibility of a pHis proteomic workflow incorporating TRIPLET to inform and direct pHis peptide MS/MS analyses, we prepared a test set of diverse pHis peptides from BSA that had been chemically phosphorylated on multiple histidine residues (pHis-BSA) using potassium phosphoramidate.11 This pHis-BSA sample was subjected to proteomic sample workup consisting of thiol reduction and alkylation, and trypsinolysis using a standard protocol slightly modified for pHis compatibility (see Supporting Information). The resulting peptides were analyzed by LC–MS/MS with CID, and the MS/MS spectra peaklist files were screened using the TRIPLET software for spectra that contained the characteristic pHis neutral loss fingerprint. These TRIPLET-positive spectra were then matched to the results of a Mascot search against a BSA protein database using the same peaklist files. Additionally, the list of precursor species associated with these TRIPLET-positive spectra was then used as a parent ion list for a second, targeted LC–MS/MS run using MSA, and this data was also subjected to a Mascot search against the BSA database. The complete set of pHis peptides identified through this process is listed in Table 1, with their corresponding initial CID-derived match and subsequent MSA-derived match Mascot ion scores. Importantly, we observed that the follow-up MSA analysis on TRIPLET-positive species markedly increased the Mascot score for nearly all of the identified pHis peptides. Indeed, some of the peptides for which no Mascot match was initially possible (above an ion score threshold of 15) based on the CID data became identifiable with substantial confidence through targeted reanalysis by MSA.
Table 1. Mascot Ion Scores of BSA pHis Peptide Matches from CID Analysis versus Those from Targeted MSA of TRIPLET-Positive Speciesb.
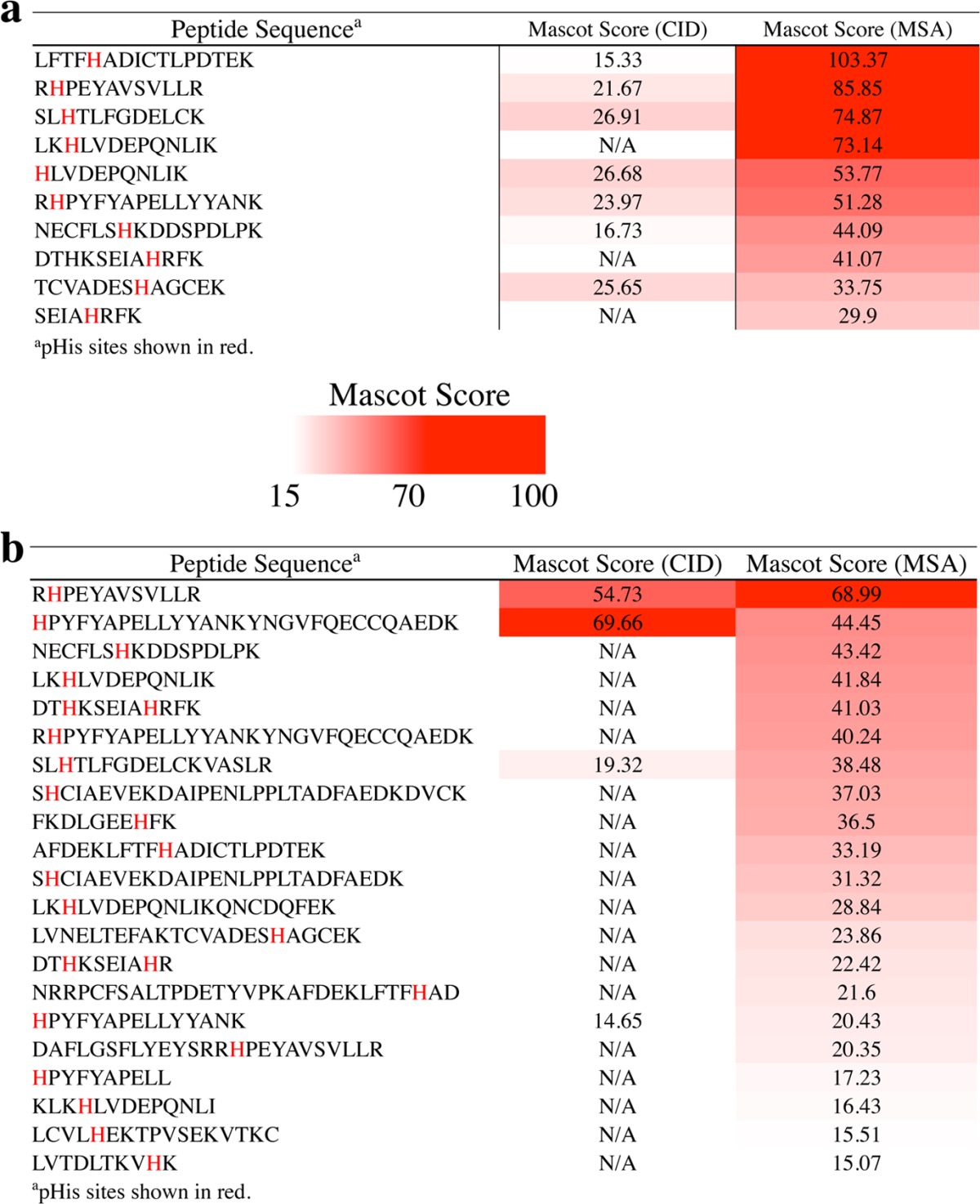
(a) 2+ ions. (b) 3+ ions. Phosphosites are indicated in red in the peptide sequences, and Mascot ion scores are highlighted according to the scale inset into the table.
Identification of Known, Previously Inferred, and Novel pHis Peptides from the E. coli Proteome
Finally, as a demonstration of the utility of our analytical methodology, we applied it to the context of a global E. coli pHis phosphoproteomics experiment. We undertook pHis proteomic analysis of lysate from nitrogen-limited, glycerol- or mannitol-fed E. coli, since those conditions led to the detection of multiple endogenous pHis-positive bands by Western blotting using the pan-pHis antibody (Figure S17). For this analysis, we subjected the lysate samples to thiol reduction and alkylation, trypsinolysis, and desalting with slight modifications to standard methods to enhance pHis compatibility (see Supporting Information). The resultant pHis containing peptides were immunoprecipitated from the lysates using the pan-α-pHis antibody and then analyzed by LC–MS/MS using CID. MS/MS peak list files were screened using TRIPLET for those containing the pHis neutral loss fingerprint. These same CID peak lists were also searched using Mascot against the SwissProt E. coli protein database. pHis peptides matched by Mascot and also found by TRIPLET to contain the pHis neutral loss fingerprint are listed in Table 2, together with their Mascot scores derived from the CID data. The peptide sample was subsequently reanalyzed by LC–MS/MS, using a parent ion list to target TRIPLET-positive peptides for fragmentation by MSA. These MSA-derived spectra were then searched against the SwissProt E. coli database using Mascot. pHis match results from this TRIPLET-targeted MSA analysis are also shown in Table 2.
Table 2. Endogenous E. coli pHis Peptides, Representing Known, Previously Inferred, and Novel pHis sites, Identified in This Study Using Our Pan-pHis Antibody Immunoprecipitation and Mass Spectrometry-Based Methodologyd.

The peptide sequences for each identified protein are listed with the pHis sites shown in red.
Identified from glycerol-grown lysates.
Identified from mannitol-grown lysates.
Displayed are peptide sequences with identified phosphosites indicated in red and Mascot ion scores from CID MS/MS matches compared to those from follow-up targeted MSA reanalysis. The table is colored to indicate whether the identified pHis site had been previously experimentally validated (blue), inferred by similarity (green), or novel (purple).
We were able to characterize 21 distinct endogenous pHis peptides (plus their derivatives, such as alternative charge states, oxidation products, etc.) representing a total of 15 pHis sites from 14 E. coli proteins (Table 2). As a negative control for this proteomic experiment, we performed immunoprecipitation without antibody (mock-IP) on glycerol-fed E. coli and conducted an identical set of LC–MS/MS and TRIPLET analyses. We were not able to match any pHis peptides from this mock-IP sample using our approach (Figure S18). Similar analyses of an acid-treated pHis IP sample also resulted in no matches to pHis peptides (Figure S18). Given the possibility that some pHis peptides might fragment productively by CID without prominent triplet neutral loss formation, we examined our CID-derived Mascot database search results for any high scoring matches to pHis peptides that did not display the characteristic pHis neutral loss triplet. We failed to find a match to any additional such peptide that had not been already identified by our TRIPLET neutral loss screening approach. Thus, all of the pHis peptides found in our samples displayed triplet neutral loss formation by CID, which led to positive identification through our strategy of targeted reanalysis.
Of the 15 pHis sites detected, 8 had been experimentally verified in the past, while five of the sites, those that we characterized on the proteins PtsP, PtsN, NagE, ManX, and FruB had only been inferred as pHis sites by analogy to other known pHis proteins.26,27 Eight of the 15 pHis protein sites were identified in both E. coli growth conditions we examined. Notably, we detected pHis modification of MtlA, a phosphotransferase protein involved in mannitol uptake, only in the context of mannitol-fed cells, suggesting that the mannitol growth conditions stimulated involvement of pHis modification of MtlA and phosphotransfer to internalized mannitol. The two novel pHis peptides we identified here, belonging to the proteins, aldehyde-alcohol dehydrogenase (AdhE) and pyruvate kinase (PykF), were only detected in the mannitol-fed E. coli growth conditions.
To further validate our novel pHis peptide assignments in the AdhE and PykF proteins, we compared the CID MS/MS spectra of our endogenously derived peptides to those of synthetic peptide standards. We found that the synthetic AdhE pHis peptide (FATpHGGYLLQGK) accurately matched the CID fragmentation pattern observed for the endogenous AdhE pHis peptide from the proteomic sample, particularly in the triplet neutral loss region (Figure 6a). To exclude the possibility that the phosphorylation site may be on the neighboring threonine residue, which is the only other potential phosphorylation site in this peptide, we also analyzed a synthetic AdhE pThr peptide (FApTHGGYLLQGK) and found it to be a poor match to the proteomic AdhE peptide, with a notable absence of the Δ80 neutral loss peak (Figure S19). We also acid-treated the proteomic sample followed by reanalysis by LC–MS/MS and, as expected for a pHis peptide, observed complete disappearance of the AhdE pHis peptide peak (Figure 6b). This acid-treatment was sufficient to cause complete loss of detection of a known pHis peptide (PtsP), but not that of non-pHis peptides that were also present in the immunoprecipitation sample (Figure S20). The acid-sensitivity of the putative AdhE pHis peptide, together with its fragmentation pattern match to that of the synthetic peptide standard, support the conclusion that this peptide is an authentic novel pHis modification of the AdhE protein.
Figure 6.

Validation of AdhE and PykF pHis MS/MS peptide assignments. (a) CID MS/MS spectrum of the endogenous E. coli AdhE pHis peptide annotated with the major matched b- and y-type ions indicated. The synthetic Adhe pHis peptide is shown in the mirror image. Inset into the spectrum is an enlargement of the neutral loss pattern for the endogenous vs the synthetic pHis peptide. (b) Extracted ion chromatogram of the [M + 2H]2+ AdhE pHis peptide ion from E. coli lysate (top) and from the acid-treated lysate (bottom, red trace). Inset into the spectrum is a high resolution MS spectrum of the precursor species at m/z 686.32679. (c) CID MS/MS spectrum of the endogenous E. coli PykF pHis peptide, with the prominent matched b- and y-type ions as indicated. The synthetic PykF pHis peptide is shown in the mirror image. Inset into the spectrum is an enlargement of the neutral loss pattern for the endogenous vs the synthetic pHis peptide. (d) Extracted ion chromatogram of the [M + 3H]3+ PykF pHis peptide ion from E. coli lysate (top) and from the acid-treated lysate (bottom, red trace). Inset into the spectrum is a high resolution MS spectrum of the precursor species at m/z 570.90574.
We observed a similar trend for the PykF pHis peptide: the endogenous peptide derived from the proteomic sample was acid-sensitive and also displayed a fragmentation pattern that matched well to that of the synthetic PykF pHis peptide standard (Figures 6c and 6d), but not to an isomeric synthetic standard peptide in which the phosphorylation site was moved to the sole serine in the sequence (Figure S21). Interestingly, while the pHis chemical phosphorylation conditions used to phosphorylate the synthetic PykF peptide, which contains 2 histidines, should result in phosphorylation of either or both histidine residues (LNFSpHGDYAEHGQR or LNFSHGDYAEpHGQR), we found the monophosphorylated form(s) of this synthetic peptide had pHis localization almost exclusively at the His5 residue, as assessed by phosphoRS28 with 100% likelihood. Thus, His5 may be a preferred site of chemical phosphorylation for this peptide, or alternatively, phosphoRS may have difficulty in distinguishing between the fragmentation pattern differences caused by modification at H5 and H11 residues. PhosphoRS showed a similarly high pHis localization confidence to the H5 residue for the endogenous PykF peptide. Consequently, while it is most likely that the authentic pHis modification of the endogenous PykF protein resides on the H5 residue of the peptide, as specified by phosphoRS, we cannot formally exclude the possibility that either of the histidines may contain the phosphorylation mark.
Discussion and Conclusions
In this paper we analyzed the behavior of pHis peptide fragmentation by CID and then used this information to develop a strategy to enhance MS/MS analysis and assignment of pHis peptides. Like other phosphopeptides, we observed that pHis-containing peptides readily undergo neutral loss upon CID fragmentation. Under our fragmentation conditions (particularly those of LIT CID), pHis peptides consistently displayed neutral losses of 98, 80, and 116 Da, with the 98 Da loss being the most dominant. We find that pSer, pThr, and pTyr peptides can also display a triplet neutral loss pattern under our CID conditions, albeit to a significantly lesser extent. However, their patterns can be distinguished from the pHis neutral loss pattern using our newly developed, open source TRIPLET software, by adjusting thresholds for the intensities and m/z error tolerances of the −116 and −80 Da peaks. Given that MS instrument type and/or instrument settings will likely factor into the CID-induced triplet neutral loss pattern of pHis peptides, user optimization of the TRIPLET software parameters may be required to effectively screen for pHis peptide MS/MS spectra. In addition, we found that filtering MS/MS spectra only for the Δ98 Da neutral loss (Figure 5b) is insufficient to uniquely distinguish pHis peptides. Relying only on a doublet neutral loss pattern (Δ98 and Δ80) for filtering of pHis peptides results in more false MS/MS spectral matches compared to triplet neutral loss filtering (data not shown), presumably due to the extra specificity afforded by the additional product ion detection criterion.
We explored the mechanisms behind the formation of the Δ98, Δ80, and Δ116 Da neutral loss species by CID for pHis peptides. The loss of 80 Da is explained by the loss of HPO3 via fragmentation of the labile P–N bond. Formation of the Δ98 and Δ116 Da neutral loss species are more perplexing and imply that, in addition to HPO3, one or two water molecules (Δ18 Da or Δ36 Da) from the peptide are involved in the neutral loss process. There are three possible sources for loss of water from a peptide: (1) loss from the side chain of a Ser, Thr, Tyr, Asp, or Glu residue, (2) loss from the peptide C-terminal carboxylate, or (3) loss from the peptide backbone itself. Using C-terminally labeled 18O-labeled peptides free of acidic residues with carboxylate side chains (TSpHTSIMAR-C18O18OH, MGpHAGAIIAGGK-C18O18OH, LIpHGQVATR-C18O18OH) we were able to show directly that the most prominent neutral loss occurred in the form of 18O-labeled phosphoric acid mediated through loss of HPO3 plus a heavy water molecule coming from the peptide α-carboxylate, rather than a light water moiety from unlabeled amino acid side chains or the peptide backbone (Figures 3b, S2, and S3). However, when an acid residue side chain carboxylate moiety was present in the context of a C-terminal 18O-labeled peptide sequence, we observed that the pHis peptide lost 98 Da most prominently, as we show for SpHEFMNK-C18O18OH, TTLTDLTpHSLK-C18O18OH, and pHGESQWNK-C18O18OH peptides (Figures 3d, S5, and S6). These observations are consistent with recent work from Schmidt and co-workers, in which they provide some evidence for water loss at the C-terminus of a pHis peptide by conducting MS3 analysis of the Δ98 Da neutral loss species.29 The Δ116 Da neutral loss species presumably originates from the loss of phosphoric acid (Δ98 Da) and an additional water molecule (Δ18 Da). Interestingly, TSpHTSIMAR-C18O18OH, MGpHAGAIIAGGK-C18O18OH, and SpHEFMNK-C18O18OH peptides gave rise to both Δ118 and Δ116 Da neutral species (Figure 3b, 3d, and S2). Loss of Δ118 can be accounted for by losing a combination of H3P18O16O3 (Δ100 Da) and light water, in the case of TSpHTSIMAR-C18O18OH and MGpHAGAIIAGGK-C18O18OH, or in the case of SpHEFMNK-C18O18OH losing a combination of H3P16O16O3 (Δ98 Da) and heavy water (Δ20 Da) or HP16O3 (Δ80) and both a heavy and light water. The Δ116 Da neutral loss species is harder to account for in TSpHTSIMAR-C18O18OH and MGpHAGAIIAGGK-C18O18OH peptides because it cannot be from the combination of heavy phosphoric acid loss (Δ100 Da in this case) and water. Conceivably, the Δ116 Da neutral loss could result from the combined loss of HPO3 and two light water molecules. Interestingly, the fact that a peptide free of hydroxyl bearing side chains (MGpHAGAIIAGGK-C18O18OH) can undergo CID-induced loss of Δ116 Da and Δ118 Da provides support for the idea that that additional water losses can come from backbone oxygens. Under our LIT CID conditions we did not observe intermolecular transfer of the phosphoryl group to a peptide of the same or different sequence leading to a doubly phosphorylated species. However, recently Gonzalez-Sanchez et al. detected low levels of CID-induced intermolecular phosphate transfer within a pHis peptide homodimer, indicating that intermolecular transfer may indeed be possible under the appropriate conditions.30
We also found that the position of the His residue in the peptide does not appear to drastically influence the production of the neutral loss pattern (Figure 2b). Since all tryptic peptides possess a free α-carboxylate, we expect that the neutral loss of phosphoric acid by CID is a generalized, sequence-independent phenomenon, at least for peptides of 2+ and 3+ charge states. As noted in Figure 3d, an amino acid side chain carboxylate is also capable of promoting neutral loss of phosphoric acid from pHis peptides. In the peptides we tested, this fragmentation pathway was favored over the α-carboxylate-mediated phosphoric acid neutral loss. This is supported by previous work where CID fragmentation of a model pHis peptide resulted in dehydration of a neighboring aspartate side chain, presumably as a result of phosphoryl transfer from pHis, followed by loss of phosphoric acid.13
On the basis of the above studies, we developed a pHis proteomics workflow which exploits the unique CID fragmentation signature of pHis-containing peptides (Figure S22). This methodology also caters to the labile chemical nature of the pHis modification itself. Thus, both high temperature and acidic conditions are minimized or avoided whenever possible. Critically, the workflow also includes a tryptic peptide enrichment step using a second generation pan-pHis antibody compatible with immunoprecipitation; this type of affinity capture is a common feature of phosphoproteomics,21−23 but until now has not been possible for pHis. The enriched sample is analyzed by LC–MS/MS and the MS/MS peak list files are then subjected to a database search to match potential pHis peptides and also subjected to additional screening for the pHis peptide neutral loss fingerprint, using the software TRIPLET. Follow-up targeted LC–MS/MS analysis is then employed with alternative fragmentation (e.g., MSA, HCD or ETD), using a parent ion list containing the TRIPLET-positive precursor species. Final database search of the targeted, alternative fragmentation data provides additional high confidence matches to pHis peptides. It is important to note that the TRIPLET algorithm could be built into the MS instrument control software, such that “on-the-fly” decision making could be carried out. Since current instrument control software is incapable of screening for multiple concurrent neutral losses, it was necessary for us to write our own software tool (TRIPLET) for this screen off-line, and then to conduct a reanalysis of samples with a second round of LC–MS/MS.
We tested our pHis proteomic approach on nitrogen-limited, glycerol- or mannitol-fed E. coli. The E. coli proteome contains 63 proteins that are either reported to have pHis sites, or that have been inferred to contain the modification based on homology to other known sites (Table S1). Of these proteins, 27 are involved in the PTS pathway, and it was expected that our growth conditions would lead to buildup of pHis on these proteins due to the absence of PTS-dependent sugars as the carbon source.31 Indeed, we were able to identify pHis on 10 unique PTS proteins, over a third of the proteins in the pathway. This is the first direct experimental evidence for pHis modification for five of these proteins, namely, PtsP, PtsN, NagE, ManX, and FruB. Given that pHis is a common enzymatic intermediate in central metabolism in both prokaryotic and eukaryotic cells,4 our results raise the intriguing possibility that pHis proteomics will provide useful information on the metabolic state of a cell, perhaps even aiding the identification of metabolic changes associated with disease.32 Indeed, E. coli grown on different carbon sources display different pHis protein band signatures as detected by Western blotting with the pHis antibody (Figure S23).
Notably, we did not detect any pHis peptides derived from histidine kinases in this study. This might seem surprising given the prevalence of multicomponent signaling systems in bacteria (29 histidine kinases in E. coli, Table S1). Conceivably, the sequence context of pHis in tryptic peptides derived from kinases might prevent recognition by our antibody. However, we have previously shown that our pan-pHis antibody can recognize phosphorylated histidine kinases in vitro,11 and we find that our pHis antibody can immunoprecipitate as little as 1 pmol of a tryptic pHis peptide derived from the histidine kinase, PhoR, arguing against this possibility. A more likely explanation, therefore, is that levels of these proteins, and/or their phosphosite occupancy, are extremely low in general or under our particular E. coli growth conditions. For example, the copy number of histidine kinase EnvZ is reported to be about 60–100 molecules per cell in E. coli, depending on growth conditions.33 Meanwhile, baseline phosphorylation levels for E. coli histidine kinases have been estimated to range from below 0.01% up to 5%,34 indicating that very few molecules of phosphorylated histidine kinases may exist per cell, resulting in pHis peptide levels below the 1 pmol detection limit of our antibody. In contrast, PTS proteins are much more abundant in E. coli (10000–20000 molecules per cell)35 and may have a higher degree of phosphorylation occupancy depending on the metabolic state. Hence, it is likely to be more challenging to detect phosphorylated histidine kinases than phosphorylated metabolic proteins, at least in E. coli.
Interestingly, in our immunoprecipitation of pHis peptides from E. coli lysate, we detected two novel pHis sites on enzymes of central carbon metabolism: on AdhE at His268 and on PykF at His36. We were able to show that the MS/MS spectra of both endogenous pHis peptides matched those of synthetic pHis peptide standards and that the pHis peptide signal for both peptides was sensitive to acid treatment. While it is not immediately clear why these two proteins would be histidine phosphorylated under mannitol-growth conditions, we note that the novel pHis sites on both AdhE and PykF are located in regions of the proteins that have been previously implicated in their function.36,37 Additional studies will be required to explore the idea that histidine phosphorylation of these proteins plays a role in regulating their activity, perhaps under specific growth conditions.
In conclusion, this study has provided a number of contributions to the field of pHis proteomics. We have investigated the particular CID fragmentation behavior of pHis peptides and exploited their distinguishing neutral loss signature to enhance pHis peptide identification by MS. To achieve this, we have created a software tool, TRIPLET, which can be used to screen MS/MS spectra for characteristic neutral losses. In addition, we have demonstrated the first use of a pan-pHis antibody in a peptide-level pHis immunoprecipitation as an enrichment step prior to LC–MS analysis. Our streamlined workflow, which combines all of these strategies, allowed us to characterize endogenous pHis sites, including two novel sites, in E. coli. We envision that this workflow can be usefully applied to study the pHis proteome of any organism. Further pHis proteomic studies, including the investigation of pHis proteins from mammalian cells, are currently underway.
Acknowledgments
We acknowledge J. Rabinowitz and M. Reaves for providing NCM 3722 cells. We thank B. Wang for stimulating discussions, S. Kyin and H. Shwe for their expert assistance in MS, and C. A. Ramirez Perez for the assistance in the synthesis of model peptides. This work was funded by the U.S. National Institutes of Health (5R01GM095880). R.C.O. and J-M.K. were supported by postdoctoral fellowships from the National Institutes of Health Research Service Award (1F32CA167901) and Damon Runyon Cancer Research Foundation (DRG-2005-09), respectively.
Supporting Information Available
Supplementary figures and tables, full methods and experimental data, including peptide synthesis and purification protocols, LC–MS characterization of peptides, preparation of tryptic pHis peptides derived from BSA, affinity purification of the pHis antibody, Dot blotting of phosphopeptides, Western blotting of glycerol-fed and mannitol-fed E. coli lysate, a detailed description of the TRIPLET software, immunoprecipitation of pHis peptides from E. coli lysate, and LC–MS/MS analysis of phosphopeptides. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States.
Author Present Address
∇ Department of Chemistry, UNIST, Ulsan 689–798, Korea.
Author Contributions
¶ R.C.O. and J-M.K. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Walsh C. T.Posttranslational Modification of Proteins: Expanding Nature’s Inventory; Roberts and Company Publishers: Greenwood Village, CO, 2006. [Google Scholar]
- Cohen P. Nat. Rev. Drug Discovery 2002, 1, 309. [DOI] [PubMed] [Google Scholar]
- Dephoure N.; Gould K. L.; Gygi S. P.; Kellogg D. R. Mol. Biol. Cell 2013, 24, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee J.-M.; Muir T. W. ACS Chem. Biol. 2012, 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood P. V.; Besant P. G.; Piggott M. J. Amino Acids 2010, 40, 1035. [DOI] [PubMed] [Google Scholar]
- Besant P. G.; Attwood P. V.; Piggott M. J. Curr. Protein Pept. Sci. 2009, 10, 536. [DOI] [PubMed] [Google Scholar]
- Boyer P. D.; Peter J. B.; Ebner K. E.; Deluca M.; Hultquist D. E. J. Biol. Chem. 1962, 237, 3306. [PubMed] [Google Scholar]
- Stock A. M.; Robinson V. L.; Goudreau P. N. Annu. Rev. Biochem. 2000, 69, 183. [DOI] [PubMed] [Google Scholar]
- Meadow N. D.; Fox D. K.; Roseman S. Annu. Rev. Biochem. 1990, 59, 497. [DOI] [PubMed] [Google Scholar]
- Kee J.-M.; Villani B.; Carpenter L. R.; Muir T. W. J. Am. Chem. Soc. 2010, 132, 14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee J. M.; Oslund R. C.; Perlman D. H.; Muir T. W. Nat. Chem. Biol. 2013, 9, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzihradszky K. F.; Phillipps N. J.; Senderowicz L.; Wang P.; Turck C. W. Protein Sci. 1997, 6, 1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis A. J.; Kjeldsen F.; Kallipolitis B.; Haselmann K. F.; Jensen O. N. Anal. Chem. 2007, 79, 7450. [DOI] [PubMed] [Google Scholar]
- Zu X. L.; Besant P. G.; Imhof A.; Attwood P. V. Amino Acids 2007, 32, 347. [DOI] [PubMed] [Google Scholar]
- Gonzalez Sanchez M. B.; Lanucara F.; Helm M.; Eyers C. E. Biochem. Soc. Trans. 2013, 41, 1089. [DOI] [PubMed] [Google Scholar]
- Boersema P. J.; Mohammed S.; Heck A. J. R. J. Mass Spectrom. 2009, 44, 861. [DOI] [PubMed] [Google Scholar]
- DeGnore J. P.; Qin J. J. Am. Soc. Mass Spectrom. 1998, 9, 1175. [DOI] [PubMed] [Google Scholar]
- Palumbo A. M.; Tepe J. J.; Reid G. E. J. Proteome Res. 2008, 7, 771. [DOI] [PubMed] [Google Scholar]
- Schnolzer M.; Jedrzejewski P.; Lehmann W. D. Electrophoresis 1996, 17, 945. [DOI] [PubMed] [Google Scholar]
- Ficarro S. B.; McCleland M. L.; Stukenberg P. T.; Burke D. J.; Ross M. M.; Shabanowitz J.; Hunt D. F.; White F. M. Nat. Biotechnol. 2002, 20, 301. [DOI] [PubMed] [Google Scholar]
- Grimsrud P. A.; Swaney D. L.; Wenger C. D.; Beauchene N. A.; Coon J. J. ACS Chem. Biol. 2010, 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J.; Moritz A.; Lee K. A.; Guo A.; Goss V. L.; Spek E. J.; Zhang H.; Zha X. M.; Polakiewicz R. D.; Comb M. J. Nat. Biotechnol. 2005, 23, 94. [DOI] [PubMed] [Google Scholar]
- Thingholm T. E.; Jensen O. N.; Larsen M. R. Proteomics 2009, 9, 1451. [DOI] [PubMed] [Google Scholar]
- Schroeder M. J.; Shabanowitz J.; Schwartz J. C.; Hunt D. F.; Coon J. J. Anal. Chem. 2004, 76, 3590. [DOI] [PubMed] [Google Scholar]
- Syka J. E.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farriol-Mathis N.; Garavelli J. S.; Boeckmann B.; Duvaud S.; Gasteiger E.; Gateau A.; Veuthey A. L.; Bairoch A. Proteomics 2004, 4, 1537. [DOI] [PubMed] [Google Scholar]
- Nucleic Acids Res. 2013, 41, D43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus T.; Kocher T.; Pichler P.; Paschke C.; Schmidt A.; Henrich C.; Mechtler K. J. Proteome Res. 2011, 10, 5354. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Ammerer G.; Mechtler K. Proteomics 2013, 13, 945. [DOI] [PubMed] [Google Scholar]
- Gonzalez Sanchez M. B.; Lanucara F.; Hardman G.; Eyers C. E. Int. J. Mass Spectrom. 2014, 367, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogema B. M.; Arents J. C.; Bader R.; Eijkemans K.; Yoshida H.; Takahashi H.; Aiba H.; Postma P. W. Mol. Microbiol. 1998, 30, 487. [DOI] [PubMed] [Google Scholar]
- Ward P. S.; Thompson C. B. Cancer Cell 2012, 21, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S. J.; Inouye M. J. Biol. Chem. 2002, 277, 24155. [DOI] [PubMed] [Google Scholar]
- Yamamoto K.; Hirao K.; Oshima T.; Aiba H.; Utsumi R.; Ishihama A. J. Biol. Chem. 2005, 280, 1448. [DOI] [PubMed] [Google Scholar]
- Postma P. W.; Lengeler J. W.; Jacobson G. R. Microbiol. Rev. 1993, 57, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Membrillo-Hernandez J.; Echave P.; Cabiscol E.; Tamarit J.; Ros J.; Lin E. C. J. Biol. Chem. 2000, 275, 33869. [DOI] [PubMed] [Google Scholar]
- Morgan H. P.; McNae I. W.; Nowicki M. W.; Hannaert V.; Michels P. A.; Fothergill-Gilmore L. A.; Walkinshaw M. D. J. Biol. Chem. 2010, 285, 12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.