

Abstract

The organometallic H-cluster at the active site of [FeFe]-hydrogenases is synthesized by three accessory proteins, two of which are radical S-adenosylmethionine enzymes (HydE, HydG) and one of which is a GTPase (HydF). In this work we probed the specific role of H atom abstraction in HydG-catalyzed carbon monoxide and cyanide production from tyrosine. The isotope distributions of 5′-deoxyadenosine and p-cresol were evaluated using deuterium-labeled tyrosine substrates in H2O and D2O. The observation of multiply deuterated 5′-deoxyadenosine and deuterated S-adenosylmethionine when the reaction is carried out in D2O provides evidence for a 5′-deoxyadenosyl radical-mediated abstraction of a hydrogen atom from a solvent-exchangeable position as a reversible event.
Hydrogen metabolism in microorganisms involves the activation of dihydrogen through complex iron–sulfur (Fe–S) active-site catalysts.1 The [FeFe]-hydrogenase H-cluster contains an unusual organometallic 2Fe subcluster in which the irons are coordinated by three carbon monoxides (CO), two cyanides (CN–), a dithiomethylamine, and a cysteine thiolate that bridges this entity to a [4Fe-4S] cluster (Figure 1).2 The 2Fe subcluster is synthesized by dedicated maturation machinery consisting of HydF, a GTPase that serves as a scaffold or carrier,3,300 and HydE and HydG, both radical S-adenosylmethionine (SAM) enzymes.5,100 While HydE has been proposed to synthesize the dithiolate ligand,6 HydG synthesizes the diatomic ligands of the H-cluster from tyrosine (Tyr) in a reaction that produces p-cresol, CO, and CN– and requires two discrete C–C bond cleavage events (Figure 2).4−9
Figure 1.

[FeFe]-hydrogenase H-cluster active site from Clostridium pasteurianum (PDB: 3C8Y).
Figure 2.

Reaction catalyzed by HydG. Top: Conversion of tyrosine to p-cresol, CO, and CN−. Concomitant with this reaction, SAM is converted to methionine and 5′-deoxyadenosine. Bottom: Two [4Fe-4S] clusters are involved. The N-terminal cluster binds and reductively cleaves SAM, and is required for the cleavage of tyrosine to produce p-cresol. The C-terminal cluster is important for diatomic ligand production, and the unique iron of this cluster may provide a site for diatomic ligand coordination prior to transfer to HydF. The cysteine motifs coordinating each cluster are shown below the cluster.
Radical SAM enzymes generally catalyze the reductive cleavage of SAM to produce methionine and a 5′-deoxyadenosyl radical intermediate (dAdo•); the latter species abstracts a hydrogen atom from substrate to produce 5′-deoxyadenosine (dAdoH).10 In the case of HydG, reductive cleavage of SAM occurs at a N-terminal site-differentiated [4Fe-4S] cluster coordinated by a CX3CX2C motif,11 producing dAdoH in slight excess over p-cresol and cyanide due to a small amount of uncoupled SAM cleavage.8 HydG also coordinates a second [4Fe-4S] cluster near the C-terminus at a CX2CX22C motif11 that is essential for catalysis.13 While the N-terminal cluster of HydG is required for reductive cleavage of SAM and tyrosine Cα−Cβ bond cleavage to produce p-cresol, the C-terminal cluster and/or residues in the C-terminal domain are essential for the production of the diatomic products.11,14,15 Recent results suggest that the diatomic products of HydG may coordinate an Fe of this C-terminal cluster, extruding the Fe to form a precursor of the 2Fe subcluster of the H cluster.16
To investigate the mechanism by which HydG initiates tyrosine cleavage, specifically deuterated Tyr substrates and/or deuterated solvent was used in enzymatic assays, and deuterium incorporation into dAdoH and SAM was monitored. Clostridium acetobutylicum HydG was expressed in Escherichia coli, was purified anaerobically, and was chemically reconstituted with Fe and S; this protein was found to exhibit visible and electron paramagnetic resonance spectroscopic properties similar to those previously reported.4,11 Assays for HydG-catalyzed dAdoH formation were carried out in an anaerobic chamber (MBraun, O2 < 1 ppm) in sealed 1.5 mL Eppendorf tubes, containing HydG (100 μM with 7.1 ± 0.2 mol Fe per mol enzyme) in buffer (50 mM tris, pH 8.1, 300 mM KCl, 5% (w/v) glycerol), 5 mM dithionite, 1 mM Tyr, and 1 mM SAM at 37 °C, similar to assays reported previously.4 Reaction products were analyzed by ESI-MS following enzyme precipitation with 1 M HCl (13% v/v).
HydG reactions carried out using Tyr deuterium (D) isotopologues ([β-D2]-Tyr, [ring-D4]-Tyr, and [α,ring-D5]-Tyr) in H2O yielded dAdoH isotope distributions comparable to natural abundance dAdoH (Figure S1), and insignificant loss of label in the p-cresol product (Figure S2). Since these isotopologues span all nonexchangeable positions on Tyr, the results indicate that the dAdo• generated upon reductive cleavage of SAM abstracts only at a solvent-exchangeable position. This hypothesis was confirmed when the HydG reaction was carried out with natural abundance Tyr in D2O buffer (50 mM tris, pD 8.1, 95% D), as significant changes in the isotope distribution of dAdoH were observed (Figure 3). The dAdoH product isolated from the reaction in D2O buffer consisted of a mixture of singly, doubly, and triply deuterated dAdoH, with 82% of dAdoH containing at least one D (Table S1). When Tyr was omitted from the assay performed in D2O, no dAdoH could be detected by LC-MS; in H2O, a small amount of dAdoH could be detected, indicative of nonproductive SAM cleavage (data not shown). A similar observation of the incorporation of deuterium from a solvent exchangeable position into dAdoH has been made for HydG from Shewanella oneidensis, however the analysis for multiple deuterium incorporations was not carried out, and thus the mechanistic implications we report here were not recognized or discussed.24
Figure 3.
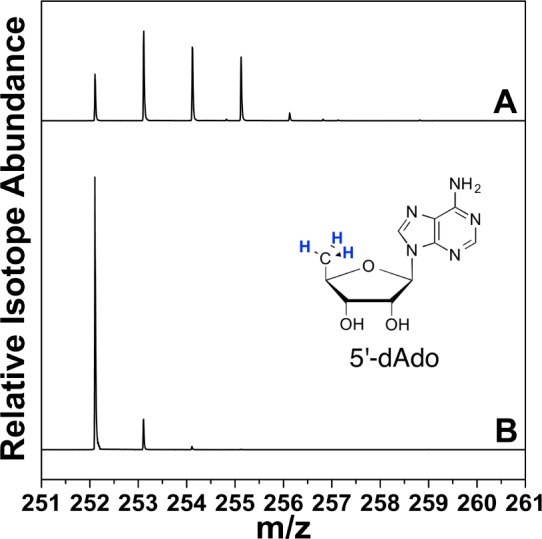
ESI-MS of HydG reaction product dAdoH for assays performed in 95% D2O buffer (50 mM tris, pD 8.1). (A) Full reaction. (B) dAdoH reference sample in H2O. Spectra are represented as normalized, extracted ion chromatograms.
Our results indicate that the D incorporation into dAdoH shown in Figure 3 requires the presence of Tyr. When HydG was assayed in D2O, Tyr analogues lacking the phenolic −OH (Phe), or the amino or carboxylate functional groups did not serve as substrates, and likewise did not mediate deuterium transfer to dAdoH (data not shown). Similarly, Phe has been described elsewhere to neither stimulate dAdoH formation,7 nor stimulate activity in hydrogenase in vitro activation experiments in H2O.17 Only samples that contained enzyme, dithionite as a reductant, SAM, and the substrate Tyr in D2O buffer resulted in significant D transfer to dAdoH. Possible sites for abstraction of a solvent-exchangeable H/D in the HydG reaction include the phenol O–H or amino of Tyr, or a protein residue in the active site. Given the requirement for Tyr to observe D incorporation, and the precedence for biological tyrosyl radicals generated by H atom abstraction from the phenolic position,18 we favor abstraction from the Tyr O–H. Although abstraction from protein residues has not been entirely ruled out, it is clear that there are no conserved Cys residues in HydG beyond the six involved in Fe–S cluster coordination; there are, however, several conserved Tyr residues.7,15
HydG consumes SAM as a cosubstrate.4,8,11 The observation of multiple D atoms being incorporated into dAdoH (Figure 3, Table S1) was surprising, since the simple mechanistic model would involve dAdo• abstracting a D from substrate to produce singly deuterated dAdoH as a primary, rate-determining event. Multiple D incorporations into the dAdoH product requires a more complex mechanism in which, after formation of dAdoH, the dAdo• is regenerated and can abstract an additional H/D. Because the incorporation of multiple D atoms into dAdoH requires the reversible generation of dAdo•, which in turn could be coupled to reversible cleavage of SAM, we investigated the possibility of D incorporation into SAM during the HydG catalyzed reaction. SAM was isolated from the optimized assay described above, and ESI-MS revealed that 56% of the SAM contained at least one D (Figure 4, Table S2). In the absence of HydG, 9% of the SAM was observed to incorporate D, consistent with previous reports of slow exchange of the C-5′ hydrogens of SAM with solvent;19−21 assays run in the absence of Tyr produced low levels of SAM deuteration similar to those observed when HydG was omitted (Figure S3), demonstrating that a stimulated incorporation of deuterium into SAM (Figure 4A) requires the presence of both enzyme and Tyr substrate. Assays were carried out with SAM in a 10-fold excess over HydG; under these conditions, 46% of the SAM underwent HydG-catalyzed exchange in the presence of Tyr (Figure S3, Table S2). Because HydG produces 4 equiv of dAdoH and 3 equiv of p-cresol under the assay conditions utilized herein,11 the extent of deuteration of both SAM and dAdoH exceeds the amount of p-cresol and diatomic products produced. Importantly, the levels of deuterium incorporation into SAM and dAdoH are appreciable, and are nearly equal. These observations point to a SAM ↔ dAdo• + Met equilibrium in which the dAdo• can be deuterated by interaction with Tyr or Tyr cleavage products as outlined below.
Figure 4.

ESI-MS of SAM for HydG assays performed in 95% D2O buffer (50 mM tris, pD 8.1). (A) Full reaction. (B) SAM reference sample in H2O. Spectra are represented as normalized, extracted ion chromatograms.
Abstraction of a hydrogen atom from substrate by dAdo• is generally accepted as a rate-limiting step in radical SAM enzymes,22 and thus substitution with D at the abstracted position should give rise to an isotope effect. In order to examine the solvent isotope effect on HydG catalysis, p-cresol production was compared in identical assays performed in tris-H2O and tris-D2O buffer (Figure 5). The rate of p-cresol production in H2O and D2O was fitted to a first-order exponential, revealing an apparent deuterium isotope effect of 1.71 (kcat-H2O/kcat-D2O). The magnitude of this kH/kD is consistent with a primary isotope effect and provides further evidence that tyrosine cleavage to produce p-cresol is initiated by H atom abstraction from a solvent-exchangeable position in a step that is partially rate-determining.
Figure 5.

Quantitative p-cresol product detection in tris-H2O (■) and 95% tris-D2O (▲). Assays contained 40 μM HydG (9.5 ± 0.2 Fe/protein), 1 mM SAM, 1 mM Tyr, 5 mM dithionite, performed at 37 °C in 50 mM tris, 300 mM KCl, pH/pD 8.1.
The results presented herein provide evidence for a mechanism of HydG-catalyzed Tyr cleavage in which a SAM-derived dAdo• abstracts a hydrogen atom from a solvent-exchangeable position as a reversible event. A single D incorporation into dAdoH when the assay is carried out in D2O would be indicative of an essentially irreversible substrate radical initiation event in which the substrate radical proceeds on in the reaction without further interaction with dAdoH. The observation of multiple D incorporations into dAdoH, and D incorporation into SAM by HydG, shows the existence of multiple reversible steps during turnover. Specifically, the tyrosyl radical resulting from abstraction at the phenolic position could either proceed forward with Cα–Cβ bond cleavage, or could reabstract a hydrogen atom from dAdoH, quenching the substrate radical to regenerate dAdo•; the dAdo• could abstract H/D from substrate or could recombine with methionine, with transfer of an electron to the [4Fe-4S] cluster, to re-form SAM (Figure 6A). Iterative abstraction–reabstraction events carried out several times prior to productive Tyr Cα–Cβ bond cleavage would result in multiple D atom incorporations into dAdoH and SAM following rapid solvent exchange at the tyrosine phenolic position. This scheme is consistent with radical SAM enzymes that catalytically regenerate SAM, but is unusual for enzymes such as HydG, that use SAM as a cosubstrate. Multiple deuterium atoms are incorporated into dAdo during turnover of [3-2H]-2-deoxy-scyllo-inosamine by the radical SAM enzyme BtrN in a mechanism that must require a reversible abstraction by dAdo• at the C(3)-H of the substrate coupled to multiple re-binding events with labeled substrate.200 In the radical SAM enzyme DesII, multiple D atoms from labeled substrate have been detected in SAM, although the dAdoH resulting from the forward reaction harbors only a single deuteron.26 Both BtrN and DesII catalyze reactions in which the dAdo• abstracts from a nonexchangeable position on substrate, unlike the abstraction from an exchangeable position catalyzed by HydG.
Figure 6.

Mechanistic proposals for observed HydG H atom abstraction—reabstraction events and dAdo• regeneration. Hydrogen atoms that have undergone exchange with solvent are colored blue.
An energetic problem with reversible abstraction at the phenolic O–H of Tyr is that the abstraction of a hydrogen atom from the 5′-methyl of dAdoH (BDE(C–H) = 100 kcal/mol)23 by a tyrosyl radical to form Tyr (BDE(O–H) = 86 kcal/mol)18 would be thermodynamically unfavorable, although the unfavorable process could be coupled to the favorable process of re-forming SAM. Alternatively, the recently characterized24p-cresol radical produced by Tyr Cα–Cβ bond cleavage could conceivably abstract a hydrogen from dAdoH to generate dAdo•, which could then abstract an additional phenolic O–H from Tyr, recombine with Met to form SAM, or undergo termination with solvent. It should be noted, however, that a mechanism involving regeneration of dAdo• by the p-cresol radical is not consistent with the roughly stoichiometric dAdoH:p-cresol product formation previously reported.8 Alternatively, the p-cresol radical could abstract an H atom on the intermediate dehydroglycine (DHG), perhaps activating it for formation of CO and CN– (Figure 6B); GC-MS data show incorporation of 1 D in p-cresol during the HydG reaction in D2O (Figure S4), consistent with abstraction at a solvent exchangeable position of either the protein or DHG (Figure 6B).
The reversible abstraction events catalyzed by HydG bear resemblance to B12 chemistry, in which adenosylcobalamin serves as a reversibly functioning “radical source”.25 Similarly, radical SAM enzymes that employ SAM as a catalytic cofactor must also generate the central dAdo• radical reversibly. HydG, however, utilizes SAM as a cosubstrate that is converted to dAdoH and Met during turnover, and yet the same B12-like reversible radical generation occurs in this reaction. Such reversibility in an enzyme utilizing SAM as a co-substrate is indicative of a fundamental interplay between SAM, dAdo•, and the substrate that reflects the central role of SAM as a radical reservoir in radical SAM reactions. Elucidating the specific role of the reversible radical chemistry in the HydG-catalyzed synthesis of CO and CN– from Tyr, and the potential interaction of the radical intermediates with the C-terminal cluster, await further investigations.
Acknowledgments
This work was supported by Department of Energy Grant DE- FG02-10ER16194 to J.B.B. and J.W.P. The Mass Spectrometry, Proteomics and Metabolomics core facility is supported by the Murdock Charitable Trust, INBRE MT Grant No. P20 RR-16455-08, and NIH Grant Nos. P20 RR-020185 and P20 RR-024237 from the COBRE Program of the National Center for Research Resources. The authors gratefully acknowledge the assistance of Jonathan Hilmer with obtaining the LC-MS data, Jesse Thomas with obtaining the GC-MS data, and Eric Shepard for critical review of the manuscript.
Supporting Information Available
Detailed experimental methods, isotope distributions, HPLC chromatograms, and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vignais P.; Billoud B. Chem. Rev. 2007, 107, 4206. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Lanzilotta W. N.; Lemon B. J.; Seefeldt L. C. Science 1998, 282, 1853. [DOI] [PubMed] [Google Scholar]
- McGlynn S. E.; Shepard E. M.; Winslow M. A.; Naumov A. V.; Duschene K. S.; Posewitz M. C.; Broderick W. E.; Broderick J. B.; Peters J. W. FEBS Lett. 2008, 582, 2183. [DOI] [PubMed] [Google Scholar]
- Shepard E. M.; McGlynn S. E.; Bueling A. L.; Grady-Smith C. S.; George S. J.; Winslow M. A.; Cramer S. P.; Peters J. W.; Broderick J. B. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10448–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posewitz M. C.; King P. W.; Smolinski S. L.; Zhang L.; Seibert M.; Ghirardi M. L. J. Biol. Chem. 2004, 279, 25711. [DOI] [PubMed] [Google Scholar]
- Shepard E. M.; Mus F.; Betz J. N.; Byer A. S.; Duffus B. R.; Peters J. W.; Broderick J. B. Biochemistry 2014, 53254090–4104. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Broderick J. B. Annu. Rev. Biochem. 2012, 81, 429. [DOI] [PubMed] [Google Scholar]
- Shepard E. M.; Duffus B. R.; George S. J.; McGlynn S. E.; Challand M. R.; Swanson K. D.; Roach P. L.; Cramer S. P.; Peters J. W.; Broderick J. B. J. Am. Chem. Soc. 2010, 132, 9247. [DOI] [PubMed] [Google Scholar]
- Pilet E.; Nicolet Y.; Mathevon C.; Douki T.; Fontecilla-Camps J. C.; Fontecave M. FEBS Lett. 2009, 583, 506. [DOI] [PubMed] [Google Scholar]
- Driesener R. C.; Challand M. R.; McGlynn S. E.; Shepard E. M.; Boyd E. S.; Broderick J. B.; Peters J. W.; Roach P. L. Angew. Chem., Int. Ed. 2010, 49, 1687. [DOI] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; George S. J.; Grady-Smith C. S.; Cramer S. P.; Swartz J. R. PLoS One 2011, 6, e20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Chem. Rev. 2014, 114, 4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driesener R. C.; Duffus B. R.; Shepard E. M.; Bruzas I. R.; Duschene K. S.; Coleman N. J.-R.; Marrison A. P. G.; Salvadori E.; Kay C. W. M.; Peters J. W.; Broderick J. B.; Roach P. L. Biochemistry 2013, 52, 8696. [DOI] [PubMed] [Google Scholar]
- King P. W.; Posewitz M. C.; Ghirardi M. L.; Seibert M. J. Bacteriol. 2006, 188, 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Martin L.; Tron C.; Fontecilla-Camps J. C. FEBS Lett. 2010, 584, 4197. [DOI] [PubMed] [Google Scholar]
- Tron C.; Cherrier M. V.; Amara P.; Martin L.; Fauth F.; Fraga E.; Correard M.; Fontecave M.; Nicolet Y.; Fontecilla-Camps J. C. Eur. J. Inorg. Chem. 2011, 1121. [Google Scholar]
- Kuchenreuther J. M.; Myers W. K.; Suess D. L. M.; Stich T. A.; Pelmenschikov V.; Shiighi S. A.; Cramer S. P.; Swartz J. R.; Britt R. D.; George S. J. Science 2014, 343, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Myers W. K.; Stich T. A.; George S. J.; NejatyJahromy Y.; Swartz J. R.; Britt R. D. Science 2013, 342, 472. [DOI] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Stapleton J. A.; Swartz J. R. PLoS One 2009, 4, e7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J.; van der Donk W. A. Chem. Rev. 1998, 98, 705. [DOI] [PubMed] [Google Scholar]
- Parks L. W.; Schlenk F. J. Biol. Chem. 1958, 230, 295. [PubMed] [Google Scholar]
- Borchardt R. T. J. Am. Chem. Soc. 1979, 101, 458. [Google Scholar]
- Hoffman J. L. Biochemistry 1986, 25, 4444. [DOI] [PubMed] [Google Scholar]
- Wang S. C.; Frey P. A. Biochemistry 2007, 46, 12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K.; Numakura M.; Kudo F.; Ohmori D.; Eguchi T. J. Am. Chem. Soc. 2007, 129, 15147–15155. [DOI] [PubMed] [Google Scholar]
- Szu P.-H.; Ruszczycky M. W.; Choi S.-H.; Yan F.; Liu H.-w. J. Am. Chem. Soc. 2009, 131, 14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore S. D.; Smith D. M.; Golding B. T.; Radom L. J. Am. Chem. Soc. 2001, 123, 7963. [DOI] [PubMed] [Google Scholar]
- Halpern J. Science 1985, 227, 869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.