

Abstract

Firefly luciferase adenylates and oxidizes d-luciferin to chemically generate visible light and is widely used for biological assays and imaging. Here we show that both luciferase and luciferin can be reengineered to extend the scope of this light-emitting reaction. d-Luciferin can be replaced by synthetic luciferin analogues that increase near-infrared photon flux >10-fold over that of d-luciferin in live luciferase-expressing cells. Firefly luciferase can be mutated to accept and utilize rigid aminoluciferins with high activity in both live and lysed cells yet exhibit 10 000-fold selectivity over the natural luciferase substrate. These new luciferin analogues thus pave the way to an extended family of bioluminescent reporters.
Introduction
Fireflies are beetles that have evolved the remarkable ability to emit visible light based on a chemical reaction. Instead of a photon of light, firefly luciferase uses adenosine triphosphate and the chemical energy of oxygen to convert its substrate d-luciferin into an excited-state molecule of oxyluciferin (Figure 1A).1 This bioluminescent reaction has been widely used as a biological reporter both in vitro2 and in vivo.3 Although bioluminescence has much lower background than fluorescence and is more sensitive for in vivo imaging, it has been limited by the relative lack of luciferases and luciferins compared to the broad palette of fluorescent probes.
Figure 1.

(A) Firefly luciferase catalyzes the adenylation and oxidation of its native substrate d-luciferin to emit a photon of light. (B) Previously synthesized aminoluciferin analogues. (C) New aminoluciferins from this work.
In nature, d-luciferin is the only luminogenic substrate for beetle luciferases. Over the past few years, many new luciferin analogues have been described, including several that yield peak light emission well into the red.4−9 Synthetic luciferins thus have the potential to extend bioluminescence imaging to wavelengths where tissue is more transparent to light. However, there is already a significant red and near-infrared component to luciferase emission with d-luciferin, and shifting the peak wavelength does not necessarily mean that the overall level of red light has actually increased.9−11 The synthetic luciferin CycLuc1 (Figure 1) performs better than d-luciferin for bioluminescence imaging in live mice, primarily due to its improved ability to access the luciferase rather than a red-shift in light emission.12 Therefore, substrates that combine this ready access to intracellular luciferase in live cells with a red-shift in total emission are expected to be candidates to further improve in vivo performance.13
Structural differences between luciferins could also potentially allow the creation of orthogonal luciferases. Although mutant firefly luciferases that exhibit luciferin selectivity have been reported, d-luciferin remains a light-emitting substrate.14 Surprisingly, recent work has identified the Drosophila fatty acyl-CoA synthetase CG6178 as a latent luciferase that emits light with CycLuc2 but not d-luciferin.15 While this demonstrates that it is possible to retain luciferase activity in beetle luciferase homologues that are unresponsive to d-luciferin, higher rates of photon emission are desirable for use as reporters.
With all of these considerations in mind, we synthesized compact, rigid aminoluciferins modeled after CycLuc1 and CycLuc2 (Figure 1).4,14 We found that these new substrates could greatly increase the total photon flux of near-IR light from live luciferase-expressing cells over d-luciferin. Moreover, high photon flux was observed from a newly identified mutant luciferase that gave virtually no light emission with the natural substrate. Chemical modification of the luciferin substrate can thus extend the scope of bioluminescence beyond what is possible with d-luciferin.13
Results and Discussion
Synthesis of New Rigid Aminoluciferins
The synthetic aminoluciferins CycLuc1 and CycLuc2 have 5′,6′-fused five-membered indoline rings (Figure 1B). To evaluate the effect of this ring fusion on bioluminescence, we synthesized new luciferin analogues with fused six-membered rings of varying composition. Luciferin analogues CycLuc3 and CycLuc4, containing a six-membered oxazine ring, were readily accessed following a slight modification of the CycLuc1 synthesis paradigm4 (Scheme 1).
Scheme 1. Synthesis of CycLuc3 and CycLuc4.

CycLuc5 and CycLuc6 were designed to emit light at longer wavelengths and to test the scope of substrates that could be accommodated by the luciferase (Scheme 2). These bulky, lipophilic analogues incorporate 2,2,4-trimethyl-dihydroquinoline, a scaffold that has been widely used to red-shift the emission of many classes of fluorophores, including coumarins,16 rhodamines,16,17 and oxazines.18 Attempted synthesis of intermediate 16 by trifluoroacetylation of the corresponding nitroarene as in Scheme 1 failed, presumably due to a combination of steric hindrance and electronic deactivation.19 We therefore synthesized the Boc-protected compound 14,20 which suffers the same steric hindrance but was anticipated to be less electronically deactivated. Gratifyingly, trifluoroacetylation of this intermediate was successful,19 and subsequent TFA deprotection readily afforded 16. Elaboration as shown in Scheme 2 afforded the desired luciferin analogues CycLuc5 and CycLuc6.
Scheme 2. Synthesis of CycLuc5 and CycLuc6.

The corresponding saturated tetrahydroquinoline aminoluciferin analogues CycLuc7 and CycLuc8 were synthesized using the same general synthetic route as CycLuc1–CycLuc4 (Figure 1C and Scheme 3).
Scheme 3. Synthesis of CycLuc7–CycLuc10.

Surprisingly, attempts to access the 5′,6′-fused ring—differing from CycLuc1 by only a single methylene—primarily yielded the 6′,7′-fused product in a >8:1 ratio. Nonetheless, elaboration of 32b to the 5′,6′-fused cyclic alkylaminoluciferins CycLuc7 and CycLuc8 proceeded uneventfully. The 6′,7′-fused product 32a was similarly converted into 6′,7′-fused cyclic alkylaminoluciferins CycLuc9 and CycLuc10. Finally, 5′,6′-fused cyclic aminoluciferins CycLuc11 and CycLuc12 were synthesized as bulkier and more lipophilic analogues of CycLuc7 and CycLuc8, where the gem-dimethyl substituents also direct exclusive formation of the 5′,6′-fused isomers (Figure 1C and Scheme 4).
Scheme 4. Synthesis of CycLuc11 and CycLuc12.

Luciferase Activity in Vitro
At the outset of this work, it was anticipated that some subset of the rigid luciferin analogues would not be well accommodated by the luciferase. However, all of the new aminoluciferin analogues are substrates, further underscoring the generality of the light emission chemistry and the tolerance of luciferase for modifications.
When purified firefly luciferase is treated with d-luciferin or an aminoluciferin, a high initial rate of light emission (burst) is observed in the first few seconds, which is then followed by a substantial decrease in the rate of sustained light output. This effect is more pronounced for high-affinity aminoluciferins than for d-luciferin itself, and all of the new substrates exhibited this same general behavior under saturating substrate conditions (Figure 2 and Supplementary Figure 3). Relative to d-luciferin with the wild-type (WT) luciferase, the initial rate of photon flux ranged from a high of 43% for CycLuc4 to a low of 2% for CycLuc11. This initial rate is high compared to luciferin analogues in which the benzothiazole has been replaced.8,21,22 However, aminoluciferins have reduced emission 1 min after substrate addition, consistent with product inhibition as the primary factor limiting the light emission in vitro (Figure 2 and Supplementary Figure 3).4,23
Figure 2.
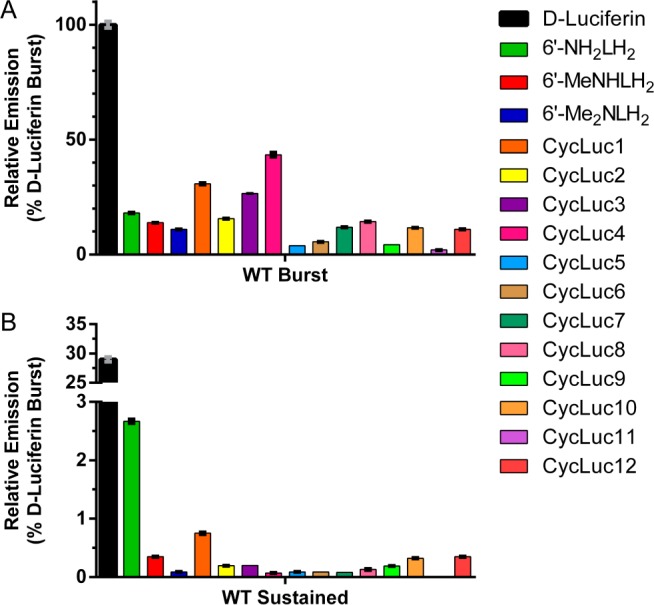
Initial and sustained bioluminescence from luciferin analogues (250 μM) with purified WT firefly luciferase (0.2 nM): (A) initial rates of emission relative to d-luciferin and (B) relative emission 1 min after substrate addition.
Bioluminescence Emission Wavelengths
Peak bioluminescence emission for the aminoluciferins ranged from 594 to 642 nm (Figure 3, Supplementary Table 1, and Supplementary Figure 1). As expected, CycLuc6 yielded strongly red-shifted bioluminescence (636 nm), exceeding that of red-emitting mutant firefly luciferases,11 railroad worm (beetle) luciferase,10 and the red-shifted emission from 6′-Me2NLH2.4 However, we were surprised to find that CycLuc10 yielded an even more red-shifted peak (642 nm). To determine whether these differences are inherent to each luciferin, we measured the fluorescence emission wavelengths of the substrates. The fluorescence and bioluminescence emission wavelengths of aminoluciferins were found to be strongly correlated, suggesting that the bioluminescence wavelength is primarily dictated by the photophysical properties of the luciferin (Supplementary Figure 2). Bioluminescence is red-shifted by 50–75 nm from the substrate fluorescence in phosphate-buffered saline, which is expected since the oxyluciferin emitter produced in the luciferase has increased conjugation with respect to the substrate.
Figure 3.

Bioluminescence emission spectra for WT luciferase with selected substrates.
The fluorescence emission of acyclic monoalkylated aminoluciferins generally ranges from 520 to 540 nm,23 more red-shifted than that of 6′-aminoluciferin (517 nm) but less so than for dialkylated substrates such as 6′-Me2NLH2 (554 nm). CycLuc6 fluoresces at still longer wavelength (567 nm), while CycLuc10 is the most red-shifted of all the luciferin analogues (576 nm). While this was initially surprising, Atkins and Bliss have described similar behavior for aminocoumarin derivatives.24
Mutation of Luciferase Modulates Emission
We have previously found that the luciferase active-site mutant R218K increases the rate of light emission from CycLuc1, CycLuc2, 6′-MeNHLH2, and 6′-Me2NLH2.14 This effect is not particularly selective, as it is observed with all of the aminoluciferins (Figures 4 and 5, Supplementary Figure 3). For instance, CycLuc7 exhibits higher initial and sustained emission rates with R218K compared to WT (Figure 4). The R218K mutant also resulted in a slight red-shift in bioluminescence for most substrates, pushing the maximal emission wavelength for CycLuc10 to 648 nm (Supplementary Table 1 and Supplementary Figure 1).
Figure 4.

Light emission from d-luciferin and CycLuc7 (250 μM) treated with equal amounts of purified WT and mutant luciferases (0.2 nM). The assay was performed in triplicate and is represented as the mean ± SEM.
Figure 5.

Initial and sustained bioluminescence from each luciferin (250 μM) with equal amounts of three different purified luciferases (0.2 nM, see text): (A) peak intensity within the first 2 s of substrate addition and (B) sustained light emission 1 min post-injection. The assay was performed in triplicate and is represented as the mean ± SEM.
Toward Orthogonal Luciferases
In principle, chemical and structural differences in luciferin substrates could be exploited to create new orthogonal luciferases. However, active-site mutations such as R218K and L286M raise the Km of d-luciferin but do not prevent its utilization as a substrate.14 A more selective point mutant, S347A, raises the Km and lowers the rate of emission from d-luciferin, possibly because it removes a hydrogen-bonding interaction with the benzothiazole nitrogen that may be more important for d-luciferin binding and orientation than for aminoluciferins.14,25 Yet d-luciferin remains a substrate for this mutant luciferase as well. Thus, previous work has not established whether high luciferase activity can be retained in luciferase mutants that do not yield light emission from d-luciferin.
Here we find that the combination of R218K, L286M, and S347A renders d-luciferin essentially inactive. The purified triple-mutant luciferase dramatically reduced both the initial and sustained rates of photon emission from d-luciferin by >10 000-fold (Figure 4 and Supplementary Figure 3). Gratifyingly, this is not due to a lack of luciferase activity, as the photon flux from CycLuc2, CycLuc7, and CycLuc11 actually increased compared to that of the WT enzyme (Figure 5 and Supplementary Figure 3). CycLuc7 is the optimal substrate for the triple-mutant luciferase in vitro, achieving 46% of the initial rate of d-luciferin with WT luciferase. This result demonstrates that high luciferase activity can be maintained in a luciferase mutant that is essentially unresponsive to the native substrate d-luciferin. Furthermore, product inhibition has largely been eliminated, as there is little diminution in flux after the initial burst (Figures 4 and 5 and Supplementary Figure 3).
Luciferin Emission in Live Luciferase-Expressing Cells
We next compared the aminoluciferins to d-luciferin in live WT luciferase-expressing Chinese Hamster Ovary (CHO) cells. Under these conditions, the luciferin substrate must cross the cell membrane to access the luciferase. In marked contrast to what is observed in vitro, almost all of the alkylated aminoluciferins yield higher flux than d-luciferin when assayed at a concentration <30 μM (Figure 6 and Supplementary Figure 4). At high substrate concentration (250 μM), the relative emission from d-luciferin is still equaled or exceeded by those of CycLuc1, CycLuc10, and CycLuc12 (Figure 6). However, if the cell membrane is removed by cell lysis, d-luciferin is the superior substrate at both high and low doses, suggesting that substrate access is the primary factor limiting luciferins in live cells (Supplementary Figure 5).
Figure 6.

Photon flux from luciferase-expressing CHO cells. (A) Live cells expressing the indicated luciferase and treated with high or low doses of luciferin. (B) Comparison of total and near-IR photon flux from WT luciferase with the indicated luciferins. (C,D) Comparison of photon flux from WT and triple-mutant luciferase with d-luciferin or CycLuc7 in live cells (C) or lysed cells (D). All assays were performed in triplicate and are represented as the mean ± SEM.
Near-IR Photon Flux in Live Cells
The peak emission wavelengths of aminoluciferins are red-shifted in vitro. To assess the near-IR emission from each luciferin in live cells, we measured the relative photon flux passing through a Cy5.5 filter (695–770 nm). All of the aminoluciferins exhibited greater relative photon flux in the near-IR than d-luciferin (Figure 6B and Supplementary Figure 6). For every substrate except 6′-aminoluciferin and CycLuc3, this translated to a higher total near-IR photon flux from live cells than d-luciferin under both low-dose and high-dose conditions (Supplementary Figure 6). CycLuc10 gave the greatest fraction of near-IR light emission: 13.9% of the total photon flux, >10-fold higher than that of d-luciferin (Figure 6B). However, total near-IR flux of CycLuc10 was slightly exceeded by that of CycLuc12. Thus, the substrate that yields the highest cellular near-IR light emission is a function of substrate access (affinity and cell permeability) as well as wavelength.
Mutant Luciferases in Live and Lysed Cells
Transfection of CHO cells with R218K luciferase instead of WT luciferase improved relative photon flux from synthetic luciferins compared to d-luciferin (Figure 6 and Supplementary Figure 4). CycLuc2 yielded the highest signal in live cells, and most alkylated aminoluciferins were superior to d-luciferin at both low and high substrate concentrations (Figure 6 and Supplementary Figure 4). Further highlighting the importance of factors other than peak emission wavelength on the total emission of red-shifted photons in live cells, CycLuc2 yielded the highest Cy5.5-filtered signal at 250 μM, while CycLuc6 was best at low concentration (Figure 6 and Supplementary Figure 6). The longer emission wavelength of CycLuc6 (Figure 3) and its higher cell permeability likely lead to its superior flux at low concentrations, while the higher maximal rate of photon emission for CycLuc2 ultimately prevails at high substrate concentration (Figure 5). Thus, the best-performing substrates are context-dependent.
No signal above background could be measured from live cells expressing the triple-mutant luciferase after treatment with d-luciferin (Figure 6C). In contrast, CycLuc2, CycLuc7, and CycLuc11 achieve high photon flux (Figure 6). The triple mutant possesses lower affinity for its substrates compared to WT or R218K luciferase, yielding reduced photon flux at low substrate concentration (Figure 6 and Supplementary Figure 4). However, in lysed cells, the lower affinity of the triple mutant improved the signal from CycLuc7 and CycLuc11 due to its lessened product inhibition (Figure 6D and Supplementary Figure 5). CycLuc7 is the best substrate in cell lysates, achieving ∼20% of the d-luciferin signal with the WT luciferase (Figure 6D and Supplementary Figure 5). Mutant and WT luciferase protein expression is equivalent in transfected cells by Western blot (Supplementary Figure 7), and the differences between luciferases in cell lysates (Supplementary Figure 5) generally mirrors what is observed with equal concentrations of purified proteins in vitro (Figure 5). The triple-mutant luciferase is thus a significant step toward orthogonal bioluminescent reporters of gene expression in both live and lysed cells, as it yields high light output with (alkylated) aminoluciferins but little to no signal with d-luciferin. We speculate that the triple mutant primarily discriminates between substrates by lowering substrate affinity and removing an interaction important for orienting the native substrate (S347). Luciferin analogues possessing high affinity for luciferase and an alternative “handle” for proper orientation remain effective substrates.14,15 Given that the mutations that comprise the triple mutant were originally identified to individually improve luciferase activity with CycLuc1,14 it is likely that further improvements in selectivity and function are possible for this broadened palette of luciferins. Ironically, the identification of a luciferase that emits strongly with d-luciferin but does not respond to synthetic luciferins has been more elusive. The development of fully orthogonal luciferin–luciferase reporter pairs will therefore require further work, perhaps by eschewing d-luciferin altogether in favor of two or more synthetic substrates.
Conclusion
There is reason to expect that substrate performance in live cells, rather than with purified protein or cell lysates, is more predictive of in vivo behavior. In recent collaborative work we have found that CycLuc1 allows dramatically improved bioluminescence imaging in live mice compared to the standard imaging conditions with d-luciferin.12 Tumor cells can be imaged with 20–200-fold less substrate than d-luciferin, and luciferase expression deep in the brain that cannot be detected with d-luciferin is detectable with CycLuc1.12 Many of the substrates described here provide higher total and red-shifted photon flux in live cells, suggesting that they may also have superior properties for in vivo imaging. Differences in substrate affinity, lipophilicity, and functionality are also anticipated to affect the pharmacokinetics of the luciferins in vivo, perhaps allowing tuning of bioluminescent half-lives and/or tissue distribution. Moreover, we have found that mutation of luciferase can essentially eliminate light output from the native d-luciferin substrate while retaining or improving light emission from one or more aminoluciferin substrates to levels comparable to or, in live cells, superior to that of d-luciferin with the WT luciferase. Thus, these synthetic luciferins and mutant luciferases not only expand the palette of luminogenic molecules but transcend the emission properties of d-luciferin and firefly luciferase. They are therefore expected to have significant potential for bioluminescence imaging applications both in vitro and in vivo.
Acknowledgments
This work was funded by the National Institutes of Health (R01EB013270 to S.C.M.).
Supporting Information Available
Experimental procedures; compound characterization, including all NMR spectra; Supplementary Table 1; and Supplementary Figures 1–7. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fraga H. Photochem. Photobiol. Sci. 2008, 7, 146. [DOI] [PubMed] [Google Scholar]
- Fan F.; Wood K. V. Assay Drug. Dev. Technol. 2007, 5, 127. [DOI] [PubMed] [Google Scholar]
- Prescher J. A.; Contag C. H. Curr. Opin. Chem. Biol. 2010, 14, 80. [DOI] [PubMed] [Google Scholar]
- Reddy G. R.; Thompson W. C.; Miller S. C. J. Am. Chem. Soc. 2010, 132, 13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura H.; Sasakura K.; Ueno T.; Urano Y.; Terai T.; Hanaoka K.; Tsuboi T.; Nagano T. Chem. Asian J. 2010, 5, 2053. [DOI] [PubMed] [Google Scholar]
- Takakura H.; Kojima R.; Urano Y.; Terai T.; Hanaoka K.; Nagano T. Chem. Asian J. 2011, 6, 1800. [DOI] [PubMed] [Google Scholar]
- Conley N. R.; Dragulescu-Andrasi A.; Rao J.; Moerner W. E. Angew. Chem., Int. Ed. 2012, 51, 3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano S.; Obata R.; Miura C.; Kiyama M.; Hama K.; Nakamura M.; Amano Y.; Kojima S.; Hirano T.; Maki S.; Niwa H. Tetrahedron 2013, 69, 3847. [Google Scholar]
- Kojima R.; Takakura H.; Ozawa T.; Tada Y.; Nagano T.; Urano Y. Angew. Chem., Int. Ed. 2013, 52, 1175. [DOI] [PubMed] [Google Scholar]
- Viviani V. R.; Bechara E. J.; Ohmiya Y. Biochemistry 1999, 38, 8271. [DOI] [PubMed] [Google Scholar]
- Branchini B. R.; Southworth T. L.; Khattak N. F.; Michelini E.; Roda A. Anal. Biochem. 2005, 345, 140. [DOI] [PubMed] [Google Scholar]
- Evans M. S.; Chaurette J. P.; Adams S. T. Jr.; Reddy G. R.; Paley M. A.; Aronin N.; Prescher J. A.; Miller S. C. Nat. Methods 2014, 11, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S. T.; Miller S. C. Curr. Opin. Chem. Biol. 2014, 21C, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood K. R.; Mofford D. M.; Reddy G. R.; Miller S. C. Chem. Biol. 2011, 18, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofford D. M.; Reddy G. R.; Miller S. C. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchuk-Voloshina N.; Haugland R. P.; Bishop-Stewart J.; Bhalgat M. K.; Millard P. J.; Mao F.; Leung W. Y.; Haugland R. P. J. Histochem. Cytochem. 1999, 47, 1179. [DOI] [PubMed] [Google Scholar]
- Belov V. N.; Bossi M. L.; Fölling J.; Boyarskiy V. P.; Hell S. W. Chemistry 2009, 15, 10762. [DOI] [PubMed] [Google Scholar]
- Pauff S. M.; Miller S. C. Org. Lett. 2011, 13, 6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson N. M.; Ward A. D. Tetrahedron 2005, 61, 155. [Google Scholar]
- Bonger K. M.; Hoogendoorn S.; van Koppen C. J.; Timmers C. M.; Overkleeft H. S.; van der Marel G. A. ChemMedChem 2009, 4, 2098. [DOI] [PubMed] [Google Scholar]
- McCutcheon D. C.; Paley M. A.; Steinhardt R. C.; Prescher J. A. J. Am. Chem. Soc. 2012, 134, 7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodroofe C. C.; Meisenheimer P. L.; Klaubert D. H.; Kovic Y.; Rosenberg J. C.; Behney C. E.; Southworth T. L.; Branchini B. R. Biochemistry 2012, 51, 9807. [DOI] [PubMed] [Google Scholar]
- Woodroofe C. C.; Shultz J. W.; Wood M. G.; Osterman J.; Cali J. J.; Daily W. J.; Meisenheimer P. L.; Klaubert D. H. Biochemistry 2008, 47, 10383. [DOI] [PubMed] [Google Scholar]
- Atkins R. L.; Bliss D. E. J. Org. Chem. 1978, 43, 1975. [Google Scholar]
- Branchini B. R.; Southworth T. L.; Murtiashaw M. H.; Boije H.; Fleet S. E. Biochemistry 2003, 42, 10429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.