

Abstract
We have studied reactions of secondary and primary alcohols with benzynes generated by the hexadehydro-Diels–Alder (HDDA) reaction. These alcohols undergo competitive addition vs dihydrogen transfer to produce aryl ethers vs reduced benzenoid products, respectively. During the latter process, an equivalent amount of oxidized ketone (or aldehyde) is formed. Using deuterium labeling studies, we determined that (i) it is the carbinol C–H and adjacent O–H hydrogen atoms that are transferred during this process and (ii) the mechanism is consistent with a hydride-like transfer of the C–H. Substrates bearing an internal trap attached to the reactive, HDDA-derived benzyne intermediate were used to probe the kinetic order of the alcohol trapping agent in the H2-transfer as well as in the alcohol addition process. The H2-transfer reaction is first order in alcohol. Our results are suggestive of a concerted H2-transfer process, which is further supported by density functional theory (DFT) computational studies and results of a kinetic isotope effect experiment. In contrast, alcohol addition to the benzyne is second order in alcohol, a previously unrecognized phenomenon. Additional DFT studies were used to further probe the mechanistic aspects of the alcohol addition process.
Introduction
o-Benzyne (2),1 characterized by its inherently low-lying LUMO,2 is among the most versatile and useful of all reactive intermediates.3 Arynes are “trapped” by many different kinds of nucleophilic species.4 It is perhaps surprising then that o-benzyne generated by the tert-butoxide-promoted 1,2-elimination of bromobenzene (1, Figure 1a) cleanly gives the [4 + 2] furan cycloadduct 3 as the major product.5 That is, neither the stoichiometric tert-butoxide reagent nor the byproduct t-BuOH reacts with o-benzyne (2) under the basic conditions. In contrast, Stiles and Miller reported that when 2 is generated by thermal decomposition of benzenediazonium-2-carboxylate (4) in the presence of tert-butanol and under neutral conditions, the alcohol adds to give tert-butyl phenyl ether (5) as the major event.6 Benzyne derivatives can be generated thermally by the cycloisomerization of triynes, a reaction pathway first identified by the groups of Johnson and Ueda.7 We recently reported that this hexadehydro-Diels–Alder (HDDA) reaction, when performed in tert-butanol solution, leads to the efficient production of t-BuOAr ethers as shown for 6 to 8 via benzyne 7 (Figure 1b).8 This reaction (along with the related example of 9 to 11 via 10) proceeds with a high degree of regioselectivity, which can be rationalized by the (computed) distorted geometry of the benzyne intermediate(s); the nucleophilic heteroatom adds to the aryne carbon having the larger internal bond angle (cf. δ+ in 7 and 10).9
Figure 1.
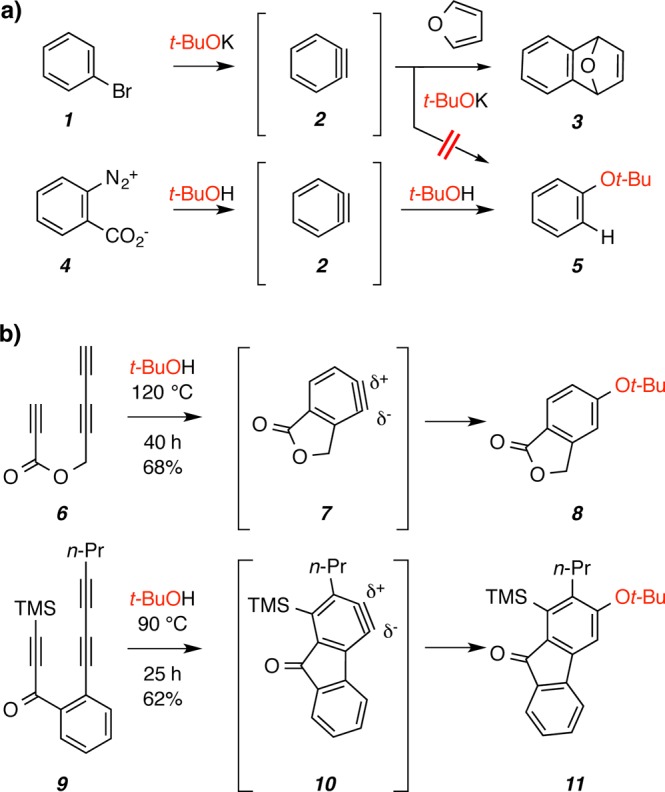
Reactions of benzynes in the presence of tert-butanol. (a) Different reactivities of o-benzyne (2) under basic5 vs neutral6 conditions. (b) HDDA-generated benzynes (7 and 10) also are trapped by neutral tert-butanol.
Results and Discussion
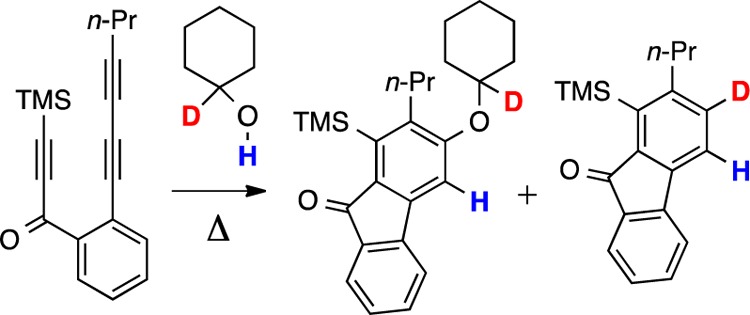
In the course of exploring other alcohols that would participate in an addition to HDDA-derived benzynes, we observed noticeably contrasting behavior between secondary and primary alcohols vis-à-vis that of tert-butanol. For example, the reaction of triyne 9 in a solution of the secondary carbinol cyclohexanol (12-hh) resulted in the isolation of the addition adduct 13 in 80% yield (Figure 2). However, we also observed the formation of a small amount of the reduced benzenoid 14-hh (13:14-hh = 12:1 in neat cyclohexanol), the result of net addition of a hydrogen atom to each of the benzyne carbon atoms. This process was also recently noted in a report by Lee and co-workers.10 To our surprise, when triyne 9 was heated in the presence of only 1.6 equiv of cyclohexanol (12-hh), the product ratio was substantially reversed, and 14-hh, the result of H2-transfer, was formed to the near exclusion of 13 (13:14-hh = 1:17 in 0.013 M cyclohexanol in CDCl3). A similar dependency of the branching ratio between the dihydrogen transfer (to give 14-hh) vs alcohol addition pathways on the bulk concentration of the trapping alcohol was also observed for both isopropanol and ethanol [see Graph S1 and Table S1 in the Supporting Information (SI)]. Together, these results indicate that in the presence of higher concentrations of trapping alcohol, relatively more addition product is formed, whereas H2-transfer is strongly dominant at low alcohol concentrations. These two reaction manifolds seemingly have different kinetic profiles.
Figure 2.

Competitive H2-transfer vs addition processes for the reaction of cyclohexanol (12-hh) with the HDDA-generated benzyne 10. The branching ratio for the pathways leading to product 13 vs 14-hh/15 is dependent on the bulk concentration of 12-hh.
When the reaction between 9 and 12-hh (1.6 equiv) was monitored directly by 1H NMR spectroscopy in CDCl3 solution, essentially equimolar amounts of 14-hh and cyclohexanone (15) were observed, demonstrating that the secondary alcohol was the source of the two hydrogen atoms appearing in the benzenoid 14-hh. We have previously reported that this same reduced product, 14-hh, arose when 9 was heated in cyclooctane solution.11 In that study we showed that concerted H2-transfer from the hydrocarbon to 10 (Figure 1) gave equimolar amounts of 14-hh and (the oxidized) cyclooctene.
To identify the sites in cyclohexanol (12-hh) from which the two hydrogen atoms were being transferred, we performed the reaction using a series of deuterated cyclohexanol derivatives. First, HDDA reaction of 9 in the presence of ca. 1.5 equiv of 2,2,6,6-tetradeuterocyclohexanol (12-d4) occurred without observable (<5%, 1H NMR and GC/MS analysis) transfer of deuterium (Figure 3a).12 That is, 14-hh and 15-d4 were the dominant products observed. This experiment established that generation of the enol 16 (and its subsequent tautomerization to 15) was not operative, indicating instead that the OH and carbinol methine hydrogen atoms in 12-hh were being transferred. We therefore made and examined the behavior of the complementary pair of monodeuterated cyclohexanols 12-hd and 12-dh (Figure 3b). Each of these labeled alcohols resulted in the formation of a monodeuterated benzenoid and the all-protio cyclohexanone (15), as would be expected for a redox process involving a simultaneous transfer of two hydrogen atoms11 from a single encounter between the benzyne and one molecule of the alcohol. Remarkably, however, each of these monodeuteration reactions was highly regioselective; that is, only 14-hd was observed using alcohol 12-hd as the donor and only 14-dh when 12-dh was used.
Figure 3.
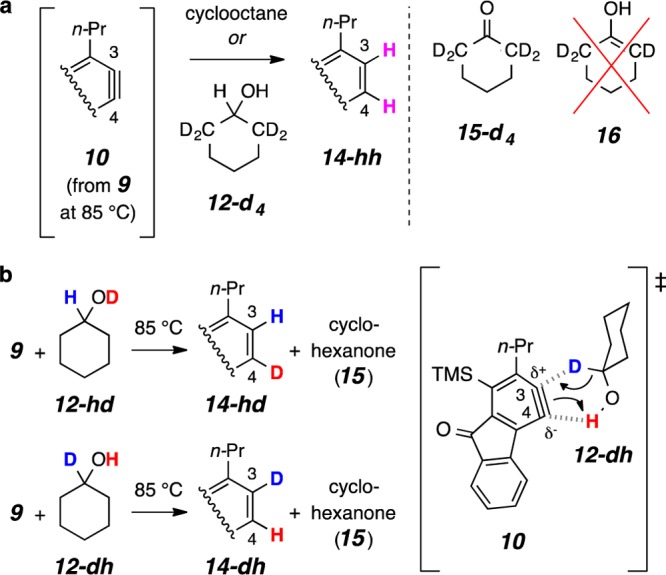
Studies with various deuterium-labeled cyclohexanols. The initial concentration of triyne 9 and alcohol 12-hh in CDCl3 for these experiments was 0.010 and 0.015 M, respectively. (a) Results from the use of tetradeuteroalcohol 12-d4 rule out the intermediacy of enol 16. (b) The H2-transfer occurs with a high degree of regioselectivity.
The sense of regioselectivity observed in the formation of 14-hd vs 14-dh can be explained by concerted H2-transfer from the H–CO–H moiety in 14, in which addition involving a hydride-like nucleophilic carbinol C–H is accompanied by a more electrophilic O–H proton transfer. This view calls to mind the Cannizzaro (or Oppenauer) class of C–H hydride transfer reduction reactions. This model, involving concerted transfer of both hydrogen atoms, accounts for the sense of the observed regioselectivity. The hydride addition as portrayed in Figure 3b for the reaction between benzyne 10 and the monodeuterated cyclohexanol 12-dh delivers the deuterium atom to C3, the more electrophilic benzyne carbon in 10 (cf. 10 to 11 in Figure 1b8).
To gain additional insight about the mechanism of the alcohol/benzyne redox process, we turned to density functional theory (DFT) computations. We first examined the H2-transfer between methanol and o-benzyne (2), to give benzene and formaldehyde, and located the transition-state (TS) structure shown as 17‡ in Figure 4a. The computed overall free energy of reaction (ΔGrxn) was = −70.8 kcal·mol–1 and the free energy of activation (ΔG‡) was found to be 13.4 kcal·mol–1. The geometry is indicative of a relatively early transition state for the reaction, consistent with its high exothermicity. The process is also considerably asynchronous, with C–H bond cleavage well advanced over that of O–H. Interestingly, the initially symmetrical o-benzyne (2) has a distorted geometry in TS structure 17‡ (internal bond angles at Ca and Cb = 132° vs 118°, respectively), which we interpret as an accommodation of a nucleophilic hydride-like transfer. The notion that aryne polarizability plays an important role in contributing to the immense versatility displayed in aryne trapping reactions4,13 perhaps merits broader consideration. Said differently, an aryne represents one of the softest and most malleable of all carbon-based electrophiles.14
Figure 4.
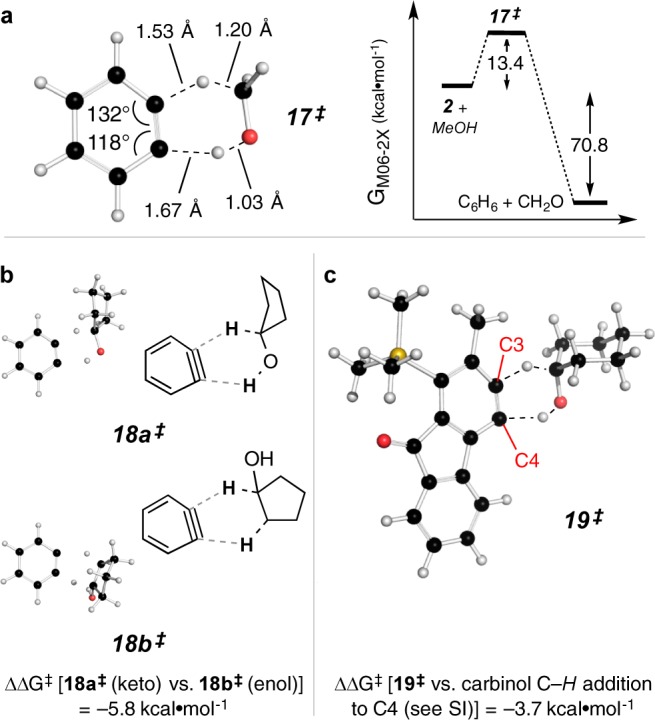
Computed TS structure geometries and energies (gas-phase) for H2-transfer from alcohols [DFT, M06-2X/6-311+G(d,p)].15 (a) Methanol to o-benzyne (17‡); (b) cyclopentanol to o-benzyne to give either cyclopentanone (18a‡) or its enol (1-hydroxycyclopentene; 18b‡) (the computed structure for each is given in the SI); and (c) cyclohexanol to an actual HDDA benzyne (19‡). The ΔΔG‡ value of −3.7 kcal·mol–1 corresponds to the two regioisomeric modes of H2-transfer from 12 to the benzyne.
We then used computation to explore the question of which two hydrogen atoms were transferred from the alcohol donor (i.e., the H–CO–H to give a ketone vs the H–CC–H to produce an enol). Here we used cyclopentanol and benzyne to locate the two relevant TS structures 18a‡ and 18b‡ (Figure 4b). These gave ΔG‡ values of 12.8 kcal·mol–1 vs 18.6 kcal·mol–1, respectively. This (computed) preference for ketone vs enol formation is consistent with the lack of deuterium incorporation in the experiments presented in Figure 3a.
Finally, we computed the TS structure geometries for the two regioisomeric modes of H2-transfer in which the carbinol methine adds to C3 vs C4 in a benzyne like 10; to simplify this computational analysis we replaced the n-propyl group in benzyne 10 by a methyl substituent. The resulting most stable TS structure is shown as 19‡ in Figure 4c. It depicts, again, an asynchronous, hydride-like addition preferentially to C3 of the now predistorted (and thereby predisposed) benzyne. The internal angles at C3 and C4 were computed to be 136° and 119°, respectively, in the benzyne acceptor, and they further distort to 138° and 112° in the TS structure 19‡. The computed ΔG‡ for the reaction via 19‡ was 11.1 kcal·mol–1. This was lower by 3.7 kcal·mol–1 than the computed ΔG‡ for the alternative mode of H2-transfer involving addition of C–H to C4 (see SI).16
Encouraged by the supportive nature of these computations, we proceeded to compute the expected kinetic isotope effect (KIE) for the reaction of 9 with 12-dh (Figure 3b). The truncated methyl-containing 19‡ was again used as the TS structure for the process. Replacement of the proton on the carbinol carbon with a deuteron gave rise to a computed KIE of 1.97 for the C−H transfer processes (see SI for additional details). This value is consistent with an expected early transition state for this H2-transfer reaction. We then measured this KIE by heating triyne 9 with a mixture of 12-hh and 12-dh and then analyzing the 1H NMR spectrum to determine the ratio of 14-hh:14-dh. The experimental KIE was 2.0, a gratifying result vis-à-vis the computed value. This bolsters the view that the redox transfer between 10 and 12-dh occurs through a species like 19‡, as portrayed in Figure 4c.
We then examined the reactivity of a variety of alcohols (cf. 23a–k, Figure 5c) to learn their propensity to participate in the H2-transfer process. The goal here was not to establish a preparative method for making either a reduced benzenoid like 14-hh (cyclooctane is a better “reagent” for that task11) or any of the ketones or aldehydes derived from 23 (a multitude of oxidants is available for that task, of course). Instead, we hoped to be able to gain further insights about mechanistic aspects of this unusual redox reaction. In situ NMR analysis proved to be a convenient method for this study. An example of such an experiment is shown in Figure 5a. A CDCl3 solution of triyne 20 (0.02 M), which bears a methyl group in place of the n-propyl substituent in triyne 9, was heated in the presence of several equivalents of cyclooctanol at 85 °C. After 14 h this reaction mixture was directly analyzed by 1H NMR spectroscopy (Figure 5b), which showed the overall cleanliness of the process. As was the case for triyne 9 when cyclized in the presence of low bulk concentration of alcohols, this reaction yielded the reduced benzenoid 21 as the dominant product together with a small amount of the alcohol addition adduct 22. Cyclooctanone was produced in an essentially equimolar amount vis-à-vis 21 as a result of this H2-transfer process [cf. integration values of the resonances for Mea (δ = 2.47 ppm) vs the C2 and C8 methylene protons (δ = 2.41 ppm) of cyclooctanone].
Figure 5.

In situ 1H NMR analysis of a variety of alcohol dihydrogen donors. (a,b) The 1H NMR spectrum of the product mixture arising from the reaction of triyne 20 with cyclooctanol (in 5-fold excess) to give products 21, 22, and cyclooctanone (ratio = 8:1:8). The vertical scale for each of the three cutouts of the NMR spectrum is identical. (c) Triyne 9 was heated [85 °C for 20 h (ca. 5 half-lives)] in the presence of 10 equiv of each of the alcohols 23a–d/f–k (2 equiv of 23e was used) in CDCl3. The product mixture was analyzed by qNMR analysis.19 The ratio of reduction to addition products is given in parentheses below each substrate. The principal product derived from each of alcohols 23a–k was identified as the corresponding ketone or aldehyde (both but-3-ynal and 2,3-butadienal observed from 23i) by analysis of the 1H NMR spectrum of the reaction mixture (see SI for the spectrum from each experiment).
Using this method of analysis, we observed that each of the alcohols 23 shown in Figure 5c was capable of donating two hydrogens to benzyne 10 (from triyne 9), albeit with different levels of efficiency. The ratio of reduction to addition products (given in parentheses in Figure 5c) varied with each alcohol. Importantly, the amount of reduced arene 14 was always very similar to that of the ketone derived from the corresponding secondary alcohol donor. Inferences that can be taken from this series of substrates include the following: (i) even highly hindered alcohols like 23a–c readily donate dihydrogen, (ii) the vicinal diol 23d did not show evidence of overoxidation or oxidative cleavage, and the internal hydrogen bond appears to inhibit the H2-addition process, (iii) the cyclopropylcarbinol 23e showed no signs of ring-opened products, which argues against a stepwise radical mechanism initiated by benzylic hydrogen atom abstraction (as well as stepwise hydride transfer to generate a transient cyclopropyl carbenium ion), and is, instead, more consistent with a simultaneous transfer of two hydrogens (cf. 17‡–19‡, Figure 4), (iv) primary alcohols (23f–j) are also functional H2-donors, (v) allylic or propargylic hydrogen atoms in substrates 23h or 23i, potential participants in ene reactions with the benzyne,8,17 do not interfere with the H2-transfer, (vi) the ease of the H2-transfer process correlates qualitatively with the bond strength of the carbinol C–H that is cleaved during the redox process, and (vii) accordingly, the poorest H2-donor is methanol (23k), the carbinol with the strongest C–H bond. Finally, the alcohol/aryne redox process is not limited to benzynes produced by an HDDA reaction. We have observed (in situ 1H NMR analysis) that o-benzyne (2) generated by the Kobayashi protocol (o-TMSC6H4OTf + CsF in CD3CN)18 in the presence of cyclohexanol produces a substantial amount of benzene (see SI).
To establish the effect of steric hindrance on the ability of an alcohol to add to benzyne 10, we performed a series of three competition experiments, each in 100% alcohol. Triyne 9 was heated in the presence of equimolar mixtures of MeOH vs EtOH, EtOH vs i-PrOH, and i-PrOH vs t-BuOH. The ratios of the two possible alcohol addition products are shown in Figure 6. It is clear that increasing steric hindrance slows the rate of addition. The extent of retardation grows exponentially in a way that is reminiscent of the nonlinear change in A-values20 across the series of Me (1.74 kcal mol–1), Et (1.78), i-Pr (2.21), and t-Bu (>4) groups, a measure of steric size that also reflects detailed conformational features of a system.
Figure 6.
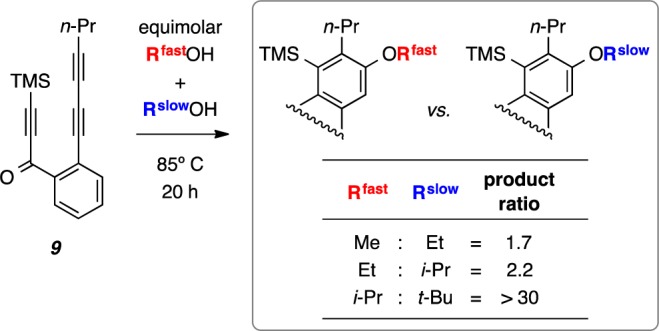
Competition experiments to assess the relative ease of addition of alcohols of differing steric bulk to the benzyne 10. An authentic sample of each of the four alkoxyarene (ROAr) products was prepared by heating 9 in the appropriate neat alcohol. The products were obtained in the following yields after chromatographic purification: R = Me (30%), R = Et (86%), R = i-Pr (83%), and R = t-BuOH (11, Figure 1b, 62%).
In order to gain additional mechanistic insights about both the alcohol addition and redox reaction pathways, we have studied kinetic aspects of the benzyne trapping events. It is challenging to obtain fundamental mechanistic information of this sort because aryne formation is the rate-limiting step for virtually every preparatively useful method of generating and trapping arynes. We have recently shown21 that a valuable protocol for probing the kinetic order of a bimolecular benzyne trapping process involves the design and use of a substrate that contains a suitably reactive, competing intramolecular trap that serves as an internal clock. By determining the ratio of products arising from the intramolecular vs bimolecular capture of the aryne as a function of concentration of the external trapping agent, the kinetic order of that agent in the product-determining reaction event can be deduced.
We selected the symmetrical tetrayne 24 as the first substrate for these kinetic studies (Figure 7). When heated alone, 24 undergoes highly efficient conversion, via an intramolecular Diels–Alder (IMDA) reaction of benzyne825, to 26 as the only product observed in the crude reaction mixture by 1H NMR spectroscopy (Figure 7a). Solutions (CDCl3) containing eight different overall concentrations of 24 and isopropanol, but always in a 1:70 starting molar ratio, were heated to 68 °C for 18 h (ca. 5 half-lives). Again 1H NMR spectroscopy was used to analyze the product ratio in each crude reaction mixture (Figure 7b,c). We were surprised to see that no aryne reduction product, i.e., 28, was detected in this experiment. This is in contrast to observations we made for the reaction between isopropanol and benzyne 10, for which the H2-transfer process outpaced alcohol addition at concentrations of isopropanol up to 2 M (see Graph S1 and Table S1 in the SI). The character of the benzyne intermediate is likely an important influence on this branching ratio. It is also noteworthy that the ratio of the two regioisomeric alcohol addition products 27a and 27b was also dependent on the bulk concentration of isopropanol (Figure 7b). That is, a relatively larger amount of 27b was formed at higher [i-PrOH]bulk. This observation suggests that different mechanisms are operating during the formation of the two regioisomeric alcohol addition products 27a and 27b. We speculated that the benzylic ether oxygen atom may be playing a role via a hydrogen bonding phenomenon similar to that shown in 29 (Figure 7d), which is influencing both (i) the proximal addition leading to 27a as well as (ii) the lack of formation of the reduced product 28.
Figure 7.
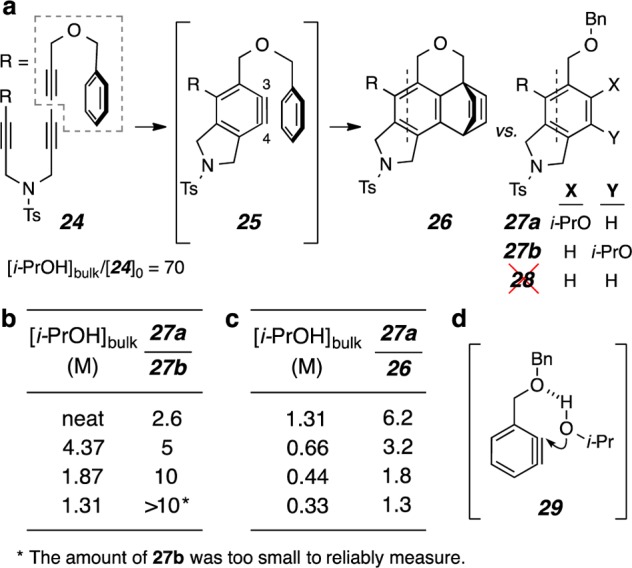
(a) Kinetic competition study involving the unimolecular IMDA reaction in benzyne 25 (to form 26) as an internal clock reaction vs various bimolecular trapping reactions with isopropanol [to give addition products 27a and 27b]. Tetrayne 24 was heated in CDCl3 at 68 °C for 18 h in the presence of the indicated bulk concentration of isopropanol; the initial ratio of reactants was [i-PrOH]bulk/[24]0 = 70 in each experiment. (b,c) Product ratios as a function of the concentration of isopropanol ([i-PrOH]bulk). (d) Possible hydrogen bond interaction influencing the reaction of an alcohol with a benzyne bearing an adjacent benzyloxymethyl group.
From a log–log plot of the ratio of the major adduct vs the clock reaction product (Figure 7c) against the bulk alcohol concentration21 [i.e., ln(27a/26) vs ln[i-PrOH]bulk, see blue data in Graph S2 in SI], we found that the kinetic order of isopropanol in the formation of 27a was 1.1, i.e., essentially first-order. However, if we adjust the value of the isopropanol concentration for the amount of isopropanol dimer present in solution, for which we assume an equilibrium value of 0.35,22 and use the resulting monomer concentration ([i-PrOH]mono), this analysis (see red data in Graph S2 in the SI) leads to an order of 1.4 for the kinetic dependence on isopropanol. Discussion of why we view this adjustment to be a more proper treatment as well as a mechanistic interpretation of the noninteger nature of the alcohol dependency are presented later, after the discussion of Figure 8. Because the ratio of the two alcohol addition products 27b vs 27a increased at higher [i-PrOH]bulk, we conclude that the formation of 27b has a different (and, in fact, higher) order of dependence on the monomeric alcohol (see below).
Figure 8.
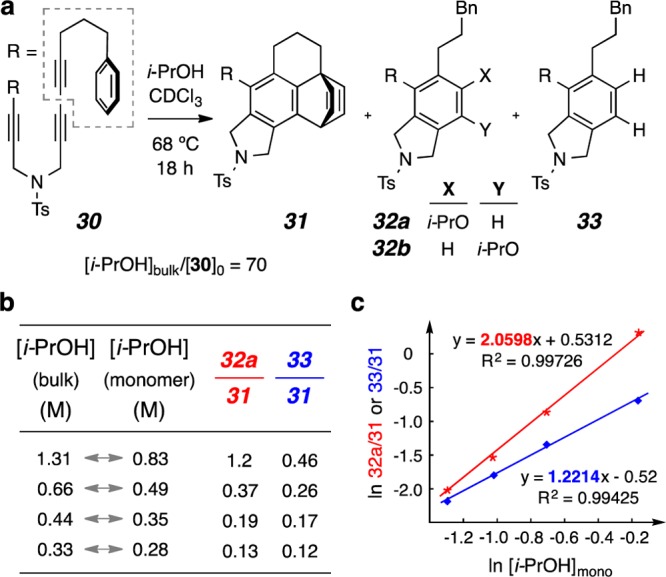
(a) Analogous kinetic competition studies using the tetrayne 30. The initial ratio of reactants was [i-PrOH]bulk/[30]0 = 70. (b) The change in product ratios as a function of the concentration of the isopropanol monomer. (c) Log–log plot of the product ratio vs concentration of isopropanol monomer. The kinetic order for the addition reaction [to give product 32a (or 32b)] vs the H2-transfer reaction (to give 33) was ca. two vs one, respectively.
To test the hypothesis that the ether oxygen in 24 is pertinent to the results just described, we prepared the tetrayne 30 containing an all-carbon linker in place of the benzylic ether. When a fully analogous set of experiments was carried out with substrate 30 (CDCl3, isopropanol, Figure 8a), competition was again observed between the intramolecular clock reaction to give the IMDA adduct 31 and the formation of products derived from engagement by isopropanol (32a/band33). In contrast to the observations with ether 24, we found that (i) the redox product 33 was now formed along with 31 and 32a/b; and (ii) the addition products 32a and 32b were always produced in the same ratio (ca. 10:1), regardless of the isopropanol concentration. From the plot of ln(32a/31) vs ln[i-PrOH]mono (cf. Figure 8c, red),21 we determined (see Graph S3 in the SI for details) that the kinetic order of isopropanol for the alcohol addition pathway was essentially 2.
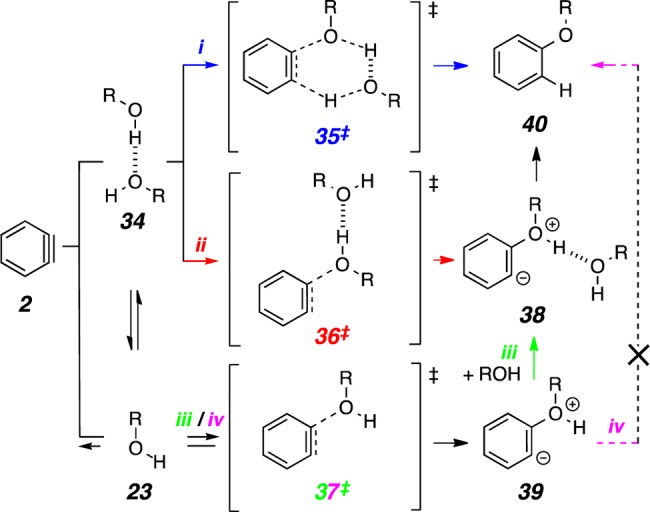
Three mechanistic interpretations for the addition of an alcohol to a benzyne, consistent with the second-order dependency on alcohol, are shown in Scheme 1. For simplicity, they are represented with o-benzyne (2) to give the alkyl phenyl ether 40. In pathway i alcohol addition occurs by a single encounter between a preformed alcohol dimer 34 and 2 via TS structure 35‡. In pathway ii dimer 34 adds to 2 to produce a discrete intermediate 38, having one new C–O bond by way of a TS structure like 36‡. Adduct 38 would then proceed to 40 by an intramolecular proton shuttling event. Assistance of intramolecular 1,3-proton migration by an external hydroxyl-containing molecule has been invoked in classical tautomerization reactions (e.g., enol ⇌ ketone and 2-hydroxypyridine ⇌ 2-pyridone). In pathway iii initial rapid and reversible addition of the first molecule of monomeric alcohol gives zwitterion 39 by way of TS structure 37‡. This is followed by a coordination with a second molecule of alcohol to intersect with intermediate 38 invoked in pathway ii. It is noteworthy that the results from our kinetic studies are inconsistent with an intramolecular proton transfer that would convert 39 directly to 40 (pathway iv) because this would show a simple first-order dependency on the alcohol concentration. Additionally, any of these mechanisms could explain the contrasting reactivity of neutral tert-butanol vs tert-butoxide presented in Figure 1a.
Scheme 1.
We have examined the reaction pathways i–iv computationally for the simple case of methanol (23k) addition to o-benzyne (2) (Figure 9). For each of the four pathways, the level of the highest energy TS structure and rate-determining step is shown in color. The relative zero of free energy corresponds to an isolated o-benzyne and a methanol dimer (all species in Figure 9 include continuum methanol solvation). The stoichiometrically equivalent combination of o-benzyne and two isolated methanol monomers was found to be 0.8 kcal·mol–1 higher in free energy.
Figure 9.

DFT calculations were performed using M06-2X/6-311+G(d,p) and the SMD solvation model (MeOH). Standard state corrections (gas phase to 1 M solution) were applied to all structures and the concentration of methanol was adjusted to 24.7 M (neat methanol). All values are of the free energies in kcal·mol–1. Minima and TS structures of species in which one vs two methanol molecules have engaged o-benzyne (2) are shown in panels (a) vs (b), respectively. The curved dashed lines represent low barrier processes in which a new hydrogen bond to methanol is being formed to convert a species in panel (a) to one in panel (b). (c) A 3D view of the geometry of each of the five TS structures; the dashed lines in each represent atom pairs between which bond order is increasing in the forward reaction direction.
The addition of methanol monomer (23k) to 2 to produce the zwitterion 39 is a step common to both pathways iii and iv (green/magenta). The associated TS structure 37‡ was found to have a relative free energy of 8.3 kcal·mol–1. The subsequent, low barrier, unimolecular proton transfer within 39 via 41‡ was suggestive of rapid conversion to product 40 to complete pathway iv, the reaction that would be first order in alcohol concentration.23 Alternatively, we envision that 39 could coordinate a second molecule of methanol24 to produce 38 by another presumably very low barrier process merely involving hydrogen bond formation (a librational motion in neat alcohol or a solvent-shell exchange reaction in a mixed solvent). Knowing the relative ease of that event vis-à-vis that passing through 41‡ is somewhat moot, however, since (i) irrespective of the barrier, a path to product having first-order dependence on methanol would be available, and, more importantly, (ii) TS structure 37‡ (green/magenta) is not associated with the overall lowest energy pathway in Figure 9.
The most favored computed pathway is ii (red), the stepwise addition of the methanol dimer (34) to 2. This proceeds through the TS structure 36‡ having a relative free energy of 7.3 kcal·mol–1. The resulting adduct 38 was located as a minimum and would be expected to proceed rapidly to product 40·MeOH through some TS structure 42‡. However, we were unsuccessful in finding a fully converged TS structure (i.e., a species having only one imaginary frequency) for this final step, presumably owing to a very flat potential-energy surface along the reaction coordinate and some numerical noise associated with nonanalytic density functional integrals determined on necessarily finite integration grids; such situations can defeat optimization algorithms in their search for a stationary TS structure. Instead, we approximately identified 42‡ (Figure 9) by manually distorting the geometry of 38 in very small increments toward that of the requisite product and computing the energy with restrained internal coordinates (see SI for details). The energy increased by only 0.3 kcal·mol–1 with respect to 38 until a final small change (0.1° in one internal angle) resulted in exergonic collapse to product upon geometry optimization. This approach has been used to identify approximate TS structures for processes for which optimization algorithms fail to converge to stationary points.25
An alternative reaction pathway involving dimer 34 (i.e., i; blue) invokes its concerted addition to 2. The requisite TS structure 35‡ shows the highest free energy (9.7 kcal·mol–1) for the rate limiting step of any of the four pathways. In summary, although it is prudent not to place excessive weight on the merit of computed energetics for reactions having activation barriers that vary by only a few kcal·mol–1, it is encouraging that the results of this study are in concert with a lowest-energy process that involves two molecules of methanol prior to its rate-limiting event.
In contrast to the second order dependency on alcohol for the addition process, we have observed that the molecularity for the H2-transfer to produce the benzenoid 33 is approximately one (1.2). The data are presented as a plot of ln(33/31) vs ln[i-PrOH]mono in Figure 8c. This result is consistent with the TS structures 17‡–19‡ discussed earlier (Figure 4). The first-order nature of the H2-transfer is also consistent with the observation that formation of the hydrogenation product is favored at lower alcohol concentration and the addition product at higher concentration (cf. Figure 2).26
Briefly let us return to the issue of the mixed-order dependence observed for the addition of isopropanol to benzyne 25 to give 27a (Figure 7a). As mentioned, using the free monomer concentration of isopropanol and plotting ln(27a/26) vs ln[i-PrOH]mono (see Graph S2 in the SI) leads to the value of 1.4 for the kinetic order of isopropanol. We suggest that the deviation of this value from a whole integer may be indicative of two competing mechanisms for formation of 27a. At high concentration of alcohol, the reaction is dominated by a pathway analogous to one of those in Scheme 1. At low concentration, the hydrogen bonding effect of the benzyl ether oxygen permits a pathway that is first-order in alcohol, one that, in fact, supersedes the reduction event that otherwise ensues for the other benzyne substrates.
Finally, we mention some anecdotal information that is relevant to and consistent with the mechanistic picture that has emerged here for this alcohol addition process. Throughout our studies of the HDDA reaction we have observed that the dryness of the solvent system does not noticeably influence the outcome of the reaction. Moreover, our attempts to directly add water using mixed aqueous organic solvent systems (e.g., 1,4-dioxane) have led to low amounts of the phenolic products. It seems reasonable to think that water addition to a benzyne would follow an analogous mechanism to that of alcohol addition. A TS structure analogous to 35‡ or 36‡ (Scheme 1) but where R = H would then be operative. A relatively rarely invoked fundamental difference between water and alcohols is the relative strength of the O–H bond. The dissociation energy of the water molecule is 119 kcal·mol–1, which stands in stark contrast to the D(RO–H) = 104–107 kcal·mol–1 for virtually all aliphatic alcohols.27 It follows, then, that a mechanism involving multiple partial O–H bond cleavages, as is the case for either 35‡ or 36‡, would be expected to be slower for water than for an alcohol because of this difference in O–H bond strengths.
Summary
We have explored various mechanistic aspects of the reactions between benzynes and nontertiary alcohols. Competing addition and H2-transfer processes are seen, the latter constituting an unusual redox process. The branching ratio between these pathways is dependent on the concentration of trapping alcohol. Deuterium labeling studies in conjunction with computational investigations have provided evidence that the H2-transfer reaction proceeds via a Cannizzaro-type mechanism in which the carbinol C–H adds to the benzyne in hydride-like and the O–H in proton-like fashion. Through kinetic studies we determined that this redox reaction is first order in the alcohol H2-donor. This is consistent with the notion of concerted, albeit asynchronous, transfer of dihydrogen. In contrast, we have found that the formation of ether product(s) from the addition of alcohol to benzynes is second-order in alcohol. This previously unrecognized mechanistic pathway has been investigated and supported by DFT computations. Among other things it provides insight into why water adds to benzynes only poorly, which happens to be a significant practical advantage in the study and use of (HDDA-derived) benzynes. The use of an internal clock reaction to uncover kinetic information about the product-forming, post-rate-determining steps in aryne trapping reactions21 is powerfully demonstrated. The fundamental insights to emerge are potentially valuable beyond the realm of aryne chemistry itself. Finally, aspects of these studies highlight the importance of the polarizability of arynes in response to their encounter with various trapping agents, thereby contributing to the tremendous versatility of arynes as reactive intermediates.
Acknowledgments
P.H.W., T.W., D.N., and M.K.H. acknowledge the support of a National Science Foundation Graduate Research Fellowship, a Wayland E. Noland Fellowship, a University of Minnesota Graduate School Doctoral Dissertation Fellowship, and a Heisig-Gleysteen Fellowship, respectively. Suggestions and interest from Professor Joseph D. Scanlon are acknowledged with appreciation. Portions of this work were performed with hardware and software resources available through the University of Minnesota Supercomputing Institute (MSI). Financial support for the research was provided by the General Medical Institute of the National Institutes of Health (GM65597).
Supporting Information Available
Experimental procedures for preparation of all new compounds; characterization data for all new compounds; description of computational methodologies; geometries and energies of species shown in Figures 4 and 9; copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Ripon College, Ripon, Wisconsin 54971, United States.
Author Present Address
‡ Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
Author Contributions
§ P.H.W. and D.N. contributed equally to the design, execution, and interpretation of the experiments and results.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- We have adopted the following terminology for use here. (i) o-Aryne: an aromatic ring containing two adjacent sp-hybridized carbon atoms, including the subfamilies of benzynes, pyridynes, indolynes, or naphthalynes. (ii) o-Benzyne (2): the parent o- or 1,2-dehydrobenzene. (iii) A benzyne derivative: any substituted derivative of o-benzyne, which may or may not have an additional, nonaromatic ring; collectively we call these benzynes.
- Rondan N. G.; Domelsmith L. N.; Houk K. N. Tetrahedron Lett. 1979, 20, 3237–3240. [Google Scholar]
- Hoffman R. W.Dehydrobenzene and Cycloalkynes; Academic: New York, 1967. [Google Scholar]
- Aspects of benzyne and aryne chemistry have been reviewed over a dozen times in the last handful of years alone. Examples that include particular emphasis on nucleophilic addition are; a Tadross P. M.; Stoltz B. M. Chem. Rev. 2012, 112, 3550–3577. [DOI] [PubMed] [Google Scholar]; b Goetz A. E.; Garg N. K. J. Org. Chem. 2014, 79, 846–851. [DOI] [PubMed] [Google Scholar]; Interested readers are directed to references therein as well.; c Bhunia A.; Yetra S. R.; Biju A. T. Chem. Soc. Rev. 2012, 41, 3140–3152. [DOI] [PubMed] [Google Scholar]
- Apeloig Y.; Arad D.; Halton B.; Randall C. J. J. Am. Chem. Soc. 1986, 108, 4932–4937. [Google Scholar]
- Stiles M.; Miller R. G.; Burckhardt U. J. Am. Chem. Soc. 1963, 85, 1792–1797. [Google Scholar]
- a Bradley A. Z.; Johnson R. P. J. Am. Chem. Soc. 1997, 119, 9917–9918. [Google Scholar]; b Miyawaki K.; Suzuki K.; Kawano T.; Ueda I. Tetrahedron Lett. 1997, 38, 3943–3946. [Google Scholar]
- Hoye T. R.; Baire B.; Niu D.; Willoughby P. H.; Woods B. P. Nature 2012, 490, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hamura T.; Ibusuki Y.; Sato K.; Matsumoto T.; Osamura Y.; Suzuki K. Org. Lett. 2003, 5, 3551–3554. [DOI] [PubMed] [Google Scholar]; b Cheong P. H. Y.; Paton R. S.; Bronner S. M.; Im G.-Y. J.; Garg N. K.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 1267–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Garr A. N.; Luo D.; Brown N.; Cramer C. J.; Buszek K. R.; VanderVelde D. Org. Lett. 2010, 12, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar R.; Yun S. Y.; Wang K.-P.; Lee D. Org. Lett. 2014, 16, 6–9. [DOI] [PubMed] [Google Scholar]
- Niu D.; Willoughby P. H.; Woods B. P.; Baire B.; Hoye T. R. Nature 2013, 501, 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We performed an entirely analogous experiment with 2,2,7,7-tetradeuterocycloheptanol and observed entirely analogous results; no deuterated benzenoid product 14 nor 2,2,7-trideuterocycloheptanone was observed.
- Kitamura T. Aust. J. Chem. 2010, 63, 987–1001. [Google Scholar]
- a Biehl E. R.; Nieh E.; Hsu K. C. J. Org. Chem. 1969, 34, 3595–3599. [Google Scholar]; b Fleming I.Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: New York, 2011. [Google Scholar]
- Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- We also located (higher energy) TS structures involving the chair-flipped form of the cyclohexanol for each of the two modes of hydride-like addition to C3 and C4 (see SI for details).
- a Jayanth T. T.; Jeganmohan M.; Cheng M.-J.; Chu S.-Y.; Cheng C.-H. J. Am. Chem. Soc. 2006, 128, 2232–2233. [DOI] [PubMed] [Google Scholar]; b Candito D. A.; Panteleev J.; Lautens M. J. Am. Chem. Soc. 2011, 133, 14200–14203. [DOI] [PubMed] [Google Scholar]; c Karmakar R.; Mamidipalli P.; Yun S. Y.; Lee D. Org. Lett. 2013, 15, 1938–1941. [DOI] [PubMed] [Google Scholar]; d Niu D.; Hoye T. R. Nat. Chem. 2013, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeshima Y.; Sonoda T.; Kobayashi H. Chem. Lett. 1983, 1211–1214. [Google Scholar]
- Pauli G. F.; Jaki B. U.; Lankin D. A. J. Nat. Prod. 2007, 70, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth H.; Everett J. R. J. Chem. Soc. Perkin 2 1980, 255–259. [Google Scholar]
- Niu D.; Wang T.; Woods B. P.; Hoye T. R. Org. Lett. 2014, 16, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alcohols are known to form aggregates in chlorinated solvents.22b The enthalpy and entropy change associated with dimerization of monomeric isopropanol in CCl4 has been determined.22c Assuming that these values would be similar to those for dimerization in CDCl3, we estimated the equilibrium constant for the dimerization [i.e., i-PrOH(monomer) + i-PrOH(monomer) × i-PrOH(dimer)] at 68 °C to be 0.35 (ΔG° = +0.95 kcal·mol–1); that is, [i-PrOH]dimer = 0.35·{[i-PrOH]mono}2; b Pimentel G. C.; McClellan A. L.. The Hydrogen Bond; W. H. Freeman and Co.: San Francisco, 1960. [Google Scholar]; c Blanks R. F.; Prausnitz J. M. J. Chem. Phys. 1963, 38, 1500–1504. [Google Scholar]
- a The possibility of a direct, single-step, O–H insertion mechanism23b to produce anisole directly from methanol and o-benzyne (not shown) was considered. Numerous attempts to locate a TS structure were unsuccessful; b Im G.-Y. J.; Bronner S. M.; Goetz A. E.; Paton R. S.; Cheong P. H.-Y.; Houk K. N.; Garg N. K. J. Am. Chem. Soc. 2010, 132, 17933–17944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xia Y.; Liang Y.; Chen Y.; Wang M.; Jiao L.; Huang F.; Liu S.; Li Y.; Yu Z.-X. J. Am. Chem. Soc. 2007, 129, 3470–3471. [DOI] [PubMed] [Google Scholar]; b Patil M. P.; Sunoj R. B. J. Org. Chem. 2007, 72, 8202–8215. [DOI] [PubMed] [Google Scholar]; c Cheong P. H.-Y.; Legault C. Y.; Um J. M.; Çelebi-Ölcüm N.; Houk K. N. Chem. Rev. 2011, 111, 5042–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hull J. F.; Balcells D.; Sauer E. L. O.; Raynaud C.; Brudvig G. W.; Crabtree R. H.; Eisenstein O. J. Am. Chem. Soc. 2010, 132, 7605–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lonsdale R.; Harvey J. N.; Mulholland A. J. Chem. Soc. Rev. 2012, 41, 3025–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To provide direct comparison, we have computed the gas-phase activation free energies for both the H2-transfer (cf. Figure 4a) and the monomeric methanol addition (pathway iv) reactions between methanol and o-benzyne at the M06-2X/6-311+G(d,p) level. The ΔΔG‡ for the two processes is predicted to be 0.0 kcal·mol–1 (see SI and cf. the data for entry 23k in Figure 5c). In addition, we have compared these same two processes using a chloroform solvation model (IEFPCM, M06-2X/aug-cc-pVTZ) to approximate the competition reactions at low alcohol concentration, which were always performed in CHCl3 or CDCl3. The solvated ΔΔG‡ is predicted to be 1.0 kcal·mol–1 in favor of alcohol addition. To within the expected accuracy of the theoretical and chemical models, which do not, for example, explore the potential effect of quantum mechanical tunneling on the rates of reactions dominated by light atom (i.e., H2-)transfer, these small differences in activation free energies are consistent with our experimental observations that these two processes are competitive with one another under various conditions.
- Blanksby S. J.; Ellison G. B. Acc. Chem. Res. 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.