

Abstract
In some natural collagen triple helices, cysteine (Cys) residues on neighboring strands are linked by disulfide bonds, enhancing association and maintaining proper register. Similarly, Cys–Cys disulfide bridges have been used to impose specific associations between collagen-mimetic peptides (CMPs). Screening a library of disulfide linkers in silico for compatibility with collagen identifies the disulfide bridge between proximal homocysteine (Hcy) and Cys as conferring much greater stability than a Cys–Cys bridge, but only when Hcy is installed in the Xaa position of the canonical Xaa–Yaa–Gly repeat and Cys is installed in the Yaa position. Experimental evaluation of CMPs that host alternative thiols validates this design: only Hcy-Cys bridges improve triple-helical structure and stability upon disulfide-bond formation. This privileged linker can enhance CMP-based biomaterials and enable previously inaccessible molecular designs.
Collagen is the most abundant protein in animals1 and is responsible for maintaining the structural integrity of animal bodies.2 Its biological significance has made collagen a common target for biomaterials engineering, encouraging the development of self-assembling synthetic peptide systems that mimic the triple-helical architecture of collagen.3 Although many of these efforts employ non-covalent means to program strand association,4 covalent cross-linking of strands remains the most robust strategy.5 Indeed, cystine “knots”—complex arrangements of interstrand Cys–Cys disulfide bridges—are found in natural fibrillar and fibril-associated collagens,6 inspiring the use of Cys–Cys bridges in synthetic collagen-like fibrillar assemblies that extend through sticky ends.7 Here, we determine the effect of this natural disulfide bridge and synthetic alternatives on triple-helix stability.
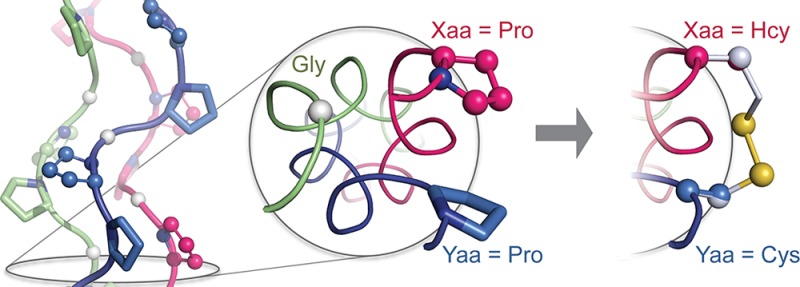
The amino-acid sequence of collagen is defined by repeating Xaa–Yaa–Gly units that feature (2S)-proline (Pro) and (2S,4R)-4-hydroxyproline (Hyp) at the Xaa and Yaa positions, which favor the formation of polyproline-type II helices.8 Collagen strands associate into triple helices with a single-residue stagger that gives rise to registers with an Xaa, Yaa, and Gly residue from each strand appearing at every cross-sectional plane along the triple helix, enabling cystines to be installed at proximal Xaa···Yaa pairs (Figure 1A).
Figure 1.
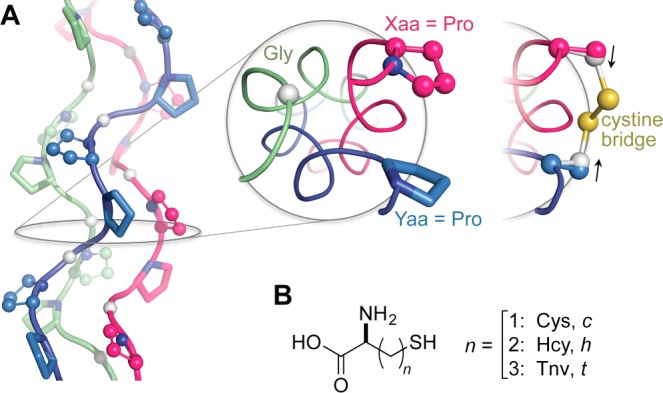
(A) (PPG)10 trimer displaying Xaa (balls and sticks), Yaa (sticks), and Gly positions (white balls). Positioning of Xaa and Yaa residues is shown in a cross-section. Application of a cystine bridge here pulls Cβ atoms inward and away from their original positions (black arrows), indicating a strained linker. (B) Cysteine analogues considered in disulfide bridges in this study. All models were generated with PyMOL v1.3, unless noted otherwise.
Examination of neighboring Xaa···Yaa pairs in a [(PPG)10]3 crystal structure (PDB entry 1kf6)9 reveals the Xaa···Yaa Cβ···Cβ distance (5 Å) to be longer than the average Cβ···Cβ distance (4 Å) predicted for a cystine.10 Thus, even neighboring Xaa and Yaa positions might not allow a geometry favorable for disulfide-bond formation. Natural cystine knots interrupt triple-helical structure,5b,6c,11 but any effect on collagen function is compensated by the length of common collagen strands, which have 103 residues. In contrast, collagen-mimetic peptides (CMPs) in typical synthetic assemblies are only ∼30 residues long and could be more susceptible to an adverse impact from the strain of a cystine linkage. “Sticky-ended” assemblies are contingent upon robust association between single- and double-stranded “overhangs” to form triple-helical segments and would be especially sensitive to linker-induced deformation.
We reasoned that relieving strain within the disulfide bridge could be the key step toward an interstrand “staple” that conforms to the collagen triple helix. Toward this end, we used molecular modeling to explore longer linkers that employ combinations of cysteine (Cys) and the homologated analogues homocysteine (Hcy) and thionorovaline (Tnv), which have one, two, and three side-chain methylene groups, respectively (Figure 1B). Neighboring Xaa and Yaa positions in the [(PPG)10]3 crystal structure were replaced with Cys, Hcy, or Tnv. All nine possible Xaa···Yaa strand pairs were created in silico. After optimization, energies were evaluated12 in a fixed triple-helical backbone, both before (Xaa···Yaa) and after (Xaa–Yaa) disulfide-bond formation. Linker strain is defined as Estrain = Ex–y – Ex·y, which is the change in energy upon disulfide-bond formation. Disulfide bridges are designated by a code that identifies the Xaa–Yaa pair: “c” for Cys, “h” for Hcy, and “t” for Tnv, such that “c–c” represents a cystine.
As expected, increasing linker length relieves the strain on the disulfide. The value of Estrain is largest for the c–c bridge, which contains only two methylene groups (Figure 2). When four or more methylene groups are present, bond and angle strain is eased substantially, and the value of Estrain decreases by ∼7 kcal/mol. Still, dihedral strain remains elevated due to eclipsed C–C or C–S torsion angles (Supporting Information Tables S1 and S2). Interestingly, the h–c disulfide (Xaa = Hcy; Yaa = Cys) falls outside this trend and is free of strained torsions. Despite being among the shortest linkers in the set, h–c forms the most stable disulfide bridge: 12 kcal/mol lower in energy than c–c. The Xaa and Yaa positions are not related by symmetry, and the c–h bridge does not show the dramatic reduction in the value of Estrain as does the h–c bridge, a mark of structural complementarity between the triple helix and the h–c bridge.
Figure 2.
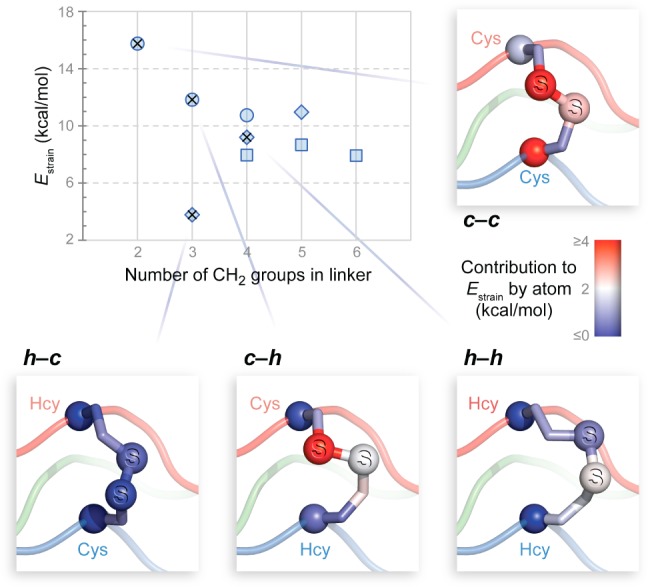
Computational design of disulfide bridges compatible with the collagen triple helix. Computed values of Estrain for disulfides are plotted with respect to linker size. Linkers having Xaa = Cys, Hcy, and Tnv are represented by circles, diamonds, and squares, respectively. Designs selected for experimental evaluation are marked with ×. Lines point to images of computational models in which Cα and S are shown as balls and colors represent the contribution of atoms to the value of Estrain.
To validate our computational predictions, we synthesized CMPs poised to form a c–c, h–c, c–h, or h–h bridge. This set includes bridges predicted to be the best (h–c) and worst (c–c). The disulfide-linked [(PPG)10]3 variants were constructed and characterized using methods established previously (Figure 3A).13,7b Of the three strands, the leading strand hosts the Xaa partner of the disulfide through either a Pro16Cys (for s1c) or a Pro16Hcy (s1h) substitution, whereas the lagging strand bearing the Yaa partner has a Pro14Cys (s3c) or Pro14Hcy (s3h) substitution. After an interstrand disulfide bond was formed by a thiol–disulfide interchange reaction (Figure 3B), a third, (PPG)10 strand (s2) was introduced to associate with the disulfide-bonded pair and thereby complete the triple helix. This setup (double strand plus single strand) recapitulates the key association event that drives sticky-ended triple helices to assemble. The placement of the linker between leading and lagging strands forces s2 to occupy the middle register, avoiding degenerate structures. The thermal stability and the oligomerization state of triple helices formed by association of s1x–s3y (x–y) pairs and s2 were assessed with circular dichroism (CD) spectroscopy and analytical ultracentrifugation (AUC).
Figure 3.
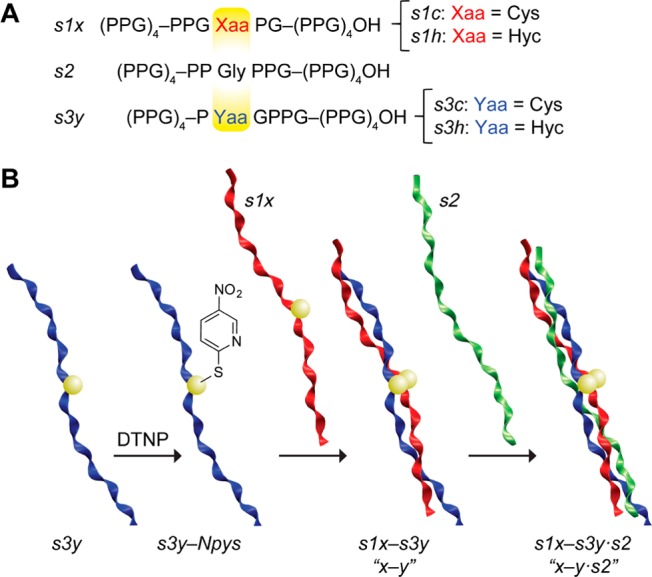
(A) Design and (B) construction of CMPs for experimental assessment of disulfide linkers. Ribbons were generated with VMD v1.9.
The disulfide-linked variants share with [(PPG)10]3 the characteristic CD signature of a collagen triple helix (Figure 4A). The variants do, however, exhibit greater mean ellipticity at 226 nm than does (PPG)10. Triple-helical association was confirmed in sedimentation equilibrium experiments with AUC. Whereas gradients formed by x–y·s2 constructs are readily described by a triple-helical model, (PPG)10 appears as a mixture of monomers and trimers. Thus, the covalent linking of strands appears not only to accommodate and promote triple-helix formation but also to increase trimer content (Supporting Information Figure S3).
Figure 4.
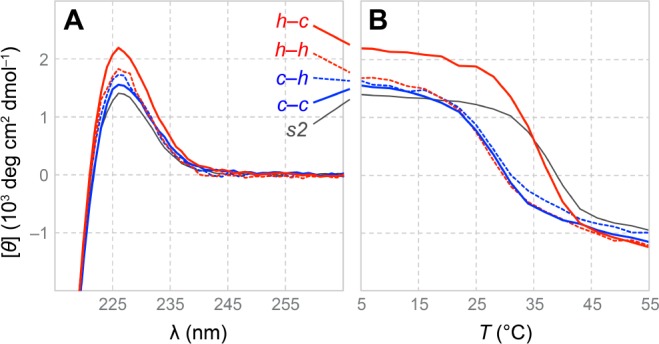
(A) CD spectra and (B) thermal denaturation data for (PPG)10 and x–y·s2 triple helices. For spectra without smoothing, see Supporting Information Figure S2.
A marked loss in the thermostability of triple helices was observed for all disulfide-linked variants, except that with an h–c bridge (Figures 4B and 5A). Use of a c–c, c–h, or h–h bridge leads to a 9 °C decrease in the value of Tm, from 37 to 28 °C. In contrast, trimers that feature an h–c bridge (Tm = 35 °C) do not experience significant destabilization, as predicted by computational analysis.
Figure 5.
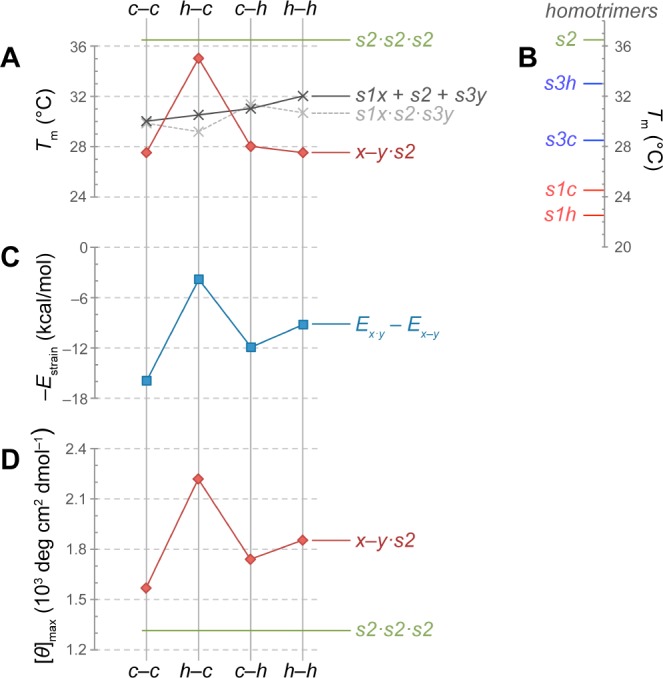
Comparison of experimental and computational data for x–y·s2 and related triple helices. (A) Experimental (red and black) and calculated (gray) Tm values for heterotrimers. (B) Experimental Tm values for homotrimers. (C) Calculated strain energy, plotted as −Estrain to simplify comparisons with experimental data. (D) [θ]max from CD spectra acquired at 4 °C. Values for s2 (green) are included in panels A, B, and D for comparison.
The large destabilizing effect of replacing a proline residue with Cys or Hcy (Figure 5B) obfuscates comparisons of a stapled triple helix (x–y·s2) with an unmodified trimer (s2·s2·s2). A more appropriate comparison would be with a trimer containing reduced Cys or Hcy (s1x·s2·s3y). A linear relationship exists between the free energy of stabilization and Tm value of CMPs.14 If interstrand interactions of reduced Cys and Hcy in a triple helix are insignificant, then the value of Tm for an s1x·s2·s3y heterotrimer can be estimated from the values for homotrimers. Values of Tm thus predicted are 30–32 °C for all s1x·s2·s3y heterotrimers. To verify this prediction, we assessed equimolar mixtures of s1x, s2, and s3y in thermal denaturation experiments. The resulting Tm values near 31 °C agree closely with predictions for reduced complexes (Figure 5A; black vs gray line). Accordingly, we conclude that c–c, c–h, and h–h bridges (Tm ≈ 28 °C) are strained and thus destabilize the triple helix, whereas only an h–c bridge is stabilizing (Tm = 35 °C) (Figure 5A; red vs black line).
Our calculations correctly predict the h–c bridge to be the least strained and thus most stabilizing linker. Still, for strained linkers the Tm values do not correlate with computational rankings (cf. Figure 5A,C). The computational models for strained linkers feature high-energy regions that cannot relax due to imposed backbone constraints. In reality, backbone distortions lower the overall energy of these structures, though rendering them less collagen-like. Indeed, we observe evidence for this relaxation in the signal intensity at the diagnostic wavelength (∼225 nm) in the CD spectrum, as the rank order of the maximum CD signal is in perfect agreement with the rank order of −Estrain for x–y·s2 triple helices (cf. Figure 5C,D). The choice of an h–c bridge over c–c increases the triple-helical content by >40%. This increase is critical, as any linker that disrupts structure would both impede the assembly of a triple helix and deter its biological recognition. With low strain, an h–c bridge reinforces triple-helical structure far better than do its strained alternatives.
We put forth the Hcy–Cys interstrand disulfide bridge as a superior alternative to Cys–Cys for collagen-like peptides and proteins. We expect facile integration of this staple into Xaa–Yaa–Gly repeats, as Hcy–Cys bridges conform well to the collagen fold. Self-assembling systems that grow through the sticky-ended assembly of triple-helical units rely on interstrand bridges and will benefit from our discovery.7a,7c,7d Hcy–Cys bridges should enhance the formation rate and thermostability of such assemblies and allow for smaller assembling units. Hence, we expect Hcy–Cys bridges to extend the reach of self-assembling collagen-like biomaterials.
Acknowledgments
We thank Drs. S. Chattopadhyay, M. D. Boersma, J. C. Lukesh, III, N. A. McGrath, and D. R. McCaslin for assistance with peptide synthesis and characterization, Prof. W. A. Goddard, III for access to computing resources, and Prof. S. H. Gellman for use of his CD spectrophotometer. The facilities of the Materials and Process Simulation Center used in this study were established with grants from DURIP-ONR, DURIP-ARO, and NSF-CSEM. This work was supported by Grant R01 AR044276 (NIH).
Supporting Information Available
Supplemental CD, AUC, and computational data; experimental and computational methods; procedures for the synthesis and purification of peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brinckmann J. In Collagen: Primer in Structure, Processing and Assembly; Brinckmann J., Notbohm H., Muller P. K., Eds.; Topics in Current Chemistry247; Springer: Berlin, 2005; pp 1–6. [Google Scholar]
- Meyers M. A.; Chen P.-Y.; Lin A. Y.-M.; Seki Y. Prog. Mater. Sci. 2008, 53, 1–206. [Google Scholar]
- a Woolfson D. N. Biopolymers 2010, 94, 118–127. [DOI] [PubMed] [Google Scholar]; b Fallas J. A.; O’Leary L. E. R.; Hartgerink J. D. Chem. Soc. Rev. 2010, 39, 3510–3527. [DOI] [PubMed] [Google Scholar]; c Przybyla D. E.; Chmielewski J. Biochemistry 2010, 49, 4411–4419. [DOI] [PubMed] [Google Scholar]; d Siebler C.; Erdmann R. S.; Wennemers H. Chimia 2013, 67, 891–895. [DOI] [PubMed] [Google Scholar]; e Chattopadhyay S.; Raines R. T. Biopolymers 2014, 101, 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cai W. B.; Kwok S. W.; Taulane J. P.; Goodman M. J. Am. Chem. Soc. 2004, 126, 15030–15031. [DOI] [PubMed] [Google Scholar]; b O’Leary L. E. R.; Fallas J. A.; Bakota E. L.; Kang M. K.; Hartgerink J. D. Nat. Chem. 2011, 3, 821–828. [DOI] [PubMed] [Google Scholar]; c Pires M. M.; Lee J.; Ernenwein D.; Chmielewski J. Langmuir 2012, 28, 1993–1997. [DOI] [PubMed] [Google Scholar]
- a Kinberger G. A.; Cai W. B.; Goodman M. J. Am. Chem. Soc. 2002, 124, 15162–15163. [DOI] [PubMed] [Google Scholar]; b Barth D.; Kyrieleis O.; Frank S.; Renner C.; Moroder L. Chem.–Eur. J. 2003, 9, 3703–3714. [DOI] [PubMed] [Google Scholar]; c Horng J.-C.; Hawk A. J.; Zhao Q.; Benedict E. S.; Burke S. D.; Raines R. T. Org. Lett. 2006, 8, 4735–4738. [DOI] [PubMed] [Google Scholar]; d Khew S. T.; Tong Y. W. Biochemistry 2008, 47, 585–596. [DOI] [PubMed] [Google Scholar]; e Byrne C.; McEwan P. A.; Emsley J.; Fischer P. M.; Chan W. C. Chem. Commun. 2011, 47, 2589–2591. [DOI] [PubMed] [Google Scholar]
- a Boulegue C.; Musiol H. J.; Gotz M. G.; Renner C.; Moroder L. Antiox. Redox Signal. 2008, 10, 113–125. [DOI] [PubMed] [Google Scholar]; b Boudko S. P.; Bächinger H. P. J. Biol. Chem. 2012, 287, 44536–44545. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wegener H.; Paulsen H.; Seeger K. J. Biol. Chem. 2014, 289, 4861–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Koide T.; Homma D. L.; Asada S.; Kitagawa K. Bioorg. Med. Chem. Lett. 2005, 15, 5230–5233. [DOI] [PubMed] [Google Scholar]; b Kotch F. W.; Raines R. T. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yamazaki C. M.; Asada S.; Kitagawa K.; Koide T. Biopolymers 2008, 90, 816–823. [DOI] [PubMed] [Google Scholar]; d Yamazaki C. M.; Kadoya Y.; Hozumi K.; Okano-Kosugi H.; Asada S.; Kitagawa K.; Nomizu M.; Koide T. Biomaterials 2010, 31, 1925–1934. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Raines R. T. Annu. Rev. Biochem. 2009, 78, 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berisio R.; Vitagliano L.; Mazzarella L.; Zagari A. Protein Sci. 2002, 11, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozhogina O. A.; Bominaar E. L. J. Struct. Biol. 2009, 168, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudko S. P.; Engel J.; Okuyama K.; Mizuno K.; Bächinger H. P.; Schumacher M. A. J. Biol. Chem. 2008, 283, 32580–32589. [DOI] [PubMed] [Google Scholar]
- a Mayo S. L.; Olafson B. D.; Goddard W. A. III J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar]; b Lim K. T.; Brunett S.; Iotov M.; McClurg R. B.; Vaidehi N.; Dasgupta S.; Taylor S.; Goddard W. A. III J. Comput. Chem. 1997, 18, 501–521. [Google Scholar]
- Rabanal F.; DeGrado W. F.; Dutton P. L. Tetrahedron Lett. 1996, 37, 1347–1350. [Google Scholar]
- Persikov A. V.; Ramshaw J. A. M.; Brodsky B. J. Biol. Chem. 2005, 280, 19343–19349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.