

Abstract
Bleomycins A5 and B2 were used to study the structural features in hairpin DNAs conducive to strong BLM–DNA interaction. Two members of a 10-hairpin DNA library previously found to bind most tightly to these BLMs were subsequently noted to share the sequence 5′-ACGC (complementary strand sequence 5′-GCGT). Each underwent double-strand cleavage at five sites within, or near, an eight base pair region of the DNA duplex which had been randomized to create the original library. A new hairpin DNA library was selected based on affinity for immobilized Fe(III)·BLM A5. Two of the 30 newly identified DNAs also contained the sequence 5′-ACGC/5′-GCGT. These DNAs bound to the Fe(II)·BLMs more tightly than any DNA characterized previously. Surface plasmon resonance confirmed tight Fe(III)·BLM B2 binding and gave an excellent fit for a 1:1 binding model, implying the absence of significant secondary binding sites. Fe(II)·BLM A5 was used to assess sites of double-strand DNA cleavage. Both hairpin DNAs underwent double-strand cleavage at five sites within or near the original randomized eight base region. For DNA 12, four of the five double-strand cleavages involved independent single-strand cleavage reactions; DNA 13 underwent double-strand DNA cleavage by independent single-strand cleavages at all five sites. DNA 14, which bound Fe·BLM poorly, was converted to a strong binder (DNA 15) by insertion of the sequence 5′-ACGC/5′-GCGT. These findings reinforce the idea that tighter DNA binding by Fe·BLM leads to increased double-strand cleavage by a novel mechanism and identify a specific DNA motif conducive to strong BLM binding and cleavage.
Introduction
The antitumor agent bleomycin (BLM) is employed clinically for the treatment of squamous cell carcinomas and malignant lymphomas.1 The clinical mixture of bleomycins known as blenoxane consists mainly of BLM A2 and BLM B2,2 and BLM A5 is also used as a single chemotherapeutic agent (Figure 1).3 While these agents are administered therapeutically in metal-free form, it is believed that Fe·BLM,4 or possibly Cu·BLM,5 formed in situ is actually responsible for the observed single- and double-strand damage to DNA.6 Double-strand DNA cleavage by Fe·BLM occurs at a frequency greater than what could be anticipated based on the random accumulation of single-strand breaks7 and has often been suggested to form the basis for the antitumor activity of bleomycin.
Figure 1.

Chemical structures of bleomycins A2, B2, and A5.
While the ability of bleomycin to mediate double-strand DNA cleavage is believed to form the basis for its antitumor activity, the specific lesion(s) that lead to tumor cell killing have not been identified. Recently, we have reported that a library of 10 hairpin DNAs8 selected for their ability to bind tightly to BLM exhibited greatly enhanced double-strand cleavage (Figure S1).9 The majority of double-strand cleavage events were of a novel type involving two independent single-strand cleavages,9 rather than the single coupled double-strand cleavage described earlier.10,11 Further, it was noted that the number of double-strand cleavages of a given hairpin DNA seemed to be in direct proportion to the affinity of that DNA for Fe(II)·BLM A5.9
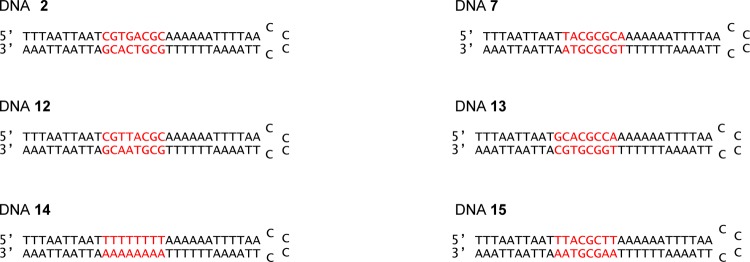
To obtain greater insight into the structural features in the hairpin DNAs which enabled their tight binding to bleomycin, we prepared a new 64-nucleotide (nt) hairpin DNA library containing eight randomized base pairs. The strategy employed was the same as that reported previously,8 with the exception that the selection for tight binders was carried out using immobilized Fe(III)·BLM A5 rather than metal free BLM A5. Thirty hairpin DNAs were isolated from this library and sequenced; none of these had the same sequence as the 10 DNAs in the original library.8 Careful inspection of the 10 DNAs studied from the original hairpin DNA library revealed that the two DNAs which bound most strongly to Fe(II)·BLM A5 (DNAs 2 and 7) shared the common sequence 5′-ACGC/5′-GCGT, albeit not in the same position within the DNAs (Figure 2). Accordingly, the 30 newly identified hairpin DNAs were inspected to determine whether any of them also had the sequence 5′-ACGC/5′-GCGT. In fact two such DNAs (12 and 13) were identified (Figure 2) and formed the basis for the current study.
Figure 2.
Structures of hairpin DNAs employed for study.
The new DNAs were found to bind to bleomycin more tightly than any species identified to date, as judged both by a competition assay and by the use of surface plasmon resonance.12 The latter technique gave results for Fe(III)·BLM B2 binding which were an excellent fit for a 1:1 binding model, arguing for a single, unique site of binding to each hairpin DNA. In spite of the unique binding site, DNA 12 underwent double-strand cleavage at five sites by Fe(II)·BLM A5, and DNA 13 also gave five sets of double-strand cleavage products. While the number of double-strand DNA cleavage sites did not increase beyond that noted in our earlier study, 9 of the 10 double-strand cleavage sites in DNAs 12 and 13 resulted from two closely spaced but independent single strand cleavage events.
To provide further evidence for the involvement of the sequence 5′-ACGC/5′-GCGT in BLM binding, we designed a hairpin DNA (14) in which the initially randomized eight base-pair sequence was 5′-TTTTTTTT/5′-AAAAAAAA (Figure 2). While this hairpin DNA had poor affinity for Fe(II)·BLM A5 as anticipated, replacement of the central four base pairs with 5′-ACGC/5′-GCGT resulted in a hairpin DNA (15) which had dramatically enhanced affinity for Fe(II)·BLM A5.
On the basis of the results obtained with hairpin DNAs containing the motif 5′-ACGC/5′-GCGT, we suggest a possible mechanism for tumor cell killing by bleomycin.
Results
As shown in Figure 3 and Table 1, hairpin DNAs 2 and 7 were capable of binding strongly to Fe(II)·BLM A5 as reported previously,8 thereby potently suppressing the cleavage of a 16-nt profluorescent hairpin DNA present at an equimolar concentration. Each of these 64-nt hairpin DNAs inhibited cleavage of the profluorescent hairpin DNA to the extent of 97% (Table 1), as reported previously.8 Newly selected hairpin DNAs 12 and 13 bound to Fe(II)·BLM A5 even more strongly, suppressing 99 and 98%, respectively, of cleavage of the profluorescent hairpin DNA (Figure 3 and Table 1). The binding specificities of DNAs 7, 12 and 13 for Fe(II)·BLM B2 were also determined and found to be quite similar (97, 97, and 99%, respectively) (Table 1).
Figure 3.
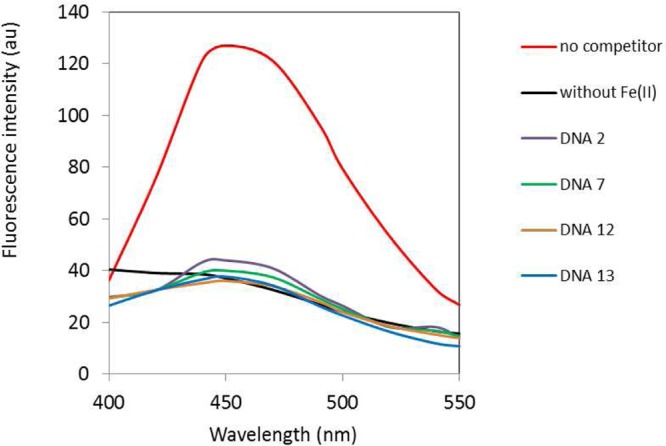
Fluorescence emission spectra resulting from treatment of 16-nt hairpin DNA-Cf15 with Fe(II)·BLM A5 in the presence or absence of 64-nt hairpin DNAs. The reaction mixture contained 0.72 μM Fe(II)·BLM A5, 0.72 μM hairpin DNA-Cf15, and 0.72 μM hairpin DNA 2, 7, 12, or 13 in 10 mM Na cacodylate buffer solution, pH 7.0, containing 100 mM NaCl. The fluorescence emission spectra were obtained following excitation at 310 nm and 25 °C. Also shown is the fluoresence emission spectrum of the 16-nt hairpin DNA-Cf15 treated with BLM A5 in the absence of any 64-nt hairpin DNA under the same conditions (“no competitor”).
Table 1. Inhibition of Fluorescence Enhancement by Selected Hairpin DNAsa.

The binding specificity (%) was calculated as the decrease in fluorescence intensity at maximum emission wavelength (450 nm) from no competitor (0%) through the reaction mixture without Fe2+ (100%).
In order to further quantify the DNA binding affinity of Fe·BLM, biosensor surface plasmon resonance (SPR) experiments were conducted with immobilized hairpin DNAs. To compare the sensorgram saturation levels, approximately the same amounts of the hairpin DNAs 7, 12, and 13 were immobilized on the surface of each sensor chip. The equilibrium constants obtained from both global kinetic fitting of the sensorgrams and the steady-state analyses were then fitted to appropriate binding models. The data are listed in Tables 2 and 3, where they are also compared with the values determined previously for hairpin DNA 2.12
Table 2. Binding of Fe(III)·BLM B2 to Hairpin DNAs 2, 7, 12, and 13 at 10 mM NaCl concentration and 15 °Ca.
| DNA | ka (× 105 M–1 s–1) | kd (× 10–2 s–1) | KA (ka/kd) (× 106 M–1) | KD (kd/ka) (× 10–9 M) | KA (× 106 M–1) (steady-state) |
|---|---|---|---|---|---|
| 2b | 1.8 ± 0.23 | 1.7 ± 0.22 | 10.5 ± 1.3 | 95 ± 12 | 9.4 ± 0.38; 0.20 ± 0.08 |
| 7 | 1.7 ± 0.12 | 1.0 ± 0.18 | 17.0 ± 2.5 | 58.8 ± 2.0 | 15.6 ± 0.28 |
| 12 | 3.3 ± 0.35 | 1.9 ± 0.28 | 17.3 ± 1.5 | 57.8 ± 1.9 | 17.5 ± 0.32 |
| 13 | 2.3 ± 0.24 | 0.75 ± 0.27 | 30.7 ± 1.8 | 32.5 ± 1.5 | 22.8 ± 0.25 |
10, 20, 40, 70, and 90 nM concentrations of Fe(III)·BLM B2 were used to determine the kinetic rate constants. Error values given in the table were obtained during the fitting of data with Biacore T200 evaluation software. Based on reproducibility of results, the errors in the strong binding constants and kinetics constants are ±15%.
Data from ref (12).
Table 3. Binding of Fe(III)·BLM B2 to Hairpin DNAs 2, 7, 12, and 13 at 10 mM NaCl concentration and 25 °C.
| DNA | KA (× 106 M–1) (steady-state) |
|---|---|
| 2 | 3.1 ± 0.20 |
| 7 | 6.8 ± 0.18 |
| 12 | 6.2 ± 0.25 |
| 13 | 8.2 ± 0.36 |
It is clear from the shape of the binding curves of Fe(III)·BLM B2 with three different hairpin DNAs that the on- and off-rates vary with changes in the position of the ACGC/GCGT and flanking sequences (Figures 4 and 5). Initially the KA values for Fe(III)·BLM B2 were determined for hairpin DNAs 7, 12, and 13 at 15 °C and 10 mM NaCl. Under these conditions, the strongest binding was observed for the Fe(III)·BLM B2 complex with hairpin DNA 13 (Table 2). The on-rate constant ka was determined as 2.3 × 105 M–1 s–1, while the off-rate kd was quite low (0.0075 s–1) (Table 2) which makes this sequence the strongest Fe(III)·BLM B2 binder in this set (KA 30.7 × 106 M–1) (Figure 4). Under the same conditions Fe(III)·BLM B2 bound to hairpin DNAs 7 and 12 with almost equal affinity (17.0 × 106 and 17.3 × 106 M–1, respectively; Table 2). Thus, the relative affinities of Fe(III)·BLM B2 with three hairpin DNAs were found to be DNA 13 > DNA 12 ≈ DNA 7 (Table 2).
Figure 4.
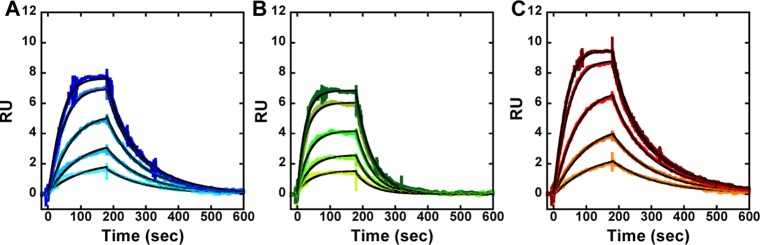
SPR sensorgrams for the interaction of Fe(III)·BLM B2 with (A) hairpin DNA 7, (B) hairpin DNA 12, and (C) hairpin DNA 13 at 10 mM NaCl concentrations and 15 °C. The individual sensorgrams (colored) represent responses at Fe(III)·BLM B2 concentrations of 10, 20, 40, 70, and 90 nM (bottom to top). Global kinetic fit (black solid lines) with a 1:1 model was performed using Biacore T200 Evaluation Software to obtain kinetic association and dissociation rate constants.
Figure 5.

SPR sensorgrams for the interaction of Fe(III)·BLM B2 with (A) hairpin DNA 7, (B) hairpin DNA 12, and (C) hairpin DNA 13 at 10 mM NaCl concentrations and 15 °C. The individual sensorgrams (colored) represent responses at Fe(III)·BLM B2 concentrations of 100, 200, 300, 500, 700, and 900 nM (bottom to top). Global kinetic fit (black solid lines) with a 1:1 model was performed using Biacore T200 Evaluation Software to obtain kinetic association and dissociation rate constants.
SPR experiments involving Fe(III)·BLM B2 with all three hairpin DNA sequences were also conducted at 25 °C and 10 mM salt concentration. Under these conditions the relative affinities of the three hairpin DNAs for Fe(III)·BLM B2 were unchanged (Table 3). It is readily apparent from the steady-state fit data (Figure 6) that the temperature had a significant effect on the absolute binding affinities (Tables 2 and 3); Fe(III)·BLM B2 exhibited 3–4-fold weaker binding for all three hairpin DNAs with increasing temperature (Figure 6; Tables 2 and 3).
Figure 6.
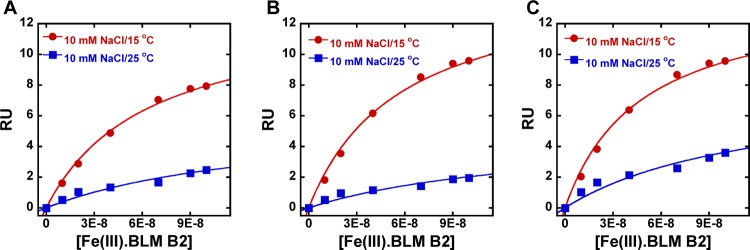
SPR equilibrium binding plots of Fe(III)·BLM B2 with (A) hairpin DNA 7, (B) hairpin DNA 12, and (C) hairpin DNA 13 at 10 mM NaCl concentrations and 15 (red) and 25 (blue) °C. The steady-state response values were fitted as a function of free ligand concentration to a single-site interaction model. The binding affinities are listed in Tables 2 and Table 3
It may be noted that hairpin DNAs 7, 12, and 13 all bound to Fe(III)·BLM B2 with significantly greater affinities than did hairpin DNA 2 (Tables 2 and 3) in spite of seemingly small differences as measured by the competition assay (Table 1). The steady-state response values were fitted as a function of free ligand concentration to a single-site interaction model. In the present case, a 1:1 affinity model provided an excellent fit, quite consistent with the kinetic fitting data. In comparison, for hairpin DNA 2, the steady-state data indicated the presence of at least one and probably multiple weaker binding sites for Fe(III)·BLM B2.12 While the competition assay was less useful than the SPR assay in differentiating between DNAs 7, 12, and 13, it was essential for quantifying the binding of DNA 14 (vide infra), which is not bound strongly enough to be analyzed by SPR. The competition assay also employs Fe(II)·BLM (as compared with Fe(III)·BLM in the SPR assay), thus measuring the behavior of the species actually involved in DNA cleavage.
We have recently reported an analysis of double-strand cleavage of a library of 10 hairpin DNAs selected for their ability to bind tightly to bleomycin.9 The analysis was carried out by modification of the strategy first described by Povirk.10 Following treatment of the alternatively 5′- and 3′-32P end-labeled hairpin DNAs with Fe(II)·BLM A5, the co-migrating bands isolated from the nondenaturing gels were analyzed on a sequencing gel. These co-migrating bands should logically represent double-strand cleavage products, differing only in the site of 32P end-labeling. In addition, the bands on the native gels that co-migrated with the uncleaved hairpin DNA were also analyzed by sequencing gel analysis to identify the location of species of type III (Figure 7), i.e., primary sites of cleavage opposite alkali-labile sites. Where these type III species occurred in the same positions as frank double-strand cleavage (species of type IV, Figure 7), identification of the strand on which cleavage had occurred to form the type III lesion permitted unambiguous identification of the primary site of cleavage.9 The absence of a unique primary site of cleavage is a hallmark of a double-strand break formed by two closely spaced single-strand cleavages.9
Figure 7.

Mechanisms of double-strand DNA cleavage induced by bleomycin.9 Activated Fe·BLM abtracts H• from C-4′ of deoxyribose at a primary cleavage site, producing either an apyrimidinic/apurinic site (I) or a single-strand break having a 3′-phosphoroglycolate terminus (II). Although the apurinic site (I) does not undergo further reaction, strand break II is a potential target for a secondary BLM cleavage reaction on the opposing DNA strand. The secondary attack of (re)activated bleomycin by abstracting the C-4′ H atom from the secondary site sugar affords either a double-strand break with 5′-phosphate and 3′-phosphoroglycolate termini (IV) or a strand break at the primary site accompanied by an AP lesion at the secondary site (III). The latter upon treatment with mild base can produce a double-strand cleavage product (V).
Hairpin DNAs 12 and 13 were alternatively 5′- and 3′-32P end-labeled and then subjected to cleavage by Fe(II)·BLM A5; the results are shown in Figures S2 and S3, respectively. A total of 16 cleavages were noted for DNA 12 and also 16 cleavages for hairpin DNA 13.
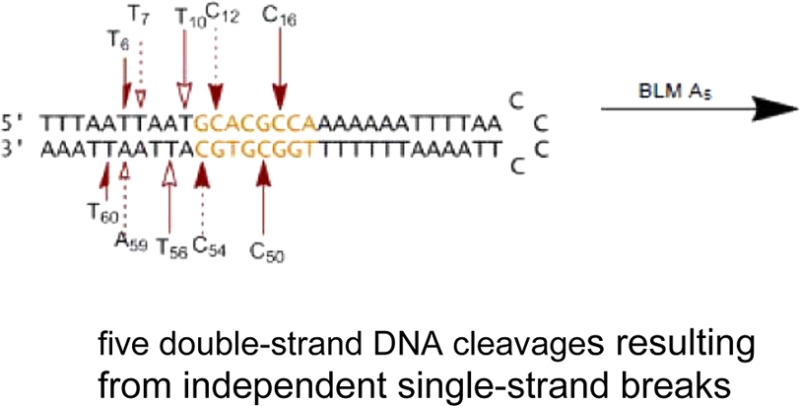
The same radiolabeled hairpin DNAs were also employed to determine the sites of double-strand cleavage. Following treatment with Fe(II)·BLM A5, the end-labeled hairpin DNAs 12 were analyzed by native gel electrophoresis. As shown in the native gel in Figure 8A, in addition to a band that co-migrated with full length hairpin DNA, five sets of bands of comparable mobility were apparent in the alternatively 5′- and 3′-32P end-labeled DNAs 12, consistent with five double-strand cleavage events. The individual bands were recovered from the native gel, and each was subjected to analysis on a sequencing gel (Figure 8B). The sequencing gel revealed the positions of double-strand DNA cleavage for each of the recovered bands. In fact, five double-strand cleavages were observed, including cleavage at T6/T60, T7/A59, T10/A55, T13/A52, and C18/C48, as summarized in Figure 10. The only primary site of cleavage observed was for T13 (Figure 8B, lane 2), indicating that the double-strand cleavage at T13/A52 was a coupled double-strand cleavage event. The other four double-strand cleavages resulted from independent single-strand breaks. All sites of cleavage of hairpin DNA 12 are also summarized in Figure S2.
Figure 8.

Analysis of bleomycin-induced double-strand cleavage of hairpin DNA 12. (A) Double-strand cleavage of [3′-32P]-end-labeled (lane 2) and [5′-32P]-end-labeled (lane 3) 64-nt hairpin DNA 12 by Fe(II)·bleomycin A5. Lane 1, [3′-32P]-end-labeled DNA alone; lane 2, 1.5 μM Fe(II)·BLM A5; lane 3, 1.5 μM Fe(II)·BLM A5; lane 4, [5′-32P]-end-labeled DNA alone. (B) Sequencing gel analysis of Fe(II)·bleomycin-induced sites of double-strand cleavage of [5′-32P]-end-labeled (lanes 1–7) and [3′-32P]-end-labeled (lanes 8–14) hairpin DNA 12. Each lane (except lanes 1 and 14) corresponds to a numbered cleavage band, shown in (A). Lane 1, Maxam–Gilbert G+A sequencing lane of [5′-32P]-end-labeled DNA 12; lane 2, band 3a; lane 3, band 3b; lane 4, band 3c; lane 5, band 3d; lane 6, band 3e; lane 7, band 3f; lane 8, band 2f; lane 9, band 2e; lane 10, band 2d; lane 11, band 2c; lane 12, band 2b; lane 13, band 2a; lane 14, Maxam–Gilbert G+A sequencing lane of [3′-32P]-end-labeled DNA 12.
Figure 10.

Summary of sites of double-strand cleavage of hairpin DNAs 12 and 13. Orange bases indicate randomized region of the original hairpin DNA library. Arrows of the same shape and color indicate paired cleavages. Black arrows correspond to coupled double-strand cleavage events, whereas red arrows indicate noncoupled double-strand cleavage events, resulting from two independent single-strand cleavages on opposite strands. Nucleotide colored in red indicates a primary site of double-strand DNA cleavage for a coupled double-strand cleavage event.
The same two-stage analysis was carried out for double-strand cleavage of hairpin DNA 13 (Figure 9). As shown in the native gel in Figure 9A, five sets of co-migrating bands were observed when the 5′- and 3′-32P end-labeled DNAs 13 were run in adjacent lanes on the native gel. Recovery of the bands from the native gel, followed by further analysis of each on a sequencing gel, permitted the positions of double-strand cleavage to be identified. As anticipated, five double-strand cleavage events were identified, and these are summarized in Figure 10. The double-strand cleavage sites included T6/T60, T7/A59, T10/T56, C12/C54 and C16/C50. No primary site of cleavage was observed, indicating that all five double-strand cleavages were produced as closely spaced single-strand cleavage events. All sites of cleavage of this hairpin DNA are summarized in Figure S3.
Figure 9.

Analysis of bleomycin-induced double-strand cleavage of hairpin DNA 13. (A) Double-strand cleavage of [3′-32P]-end-labeled (lane 2) and [5′-32P]-end-labeled (lane 3) 64-nt hairpin DNA 13 by Fe(II)·bleomycin A5. Lane 1, [3′-32P]-end-labeled DNA alone; lane 2, 1.5 μM Fe(II)·BLM A5; lane 3, 1.5 μM Fe(II)·BLM A5; lane 4, [5′-32P]-end-labeled DNA alone. (B) Sequencing gel analysis of sites of Fe·bleomycin-induced double-strand cleavage of [5′-32P]-end-labeled (lanes 1–7) and [3′-32P]-end-labeled (lanes 8–14) hairpin DNA 13. Each lane (except lanes 1 and 14) corresponds to a numbered cleavage band, shown in (A). Lane 1, Maxam–Gilbert G+A sequencing lane of [5′-32P]-end-labeled DNA 13; lane 2, band 3a; lane 3, band 3b; lane 4, band 3c; lane 5, band 3d; lane 6, band 3e; lane 7, band 3f; lane 8, band 2f; lane 9, band 2e; lane 10, band 2d; lane 11, band 2c; lane 12, band 2b; lane 13, band 2a; lane 14, Maxam–Gilbert G+A sequencing lane of [3′-32P]-end-labeled DNA 13.
In summary, for DNA 12, there was a single site at which double-strand cleavage involved a coupled process, namely T13/A52. The primary cleavage site was at T13. The remaining four double-strand cleavages each resulted from two independent cleavage events in close proximity. In the case of hairpin DNA 13, all five observed double-strand cleavage events resulted from closely spaced single-strand cleavage.
Of special interest were the cleavage events involving the 5′-ACGC/5′-GCGT sequences common to hairpin DNAs 2, 7, 12, and 13. For hairpin DNA 2, these included A15/T50, C16/C48, and C18/(T46), all three of which involved coupled double-strand cleavage events. For hairpin DNA 7, the sites of cleavage included (T11)/T53, C13/C51, and C15/(C49), only two of which involved coupled double-strand cleavage. For hairpin DNA 12, C18/C48 was the only double-strand cleavage site, and for DNA 13, C16/C50 was the only double-strand cleavage site; neither of these represented coupled events. Thus, the only common feature of the cleavages within the common 5′-ACGC/5′-GCGT sequences was that the cytidine moiety in the 5′-GCGT sequence was cleaved in each case.
The assumption that the 5′-ACGC/5′-GCGT sequence common to hairpin DNAs 2, 7, 12, and 13 was responsible for the exceptionally strong binding of these DNAs to Fe·BLM was tested directly. The randomized region of the 64-nt hairpin DNA was substituted with 5′-TTTTTTTT/5′-AAAAAAAA (hairpin DNA 14, Figure 2). When this hairpin DNA was employed in the competition assay with an equimolar amount of the profluorescent 16-nt hairpin DNA, the reduction in release of the fluorescent nucleobase from the 16-nt DNA was only 21% (Figure 11 and Table 1), indicating quite weak binding by Fe(II)·BLM A5. Substitution of nucleosides 13–16 by 5′-ACGC (and concomitant substitution of nucleotides 49–52 by 5′-GCGT) afforded hairpin DNA 15. Although the strategy that lead to the preparation of this hairpin DNA did not involve any selection whatsoever by an immobilized BLM, it was found to have a binding specificity of 93% for Fe(II)·BLM A5 (Table 1).
Figure 11.
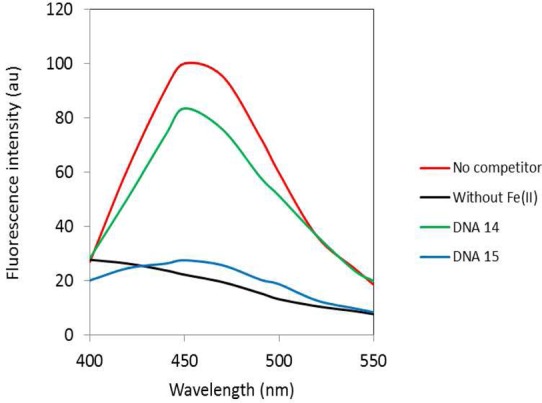
Fluorescence emission spectra resulting from treatment of 16-nt hairpin DNA-Cf15 with Fe(II)·BLM A5 in the presence or absence of 64-nt hairpin DNAs 14 and 15. The procedure employed was the same as that described in the legend to Figure 3.
The uniqueness of the 5′-ACGC/5′-GCGT motif was studied by searching for other tetranucleotide sequences that occurred multiple times in the two hairpin DNA libraries. A number of sequences occurred at least three times, and seven of these were investigated to identify other motifs associated with strong BLM binding. These included 5′-TCCG/5′-CGGA which was present in seven hairpin DNAs. Three of these hairpin DNAs were tested for Fe(II)·BLM A5 binding, and one had a binding specificity of 94%; however, the binding specificities of the other two were 64% and 86%. The motif 5′-GGGC/5′-GCCC was present in five hairpin DNAs. Three of these were tested using the competition assay, but the binding specificities of these DNAs for Fe(II)·BLM were relatively modest (82%, 64%, and 89%). The motif 5′-CGGG/5′-CCCG occurred four times in the hairpin DNA library. Testing of three of these DNAs indicated Fe(II)·BLM binding specificities of 82%, 64%, and 89%. Other motifs which appeared at least three times included 5′-TTGA/5′-TCAA, 5′-GGCC/5′-GGCC, 5′-ATCC/5′-GGAT, and 5′-GCGC/5′-GCGC. The assay of representative hairpin DNAs containing these motifs again failed to provide evidence of their ability to confer strong Fe(II)·BLM binding specificity. Thus, the results obtained with the motif 5′-ACGC/5′-GCGT appeared to be quite unique within these hairpin DNA libraries.
Discussion
The bleomycins are clinically used as antitumor agents whose mechanism of action is believed to involve DNA cleavage. The bleomycins mediate sequence selective DNA cleavage, and about 20% of the lesions that they produce involve double-strand DNA cleavage.6,7,10 The double-strand DNA breaks have attracted significant attention, as they are presumably difficult to repair at a cellular level and may well form the basis for the antitumor activity of this class of compounds.
The study of double-strand DNA cleavage was studied initially by the Povirk laboratory, who demonstrated that the most commonly monitored process involved a coupled event in which initial cleavage on one DNA strand was followed by oxidative damage in close proximity on the opposite strand.7,10,13 In one study, these workers analyzed the cleavage of three linear DNA duplexes. Samples of each of the duplexes were alternatively 5′- and 3′-32P end-labeled, and then treated with Fe(II)·BLM A2 and separated on a nondenaturing polyacrylamide gel to permit identification of the (co-migrating) bands resulting from double-strand cleavage.10 Each of the bands was employed for DNA sequence analysis. This analysis indicated that all of the double-strand breaks involved a 5′-G-Py sequence on one strand, but there were a variety of sequences involved in the breakage on the opposite strand. It was assumed that the shared G-Py sequence represented the primary cleavage site. The orientation of the breaks afforded products with blunt ends and 1-nt 5′-extensions, but no product with 3′-extensions.10 In addition to the double-strand breaks, alkali-labile lesions were also noted at the secondary cleavage sites on the strand opposite the primary cleavage site. The alkali-labile lesion is believed to form by an alternative pathway from the same (C4′ deoxyribose) radical intermediate that leads to frank strand scission.2,6 Critically, because these experiments were carried out under conditions of single-hit kinetics, the authors concluded that the double-strand breaks must have been mediated by a single Fe·BLM molecule.
The conclusion that the double-strand breaks were coupled mechanistically has been supported by studies from other laboratories. This has included the finding of enhanced cleavage opposite an initial BLM-induced nick11a and more detailed studies by Absalon et al.11b,11c utilizing hairpin DNAs bearing an internal 32P-labeled phosphate group, which enabled measurement of the ratio of single- to double-strand cleavage to be measured at individual sites. An earlier study from our laboratory documented the existence of a highly efficient double-strand cleavage site in a hairpin DNA, which was shown to be a coupled event by studying the relative intensities of cleavage at the two sites following alternative 5′- or 3′-32P end-labeling.11d All of these studies supported the idea that the double-strand cleavage observed was mediated by a single BLM molecule.
Analysis of the 10 hairpin DNAs revealed the presence of a total of 31 double-strand cleavage sites. Unexpectedly, only 14 of these sites resulted from coupled double-strand cleavage. The remaining 17 double-strand cleavages lacked a unique primary cleavage site and appeared to have arisen from two independent single-strand cleavage events.9 The occurrence of this novel type of cleavage was attributed to the exceptional affinity of BLM for the hairpin DNAs in the library. Consistent with this interpretation was the finding that those DNAs in the library which bound most avidly to Fe(II)·BLM A5 underwent the greatest number of double-strand cleavages.
A study of the dynamic behavior of Fe·BLMs in the presence of three of these hairpin DNAs by surface plasmon resonance also indicated that there was a single strong binding site for each, along with one (or more likely more than one) much weaker binding sites.12 In spite of the single strong binding site, each of these DNAs was cleaved strongly at multiple sites, and the Fe(II)·BLM bound to the hairpin DNAs was unavailable for cleavage of a second competitor DNA known to be a good substrate for cleavage by Fe·BLM in the absence of the 64-nt hairpin DNA.12 This finding is fully consistent with the observation of double-strand cleavages resulting from independent, closely spaced single-strand cleavage events.
The foregoing studies verified the existence of a number of hairpin DNAs able to bind tightly to BLM. These DNAs exhibit unusual sequence selectivity of DNA cleavage and undergo numerous single- and double-strand cleavage events,8,9,14 the latter of which seem more numerous than in arbitrarily chosen duplex DNAs7,10 and form via two distinct mechanisms.9 However, none of these studies suggested which specific structural elements in the hairpin DNAs might be responsible for the strong binding of BLM. Inspection of the 10 hairpin DNAs employed revealed that the two which bound the most strongly to Fe·BLM (DNAs 2 and 7, Figure 1) had the common sequence 5′-ACGC (and its complement 5′-GCGT). The present study was designed to determine whether this sequence was responsible for the strong binding of hairpin DNAs 2 and 7 to BLM.
A new randomized library of hairpin DNAs was prepared as described previously8 and used to select additional DNAs that bound strongly to BLM. In this experiment, the selection was carried out using immobilized Fe(III)·BLM A5 rather than metal free BLM A5. Thirty new hairpin DNAs were identified in this selection and sequenced, and two of them (hairpin DNAs 12 and 13) were found to contain the sequence 5′-ACGC/5′-GCGT. As shown in Figure 3 and Table 1, these two DNAs were found to bind more strongly to Fe(II)·BLM A5 than any previously characterized DNA, as judged by a competition binding assay. The new DNAs were also characterized for their interaction with Fe(III)·BLM B2 by surface plasmon resonance (Figures 4–6, Tables 2 and 3). Hairpin DNA 12 had binding properties not dissimilar to hairpin DNA 7, while hairpin DNA 13 clearly bound to Fe(III)·BLM B2 more avidly than any species tested to date. Interestingly, unlike the hairpin DNAs characterized previously,12 hairpin DNAs 12 and 13 gave steady-state response values fully consistent with a 1:1 affinity model.
Hairpin DNAs 12 and 13 were utilized as substrates for cleavage by Fe(II)·BLM A5, and both gave multiple double-strand DNA cleavages. For both hairpin DNAs, five double-strand cleavages were observed. Interestingly, unlike the cleavage patterns observed in earlier studies,9,10 the orientation of the breaks afforded products with blunt ends and 1-nt 3′-extensions, the latter of which had not been observed previously as a common feature.10 In particular, hairpin DNA 12 gave two double-strand cleavage products with blunt ends and three having 1-nt 3′-extensions, while hairpin DNA 13 afforded products all of which had 1-nt 3′-extensions. In fact, analysis of the mechanism of double-strand cleavage indicated that only one of the 10 double-strand cleavage reactions (involving products with blunt ends) occurred by a coupled double-strand cleavage mechanism.
To determine whether the presence of the 5′-ACGC/5′-GCGT sequence in an arbitrarily chosen hairpin DNA sequence would be sufficient to confer strong binding to Fe·BLM, we prepared hairpin DNA 14, in which the eight base pair randomized region consisted of a sequence (5′-TTTTTTTT/5′-AAAAAAAA) which we felt would be unlikely to contribute to efficient binding of Fe·BLM. In fact, hairpin DNA 14 exhibited a binding specificity of only 21% in our competition binding assay (Figure 11 and Table 1). In contrast, when the sequence 5′-ACGC/5′-GCGT was substituted in the middle of this eight base pair region, the binding specificity for Fe(II)·BLM A5 increased to 93%, providing additional evidence of the importance of this sequence in BLM binding.
In spite of the importance of the 5′-ACGC/5′-GCGT sequence for BLM binding to the hairpin DNAs and the SPR evidence for unique strong Fe·BLM binding sites in DNAs 12 and 13, there was a surprising lack of consistency of the patterns of cleavage within this sequence. The majority of cleavage sites were external to the common four base pair sequence associated with tight Fe·BLM binding, suggesting a transient scanning mechanism sufficient to allow double-strand cleavage at susceptible sites external to the 5′-ACGC/5′-GCGT sequence. Since the binding of Fe·BLM to DNAs 12 and 13 is consistent with a 1:1 affinity model, cleavage at sites distant from the 5′-ACGC/5′-GCGT sequence must logically involve transient association with sites on the DNA highly susceptible to cleavage when they are populated. This type of behavior is exemplified by netropsin, which was demonstrated to undergo end-to-end flipping without dissociating from a DNA to which it was strongly bound.15 It may be noted that for all four hairpin DNAs containing 5′-ACGC/5′-GCGT (DNAs 2, 7, 12, and 13), the 5′-ACGC sequence was always on the 5′-arm of the hairpin and the 5′-GCGT sequence on the 3′-arm. Most of the cleavage outside this common tetranucleotide domain occurred toward the ends of the hairpin DNA, although the number of examples is insufficient to permit any firm conclusion to be drawn as to the possible directionality of the putative scanning process. Plausibly, the tight association of Fe·BLM with these hairpin DNAs as a consequence of the presence of the 5′-ACGC/5′-GCGT sequences may enable multiple double-strand cleavages of individual DNAs sufficient to render DNA repair problematic.
The 30 hairpin DNAs isolated using immobilized Fe(III)·BLM A5 for the selection procedure have sequences all of which differ from the 10 hairpin DNAs described to date.8 The new DNAs also include some that bind quite strongly to BLM. It seems likely that some of these have sequences which are analogous to 5′-ACGC/5′-GCGT, in that their presence is sufficient to confer strong BLM binding properties to DNAs in which they are present. Further, the absence of sequence duplication within any of the hairpin DNAs isolated to date suggests that there must be significantly larger numbers of sequences capable of mediating strong binding to BLM. It seems possible that some of these could prove to be more efficient than 5′-ACGC/5′-GCGT in conferring strong BLM binding properties.
Nonetheless, even in the absence of such alternative tight binding sequences it is worthy of note that the NCBI-BLAST program16 indicates that the 16-nt sequence 5′-ACGCACGCACGCACGC occurs 32 times in the human genome17 at sites within or close to protein coding regions of the genome; these could well constitute sites of enhanced cleavage by Fe·BLM. In fact, a 48-nt sequence consisting of 12 tandem repeats of ACGC occurs to the 5′-side of gene B3GAT1 (Gene ID 27087) on chromosome 11 which encodes glucuronosyltransferase P. Other long tandem repeats of ACGC, or close variants thereof, occur within the genes for the IQ domain-containing protein G (Gene ID 84223), cyclic AMP-responsive element-binding protein 5 (Gene ID 9586), ribonuclease T2 precursor (Gene ID 8635), FERM domain-containing protein 4A (Gene ID 55691), and SET-binding protein isoform (Gene ID 26040). This frequency of occurrence for tandem repeats of ACGC compared quite favorably with that found for other similar but randomly chosen tetranucleotide sequences.
The study of Fe·BLM-mediated cleavage of strongly bound hairpin DNAs has already enabled a new mechanism of double strand DNA cleavage to be identified.9 The present analysis of hairpin DNAs containing the tightly bound motif 5′-ACGC/5′-GCGT further suggests a mechanism by which multiple double-strand cleavage reactions involving a single substrate DNA could lead to the formation of gaps in a DNA structure, which would likely be quite difficult to repair. A further potential benefit to the identification of DNA motifs conducive to tight binding by Fe·BLM is related to the likelihood that such motifs are involved in tumor cell killing by bleomycin. Previous structural studies of BLM–DNA interaction carried out by NMR18 and X-ray crystallographic analysis19 have provided structural insights, but inconsistency from one study to another, underscoring the need for the use of an appropriate DNA substrate. The current findings for the 5′-ACGC/5′-GCGT motif suggest that DNAs containing this motif may constitute more relevant species for structural studies of BLM interaction.
Conclusions
A comparison of sequences common among members of two hairpin DNA libraries revealed that the motif 5′-ACGC/5′-GCGT was present in four hairpin DNAs, all of which bound Fe·BLM A5 exceptionally strongly. Newly identified hairpin DNAs 12 and 13, containing the above motif, were characterized in some detail. DNA 13, which has a KA of about 30 × 106 M–1 for Fe(III)·BLM B2, binds the drug more tightly than any DNA previously identified. In keeping with the earlier finding that DNAs which bind Fe·BLM strongly exhibit enhanced double-strand cleavage, and do so by a novel mechanism involving closely spaced DNA breaks, DNA 12 was found to undergo five double-strand DNA breaks when treated with Fe·BLM A5 and four of these occurred by the newly recognized mechanism. For DNA 13, all five double-strand breaks involved closely spaced single-strand breaks. It is interesting that tandem repeats of the motif 5′-ACGC/5′-GCGT, some up to 48 nucleotides in length, occur abundantly in the human genome and may possibly constitute cellular targets for bleomycin.
Experimental Methods
Materials
[γ-32P]ATP and [α-32P]ddATP were purchased from PerkinElmer. DNA polymerase (Klenow fragment) and restriction endonucleases AseI, ApoI and T4 polynucleotide kinase were obtained from New England Biolabs. T4 ligase was purchased from Fermentas. Recombinant terminal deoxynucleotidyl transferase was obtained from Roche. The vector pUC19, plasmid Mini kits, and competent cells DH5α were from Invitrogen. Fe(SO4)2(NH4)2·6H2O was purchased from Sigma-Aldrich and was used to prepare fresh aqueous solutions for admixture to BLM A5 immediately prior to use. Chelex 100 was purchased from Sigma-Aldrich and used to remove adventitious Fe2+ from solutions prior to experiments. Oligonucleotides were purchased from Integrated DNA Technology, Inc.
Synthesis of a 64-nt DNA Hairpin Library by Klenow Fragment
The 41-nt template containing 8-nt random sequences (2 μg/μL) was self-annealed in annealing buffer (10 mM sodium cacodylate, pH 7.0, containing 100 mM NaCl) at 75 °C for 15 min. The annealed 41-nt template (10 μg) was then treated with 12.5 μL of DNA polymerase Klenow fragment (62.5 U) and 2.1 μL of 10 mM dNTPs in 10 mM Tris-HCl, pH 7.9, containing 50 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol. The reaction mixture was incubated at 37 °C for 30 min and then heated at 75 °C for 20 min.
Preparation of Fe(III)·BLM A5
An aqueous solution containing 2.0 mg (1.4 μmol) of BLM A5 was treated with 0.4 mg (1.4 μmol) of FeCl3·6H2O, and the combined solution was stirred at room temperature for 5 h and then lyophilized to afford a solid.
Coupling of Fe(III)·BLM A5 to Sepharose 4B
Twenty-five mg of Sepharose 4B beads were added to 1.0 mL of 0.1 M sodium bicarbonate, pH 8.3, containing 2.0 mg (1.4 μmol) of Fe(III)·BLM A5, and the resulting suspension was stirred at 0–4 °C. The solution was monitored at 292 nm by ultraviolet spectroscopy. After 36 h, the coupled Fe(III)·BLM A5–Sepharose 4B was washed with 4.0 mL of 0.1 M sodium bicarbonate, pH 8.3, and subsequently washed extensively with water to remove any traces of free Fe(III)·BLM A5. The extent of bead derivatization was determined from the supernatant buffer after the coupling reaction was complete; the absorption maximum at 292 nm is characteristic for bleomycin, and the known molar absorptivity (14500 M–1cm–1) permitted calculation of the amount of bleomycin that had failed to undergo coupling. The remainder of the material was assumed to have undergone coupling.
Binding to 64-nt Hairpin DNA by Resin Bound Fe(III)·BLM A5
Resin bound Fe(III)·BLM A5 (2.0 nmol) was incubated with 1.0 nmol of 64-nt hairpin DNA in 20 μL (total volume) of 20 mM Tris-HCl buffer, pH 7.4, at room temperature for 20 min. The mixture was washed once with 20 μL of 20 mM Tris-HCl buffer. Then the hairpin DNA still bound to Fe(III)·BLM A5 was isolated from the solid support by washing with 1 M NaCl and desalted by Amicon ultracentrifugal filtration (Millipore).
Digestion of the Eluted 64-nt Hairpin DNAs with Restriction Enzymes AseI and ApoI
The mixture of hairpin DNAs eluted from resin bound Fe(III)·BLM A5 (250 ng) was digested with 5 U of restriction endonuclease AseI in 25 μL of 50 mM Tris-HCl, pH 7.9, containing 100 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol. The reaction mixture was incubated at 37 °C for 1 h and then incubated with 5 U of ApoI at 50 °C for 1 h. The enzymes were inactivated by heating the solution at 80 °C for 20 min.
Digestion of pUC19 with NdeI and EcoRI and Purification
The plasmid vector pUC19 (4.5 μg) was digested with 20 U of NdeI and EcoRI-HF at 37 °C for 3 h in 50 μL of 20 mM Tris-acetate, pH 7.9, containing 50 mM potassium acetate, 10 mM magnesium acetate, and 1 mM dithiothreitol. The incubation mixture was heated at 70 °C for 20 min to inactivate the enzymes. The digested pUC19 was isolated from a 1% agarose gel and purified using a QIA quick gel extraction kit (QIAGEN).
Ligation of the DNA Oligomers to the Digested pUC19
The AseI/ApoI digested 64-nt hairpin DNAs were incubated with the NdeI/EcoRI digested plasmid pUC19 in a 20 μL reaction mixture having 5 U of T4 DNA ligase in 50 mM Tris-HCl, pH 7.5, containing 10 mM MgCl2, 10 mM dithiothreitol, and 1 mM ATP. The reaction mixture was maintained at room temperature for 1 h.
Bacterial Transformation and Growth
Five μL (21 ng) of recombinant DNA was added to 50 μL of competent cells DH5α preparation. The incubation mixture was maintained on ice for 30 min, followed by heating at 42 °C for 20 s and cooling on ice for 3 min. The mixture was diluted with 1 mL of LB medium (10 mg/mL trypton, 5 mg/mL yeast extract, 10 mg/mL NaCl, pH 7.4) and incubated at 37 °C with shaking at 150 rpm for 1 h. The cell suspension was cultured on LB agar plates including 100 μg/mL ampicillin, 30 μg/mL X-gal, and 1 mM IPTG at 37 °C overnight. The white colonies were transferred to LB broth including ampicillin (100 μg/mL) and incubated at 37 °C overnight.
Isolation and Sequencing of Recombinant Plasmid DNA
The isolation and purification of recombinant plasmid DNA were carried out using Invitrogen plasmid Mini kits. DNA sequencing was carried out in the DNA Laboratory of the School of Life Sciences at Arizona State University. The sequences so determined enabled the identification of the 64-nt hairpin DNAs selected from the original library; these were prepared for study by chemical synthesis.
Fluorescence Inhibition Assay of 64-nt Hairpin DNAs
A solution formed by admixture of 1.2 μL of 30 μM 64-nt hairpin DNA and 0.25 μL of 144 μM BLM A5 was pre-incubated for 20 min. The solution containing the 64-nt hairpin DNA and BLM A5 was added to a 16-nt hairpin DNA-Cf15 solution prepared by addition of 1.2 μL of 30 μM hairpin DNA-Cf15 to 46.85 μL of 10 mM Na cacodylate buffer, pH 7.0, containing 100 mM NaCl. The reaction mixture was maintained at room temperature for 1 min followed by the addition of 0.5 μL of 72 μM freshly prepared aqueous Fe(NH4)2(SO4)2·6H2O. The same volume of buffer solution was added to the control sample without Fe2+ and 64-nt hairpin DNA. The final concentrations of 16-nt hairpin DNA-Cf15, 64-nt hairpin DNA, and Fe(II)·BLM A5 were all 0.72 μM (total volume 50 μL). The reaction mixture was incubated at room temperature for 30 min. The fluorescence emission was measured at 25 °C. The samples were excited at 310 nm, and the emission signal was measured from 400–550 nm using an excitation slit width of 10 nm and an emission slit width of 10 nm.
[5′-32P]-End Labeling/[3′-32P]-End Labeling and Purification of Hairpin DNAs
Hairpin DNAs were 5′-end labeled using [γ-32P]-ATP + T4 polynucleotide kinase and 3′-end labeled with [α-32P]-ATP + terminal deoxynucleotidyltransferase. Ten pmol of 64-nt hairpin DNAs was [5′-32P]-end labeled by incubation with 20 units of T4 polynucleotide kinase and 0.06 mCi [γ-32P]-ATP (specific activity 6000 Ci (222 TBq)/mmol) in 50 μL (total volume) of 70 mM Tris-HCl buffer, pH 7.6, containing 10 MgCl2 and 5 mM dithiothreitol. The reaction mixture was incubated at 37 °C for 1 h followed by purification of DNA by 16% polyacrylamide gel electrophoresis carried out at 2000 V for 2 h. The 3′-end labeling was done by incubating 10 pmol of hairpin DNA with 20 units of terminal deoxynucleotidyltransferase and 0.06 mCi [α -32P]-ATP (specific activity 6000 Ci (222 TBq)/mmol) in 50 μL (total volume) of 70 mM Tris-HCl buffer, pH 7.6, containing 10 MgCl2, 10 mM CoCl2, and 5 mM dithiothreitol. The reaction mixture was incubated at 37 °C for 1 h followed by DNA purification using 16% polyacrylamide gel electrophoresis carried out at 2000 V for 2 h.
Double-Strand Cleavage of [5′-32P]- and [3′-32P]-End Labeled Hairpin DNAs by Bleomycin A5
Fe·Bleomycin mediated cleavage of [5′-32P]- and [3′-32P]-end-labeled hairpin DNAs was carried out by incubating the hairpin DNA (∼60000–80000 cpm) with 0–1.5 μM Fe(II)·bleomycin A5 at 25 °C for 30 min in a solution of 5 μL of 10 mM Na cacodylate buffer, pH 7.0. Two μL of native gel loading buffer containing 0.25% bromophenol blue, 0.25% xylene cyanol, and 40% sucrose were added to the bleomycin reaction mixture, which was resolved on a 20% native polyacrylamide gel (260 V at 4 °C for 20 h). Double-strand cleavage sites were confirmed by visualizing gels using a phosphorimager (Molecular Dynamics). Co-migrating bands derived from an alternatively [5′-32P]- and [3′-32P]-end-labeled hairpin DNA were presumed to arise from double-strand cleavage.
Denaturing Gel Electrophoresis of DNA Cleavage Products
The [5′-32P]- and [3′-32P]-end-labeled double-strand DNA cleavage bands visualized by native gel electrophoresis were extracted from the gel. Each gel slice was cut into several pieces, placed in 0.3 mL of H2O, and incubated at 4 °C overnight to elute the DNA, which was then concentrated under diminished pressure. Each concentrated DNA solution was admixed with 5 μL of denaturing loading buffer containing 80% formamide, 2 mM EDTA, 1% bromophenol blue, and 1% xylene cyanol and heated at 90 °C for 10 min. Five μL of the final solutions were chilled on ice and resolved in a 16% denaturing polyacrylamide gel containing 7 M urea and run at 2000 V for 2.5 h. Cleavage sites were confirmed by comparison with the reaction products obtained through the Maxam–Gilbert G + A sequencing protocol. Gels were visualized using a phosphorimager (Molecular Dynamics).
Maxam–Gilbert Sequencing Reactions20
Ten μL of [5′-32P]- or [3′-32P]-end-labeled DNAs (∼20000 cpm) recovered from the native gel was treated with 25 μL of formic acid and incubated at 25 °C for 4–5 min. The reaction was stopped by treatment with 200 μL of 0.3 M NaOAc, pH 7.0, containing 0.1 mM EDTA and 25 μg/mL of tRNA. The resulting solution was mixed with 700 μL of ethanol, and the DNA was precipitated. The DNA pellet was washed twice with 70% ethanol, and the pellet was resuspended in 75 μL of 10% piperidine. The reaction mixture was incubated at 90 °C for 30 min, and the cooled supernatant was concentrated under diminished pressure. The DNA pellet was washed with small amounts of water to remove residual piperidine and mixed with denaturing loading buffer containing 80% formamide, 2 mM EDTA, 1% bromophenol blue, and 1% xylene cyanol. The combined solution was heated at 90 °C for 10 min and applied to a sequencing gel to compare [5′-32P]-end and [3′-32P]-end-labeled DNAs by denaturing polyacrylamide gel electrophoresis.
Biosensor SPR
SPR measurements were performed with a four-channel Biacore T200 optical biosensor system. Flow cell 1 was left blank, while flow cells 2–4 were immobilized with 5′-biotin-labeled DNA sequences (hairpin DNAs 7, 12 and 13).21 The SPR experiments were performed at 15 and 25 °C in degassed and filtered cacodylate buffer (10 mM cacodylate, 0.1 mM EDTA, and 10 mM NaCl, pH 7.2). Solutions of different known Fe(III)·BLM B2 concentrations were injected over the immobilized DNA surface at a flow rate of 75 μL/min until a constant steady-state response was obtained. Compound solution flow was then replaced by buffer flow resulting in dissociation of the complex. After each cycle, the sensor chip surface was regenerated with running buffer for 30 s followed by three buffer injections (each 60 s) to yield unbound DNA and a stable baseline for the following cycles. The reference response from the blank cell was subtracted from the response in each flow cell containing DNA to give a signal (RU, response units) that is directly proportional to the amount of bound compound. The predicted maximum response per bound compound in the steady-state region (RUmax) was determined from the DNA molecular weight, the amount of DNA on the flow cell, the compound molecular weight, and the refractive index gradient ratio of the compound and DNA, as previously described.22a RU was plotted as a function of free ligand concentration (Cfree), and the equilibrium binding constants were determined with a one-site binding model (K2 = 0).
| 1 |
where r represents the moles of bound compound per mole of DNA hairpin duplex, K1 and K2 are macroscopic binding constants (for a single-site model K2 = 0), and Cfree is the free compound concentration in equilibrium with the complex. RUmax in the equation was used as a fitting parameter, and the obtained value was compared to the predicted maximal response per bound ligand to evaluate the stoichiometry. Kinetic analysis was performed by globally fitting the binding results for the entire concentration series using a standard 1:1 kinetic model with integrated mass transport-limited binding parameters as described previously.21,22
Acknowledgments
This work was supported by National Institutes of Health Research Grant CA140471, awarded by the National Cancer Institute.
Supporting Information Available
High-resolution polyacrylamide gels illustrating all sites of Fe(II)·BLM A5-mediated cleavage for hairpin DNAs 12 and 13 as well as a summary of cleavage sites for hairpin DNAs 1 – 10. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Sikic B. I.; Rozencweig M.; Carter S. K.. Bleomycin Chemotherapy; Academic Press: New York, 1985. [Google Scholar]; b Levi J. A.; Raghavan D.; Harvey V.; Thompson D.; Sandeman T.; Gill G.; Stuart-Harris R.; Snyder R.; Byrne M.; Kerestes Z. J. Clin. Oncol. 1993, 11, 1300. [DOI] [PubMed] [Google Scholar]
- Hecht S. M. In Cancer Chemotherapeutic Agents; Foye W. O., Ed.; American Chemical Society: Washington, DC, 1995; p 369. [Google Scholar]
- a Lin F. T.; Li D. D.; Yang X. P.; Li Q.; Xue Y. C.; Zhen Y. S. Zhonghua Zhongliu Zazhi 1979, 1, 161. [PubMed] [Google Scholar]; b Zheng J. W.; Yang X. J.; Wang Y. A.; He Y.; Ye W. M.; Zhang Z. Y. Oral Oncol. 2009, 45, 872. [DOI] [PubMed] [Google Scholar]
- a Ishida R.; Takahashi T. Biochem. Biophys. Res. Commun. 1975, 66, 1432. [DOI] [PubMed] [Google Scholar]; b Sausville E. A.; Peisach J.; Horwitz S. B. Biochemistry 1978, 17, 2740. [DOI] [PubMed] [Google Scholar]; c Sausville E. A.; Stein R. W.; Peisach J.; Horwitz S. B. Biochemistry 1978, 17, 2746. [DOI] [PubMed] [Google Scholar]
- a Ehrenfeld G. M.; Rodriguez L. O.; Hecht S. M.; Chang C.; Basus V. J.; Oppenheimer N. J. Biochemistry 1985, 24, 81. [DOI] [PubMed] [Google Scholar]; b Ehrenfeld G. M.; Shipley J. B.; Heimbrook D. C.; Sugiyama H.; Long E. C.; van Boom J. H.; van der Marel G. A.; Oppenheimer N. J.; Hecht S. M. Biochemistry 1987, 26, 931. [DOI] [PubMed] [Google Scholar]
- a Burger R. M. Chem. Rev. 1998, 98, 1153. [DOI] [PubMed] [Google Scholar]; b Claussen C. A.; Long E. C. Chem. Rev. 1999, 99, 2797. [DOI] [PubMed] [Google Scholar]; c Hecht S. M. J. Nat. Prod. 2000, 63, 158. [DOI] [PubMed] [Google Scholar]; d Chen J.; Stubbe J. Nat. Rev. 2005, 5, 102. [DOI] [PubMed] [Google Scholar]
- Povirk L. F. In Molecular Aspects of Anti-Cancer Drug Action; Neidle S., Waring M., Eds.; Macmillan: London, 1983; Vol. 3. [Google Scholar]
- a Akiyama Y.; Ma Q.; Edgar E.; Laikhter A.; Hecht S. M. J. Am. Chem. Soc. 2008, 130, 9650. [DOI] [PubMed] [Google Scholar]; b Ma Q.; Akiyama Y.; Xu Z.; Konishi K.; Hecht S. M. J. Am. Chem. Soc. 2009, 131, 2013. [DOI] [PubMed] [Google Scholar]
- Roy B.; Hecht S. M. J. Am. Chem. Soc. 2014, 136, 4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povirk L. F.; Han Y.-H.; Steighner R. J. Biochemistry 1989, 28, 5808. [DOI] [PubMed] [Google Scholar]
- a Keller T. J.; Oppenheimer N. J. J. Biol. Chem. 1987, 262, 15144. [PubMed] [Google Scholar]; b Absalon M. J.; Kozarich J. W.; Stubbe J. Biochemistry 1995, 34, 2065. [DOI] [PubMed] [Google Scholar]; c Absalon M. J.; Wu W.; Kozarich J. W.; Stubbe J. Biochemistry 1995, 34, 2076. [DOI] [PubMed] [Google Scholar]; d Keck M. V.; Manderville R. A.; Hecht S. M. J. Am. Chem. Soc. 2001, 123, 8690. [DOI] [PubMed] [Google Scholar]
- Bozeman T. C.; Nanjunda R.; Tang C.; Liu Y.; Segerman Z. J.; Zaleski P. A.; Wilson W. D.; Hecht S. M. J. Am. Chem. Soc. 2012, 134, 17842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Povirk L. F.; Houlgrave C. W. Biochemistry 1988, 27, 3850. [DOI] [PubMed] [Google Scholar]; b Steighner R. J.; Povirk L. F. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 8350. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Povirk L. F.; Bennett R. A. O.; Wang P.; Swerdlow P. S.; Austin M. J. F. J. Mol. Biol. 1994, 243, 216. [DOI] [PubMed] [Google Scholar]; d Charles K.; Povirk L. F. Chem. Res. Toxicol. 1998, 11, 1580. [DOI] [PubMed] [Google Scholar]
- a Giroux R. A.; Hecht S. M. J. Am. Chem. Soc. 2010, 132, 16987. [DOI] [PubMed] [Google Scholar]; b Segerman Z. J.; Roy B.; Hecht S. M. Biochemistry 2013, 52, 5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig M.; Germann M. W.; Ismail M. A.; Batista-Parra A.; Munde M.; Boykin D. W.; Wilson W. D. J. Phys. Chem. B 2012, 116, 5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Madden T. L.; Schäffer A. A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. J. Nucleic Acids Res. 1997, 25, 3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wootton J. C.; Federhen S. Comput. Chem. 1993, 17, 149. [Google Scholar]; b Hancock J. M.; Armstrong J. S. Comput. Appl. Biosci. 1994, 10, 67. [DOI] [PubMed] [Google Scholar]; c Wootton J. C.; Federhen S. Methods Enzymol. 1996, 266, 554. [DOI] [PubMed] [Google Scholar]
- a Wu W.; Vanderwall D. E.; Stubbe J.; Kozarich J. W.; Turner C. J. J. Am. Chem. Soc. 1994, 116, 10843. [Google Scholar]; b Manderville R. A.; Ellena J. F.; Hecht S. M. J. Am. Chem. Soc. 1994, 116, 10851. [Google Scholar]; c Manderville R. A.; Ellena J. F.; Hecht S. M. J. Am. Chem. Soc. 1995, 117, 7891. [Google Scholar]; d Wu W.; Vanderwall D. E.; Lui S. M.; Tang X. J.; Turner C. J.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1996, 118, 1268. [Google Scholar]; e Wu W.; Vanderwall D. E.; Turner C. J.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1996, 118, 1281. [Google Scholar]; f Lui S. M.; Vanderwall D. E.; Wu W.; Tang X.-J.; Turner C. J.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1997, 119, 9603. [DOI] [PubMed] [Google Scholar]; g Vanderwall D. E.; Lui S. M.; Turner C. J.; Kozarich J. W.; Stubbe J. Chem. Biol. 1997, 4, 373. [DOI] [PubMed] [Google Scholar]; h Caceres-Cortes J.; Sugiyama H.; Ikudome K.; Saito I.; Wang A. H. J. Biochemistry 1997, 36, 9995. [DOI] [PubMed] [Google Scholar]; i Sucheck S. J.; Ellena J. F.; Hecht S. M. J. Am. Chem. Soc. 1998, 120, 7450. [Google Scholar]; j Wu W.; Vanderwall D. E.; Teramoto S.; Lui S. M.; Hoehn S. T.; Tang X.-J.; Turner C. J.; Boger D. L.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1998, 120, 2239. [Google Scholar]; k Hoehn S. T.; Junker H. D.; Bunt R. C.; Turner C. J.; Stubbe J. Biochemistry 2001, 40, 5894. [DOI] [PubMed] [Google Scholar]; l Wu W.; Vanderwall D. E.; Turner C. J.; Hoehn S.; Chen J. Y.; Kozarich J. W.; Stubbe J. Nucleic Acids Res. 2002, 30, 4881. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Zhao C. Q.; Xia C. W.; Mao Q. K.; Forsterling H.; DeRose E.; Antholine W. E.; Subczynski W. K.; Petering D. H. J. Inorg. Biochem. 2002, 91, 259. [DOI] [PubMed] [Google Scholar]
- Goodwin K. D.; Lewis M. A.; Long E. C.; Georgiadis M. M. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxam A. M.; Gilbert W. Methods Enzymol. 1980, 65, 499. [DOI] [PubMed] [Google Scholar]
- a Nguyen B.; Tanious F. A.; Wilson W. D. Methods 2007, 42, 150. [DOI] [PubMed] [Google Scholar]; b Liu Y.; Wilson W. D. Methods Mol. Biol. 2012, 613, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nanjunda R.; Munde M.; Liu Y.; Wilson W. D. In Methods for Studying DNA/Drug Interactions; Wanunu M., Tor Y., Eds.; CRC Press-Taylor & Francis Group: Boca Raton, FL, 2011; Chapter 4. [Google Scholar]; b Liu Y.; Chai Y.; Kumar A.; Tidwell R. R.; Boykin D. W.; Wilson W. D. J. Am. Chem. Soc. 2012, 134, 5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.