

Abstract
In vivo imaging of αvβ3 expression has important diagnostic and therapeutic applications. Multimeric cyclic RGD peptides are capable of improving the integrin αvβ3–binding affinity due to the polyvalency effect. Here we report an example of 18F-labeled tetrameric RGD peptide for PET of αvβ3 expression in both xenograft and spontaneous tumor models.
Methods
The tetrameric RGD peptide E{E[c(RGDyK)]2}2 was derived with amino-3,6,9-trioxaundecanoic acid (mini-PEG; PEG is poly(ethylene glycol)) linker through the glutamate α-amino group. NH2-mini-PEG-E{E[c(RGDyK)]2}2 (PRGD4) was labeled with 18F via the N-succinimidyl-4-18F-fluorobenzoate (18F-SFB) prosthetic group. The receptor-binding characteristics of the tetrameric RGD peptide tracer 18F-FPRGD4 were evaluated in vitro by a cell-binding assay and in vivo by quantitative microPET imaging studies.
Results
The decay-corrected radiochemical yield for 18F-FPRGD4 was about 15%, with a total reaction time of 180 min starting from 18F-F−. The PEGylation had minimal effect on integrin-binding affinity of the RGD peptide. 18F-FPRGD4 has significantly higher tumor uptake compared with monomeric and dimeric RGD peptide tracer analogs. The receptor specificity of 18F-FPRGD4 in vivo was confirmed by effective blocking of the uptake in both tumors and normal organs or tissues with excess c(RGDyK).
Conclusion
The tetrameric RGD peptide tracer 18F-FPRGD4 possessing high integrin-binding affinity and favorable biokinetics is a promising tracer for PET of integrin αvβ3 expression in cancer and other angiogenesis related diseases.
Keywords: microPET, integrin αvβ3, tetrameric RGD peptide, PEGylation, 18F
Integrins constitute an important family of transmembrane receptors involved in cell–cell or cell–matrix interactions and are central players in outside-in and inside-out signal transduction pathways (1). The αvβ3 heterodimer, which is not readily detectable in quiescent vessels but becomes highly expressed in angiogenic vessels and tumor cells, has been extensively studied (2). Integrin αvβ3 is necessary for the formation, survival, and maturation of newly formed blood vessels (3), and its expression correlates with tumor grade and histologic type in several cancer types, including melanoma, glioma, and ovarian and breast cancers (4–6). Thus, it would be highly advantageous to develop imaging agents that can be used to visualize and quantify integrin αvβ3 expression level, to more appropriately select patients considered for antiintegrin αvβ3 treatment, and to monitor antiintegrin treatment efficacy in αvβ3-positive patients.
Various imaging techniques—such as PET, SPECT, near-infrared fluorescence (NIRF) imaging, MRI, and ultrasound accompanied by appropriate imaging probes—have been applied to image integrin αvβ3 (7–9). For PET, Haubner et al. reported in 2001 that 18F-galacto-RGD exhibited αvβ3-specific tumor uptake in an integrin-positive M21 melanoma xenograft model (10). Further tests in humans also indicated that the intensity of 18F-galacto-RGD uptake correlates with αvβ3 expression (11–14). Recently, we and others found that dimeric and multimeric RGD peptides have significantly higher integrin affinity and, thus, significantly improved tumor targeting than the monomeric RGD analogs (15–20). For example, 18F-fluorobenzoyl-E[c(RGDyK)]2 (18F-FB-E[c(RGDyK)]2, denoted as 18F-FRGD2), exhibited higher tumor uptake and more favorable in vivo pharmacokinetics than 18F-FB-c(RGDyK) (18F-FRGD) (16,19). It was hypothesized that the receptor binding of one RGD peptide significantly enhances the “local concentration” of the other RGD peptides in the vicinity of the receptor, which may lead to a faster rate of receptor binding or a slower rate of dissociation of radiolabeled RGD dimer from the integrin αvβ3, resulting in higher uptake and longer retention time in the tumor (21). Recently, we reported that 64Cu-labeled tetrameric RGDfK peptide (64Cu-DOTA-E{E[c(RGDfK)]2}2) had significantly higher tumor uptake and slower tumor washout rate compared with 64Cu-labeled dimeric RGDfK peptide (64Cu-DOTA-E[c(RGDfK)]2) in a subcutaneous U87MG xenograft model (22). It was also found that replacing D-Phe (f) with D-Tyr (y) increased the hydrophilicity of the RGD peptides and resulted in increased integrin αvβ3–mediated tumor uptake and more favorable biokinetics in an orthotopic MDA-MB-435 breast cancer model (23). On the basis of these findings, we believe that 18F-labeled tetrameric RGDyK peptide(18F-FB-E{E[c(RGDyK)]2}2, 18F-FRGD4) might provide much higher receptor-binding affinity and tumor uptake than the corresponding dimeric and monomeric RGD peptide counterparts. However, because of the increased molecular size and spatial hindrance, direct labeling of RGD tetramer E{E[c(RGDyK)]2}2 with N-succinimidyl-4-18F-fluorobenzoate (18F-SFB) resulted in extremely low yield and, thus, was impractical for imaging applications.
In this study, we labeled PEGylated tetrameric RGD peptide NH2-mini-PEG-E{E[c(RGDyK)]2}2 (PEG is poly (ethylene glycol)) with 18F in reasonable yield and compared the tumor targeting efficacy and in vivo kinetics of the RGD tetramer with those of the RGD dimer analogs.
MATERIALS AND METHODS
All chemicals obtained commercially were of analytic grade and used without further purification. No-carrier-added 18F-F− was obtained from an in-house PETtrace cyclotron (GE Healthcare). Reversed-phase extraction C-18 Sep-Pak cartridges were obtained from Waters and were pretreated with ethanol and water before use. The syringe filter and polyethersulfone membranes (pore size, 0.22 μm; diameter, 13 mm) were obtained from Nalge Nunc International. 125I-Echistatin, labeled by the lactoperoxidase method to a specific activity of 74,000 GBq/mmol (2,000 Ci/mmol), was purchased from GE Healthcare. Analytic as well as semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC) was performed on a Dionex 680 chromatography system with a UVD 170U absorbance detector and model 105S single-channel radiation detector (Carroll & Ramsey Associates). The recorded data were processed using Chromeleon version 6.50 software. Isolation of peptides and 18F-labeled peptides was performed using a Vydac protein and peptide column (218TP510; 5 μm, 250 × 10 mm). The flow was set at 5 mL/min using a gradient system starting from 95% solvent A (0.1% trifluoroacetic acid [TFA] in water) and 5% solvent B (0.1% TFA in acetonitrile [ACN]) (0–2 min) and ramped to 35% solvent A and 65% solvent B at 32 min. The analytic HPLC was performed using the same gradient system, but with a Vydac column (218TP54, 5 μm, 250 × 4.6 mm) and a flow of 1 mL/min. The ultraviolet (UV) absorbance was monitored at 218 nm and the identification of the peptides was confirmed based on the UV spectrum acquired using a photodiode array detector.
Preparation of NH2-Mini-PEG-E{E[c(RGDyK)]2}2 (PRGD4)
The E{E[c(RGDyK)]2}2 (denoted as RGD4) was prepared from cyclic RGD dimer E[c(RGDyK)]2 according to our previously reported procedure (17). To a solution of Boc-11-amino-3,6,9-trioxaundecanoic acid (Boc-NH-mini-PEG-COOH, 40 mg, 0.13 mmol) and 20 μL N,N-diisopropylethylamine (DIPEA) in ACN was added O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TSTU, 27 mg, 0.09 mmol). The reaction mixture was stirred at room temperature for 0.5 h and then added to E{E[c(RGDyK)]2}2 (10 mg, 3.6 μmol) in N,N′-dimethylformamide (DMF). The reaction was stirred at room temperature for another 2 h and the desired product, Boc-NH-mini-PEG-E{E[c(RGDyK)]2}2, was isolated by semipreparative HPLC. The collected fractions were combined and lyophilized to give a fluffy white powder (60% yield). The Boc-group was readily removed by treating Boc-NH-mini-PEG-E{E[c(RGDyK)]2}2 with anhydrous TFA for 5 min at room temperature. The crude product was purified by HPLC. The collected fractions were combined and lyophilized to afford NH2-mini-PEG-E{E[c(RGDyK)]2}2 (denoted as PRGD4) as a white powder (90%). Analytic HPLC (retention time [Rt] = 13 min) and mass spectrometry (matrix-assisted laser desorption/ionization mass spectrometry [MALDI-TOF-MS]: m/z 3,001.0 for [MH]+ (C131H194N40O42, calculated molecular weight [MW] 3,001.1)) confirmed the identity of the purified product.
Preparation of FB-NH-Mini-PEG-E{E[c(RGDyK)]2}2 (FPRGD4)
SFB (4 mg, 16.8 μmol) and PRGD4 (2 mg, 0.67 μmol) were mixed in 0.05 M borate buffer (pH 8.5) at room temperature. After 2 h, the desired product FB-NH-mini-PEG-E{E[c(RGDyK)]2}2 (denoted as FPRGD4) was isolated by semipreparative HPLC with a 65% yield. Analytic HPLC (Rt = 15.7 min) and mass spectrometry (MALDI-TOF-MS: m/z 3,123.4 for [MH]+ (C138H197FN40O43, calculated [MW] 3,123.3) analyses confirmed product identification.
Radiochemistry
18F-SFB was synthesized according to our previously reported procedure (24). Recently, we adapted the procedure into a commercially available synthesis module (GE TRACERlab FXFN; GE Healthcare). The purified 18F-SFB was rotary evaporated to dryness, reconstituted in dimethyl sulfoxide (DMSO; 200 μL), and added to a DMSO solution of PRGD4 (300 μg, 0.1 μmol) with DIPEA (20 μL). The peptide mixture was incubated at 60°C for 30 min. After dilution with 700 μL of water with 1% TFA, the mixture was purified by semipreparative HPLC. The desired fractions containing 18F-FPRGD4 (Fig. 1) were combined and rotary evaporated to remove the solvent. 18F-FPRGD4 was then formulated in normal saline and passed through a 0.22-μm Millipore filter into a sterile multidose vial for in vivo experiments.
FIGURE 1.

(Upper) Radiosynthesis scheme of 18F-FPRGD4. (Lower) Chemical structure of 18F-FPRGD4.
Octanol–Water Partition Coefficient
Approximately 111 kBq of 18F-FPRGD4 in 500 μL of phosphate-buffered saline (PBS, pH 7.4) were added to 500 μL of octanol in an Eppendorf microcentrifuge tube. The mixture was vigorously vortexed for 1 min at room temperature. After centrifugation at 12,500 rpm for 5 min in an Eppendorf microcentrifuge (model 5415R; Brinkman), 200-μL aliquots of both layers were measured using a γ-counter (Packard Instruments). The experiment was carried out in triplicates.
Cell Line and Animal Model
Animal procedures were performed according to a protocol approved by the Stanford University Institutional Animal Care and Use Committee. The U87MG tumor model was generated by subcutaneous injection of 5 × 106 cells into the front flank of female athymic nude mice (Harlan). The MDA-MB-435 tumor model was established by orthotopic injection of 5 × 106 cells into the left mammary fat pad of female athymic nude mice. The DU145 tumor model was established by subcutaneous injection of 5 × 106 cells into the left front flank of male athymic nude mice. The mice were subjected to microPET studies when the tumor volume reached 100–300 mm3 (3–4 wk after inoculation) (20). The c-neu oncomouse (Charles River Laboratories) is a spontaneous tumor-bearing model that carries an activated c-neu oncogene driven by a mouse mammary tumor virus (MMTV) promoter (25). Transgenic mice uniformly expressing the MMTV/c-neu gene develop mammary adenocarcinomas between 4 and 8 mo postpartum that involve the entire epithelium in each gland. These mice were subjected to microPET scans at about 8 mo of age, and the tumor volume was about 300–500 mm3.
Cell Integrin Receptor-Binding Assay
In vitro integrin αvβ3–binding affinities and specificities of RGD4, PRGD4, and FPRGD4 were assessed via displacement cell-binding assays using 125I-echistatin as the integrin αvβ3-specific radioligand. Experiments were performed on U87MG human glioblastoma cells by the method previously described (17,20). The best-fit 50% inhibitory concentration (IC50) values for the U87MG cells were calculated by fitting the data with nonlinear regression using GraphPad Prism (GraphPad Software, Inc.). Experiments were performed with triplicate samples.
microPET Studies
PET scans and image analysis were performed using a micro-PET R4 rodent model scanner (Siemens Medical Solutions USA, Inc.) as previously reported (17,19). For the U87MG tumor model, mice (n = 3) were tail-vein injected with about 3.7 MBq (100 μCi) of 18F-FPRGD4 under isoflurane anesthesia and then subjected to a 30-min dynamic scan (1 × 30 s, 4 × 1 min, 1 × 1.5 min, 4 × 2 min, 1 × 2.5 min, 4 × 3 min; total of 15 frames) starting 1 min after injection. Five-minute static PET images were also acquired at 1, 2, and 3 h after injection. The images were reconstructed by 2-dimensional ordered-subsets expectation maximization (OSEM) algorithm. No attenuation or scatter correction was applied. For the receptor-blocking experiment, a U87MG tumor mouse was coinjected with 10 mg/kg mouse body weight of c(RGDyK) and 3.7 MBq of 18F-FPRGD4. The 5-min static PET scans was then acquired at 30 min and 1 h after injection. Multiple time-point static scans were also obtained for orthotopic MDA-MB-435, c-neu oncomouse, and subcutaneous DU145 tumor models after tail-vein injection with 3.7 MBq of 18F-FPRGD4.
For each microPET scan, regions of interest (ROIs) were drawn over the tumor, normal tissue, and major organs by using vendor software (ASI Pro 5.2.4.0; Siemens Medical Solutions) on decay-corrected whole-body coronal images. The maximum radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume, which were converted to counts/mL/min by using a conversion factor. Assuming a tissue density of 1 g/mL, the ROIs were converted to counts/g/min and then divided by the administered activity to obtain an imaging ROI–derived percentage injected dose per gram tissue (%ID/g).
Immunofluorescence Staining of c-neu Oncomice
Frozen tumor and organ tissue slices (5-μm thickness) were fixed with ice-cold acetone for 10 min and dried in air for 30 min. The slices were rinsed with PBS for 3 min and blocked with 10% goat serum for 30 min at room temperature. The slices were incubated with rat antimouse CD31 antibody (1:100; BD Biosciences) and hamster anti-β3 antibody (1:100; BD Biosciences) for 3 h at room temperature and then visualized with Cy3-conjugated goat antihamster and fluorescein isothiocyanate (FITC)-conjugated goat antirat secondary antibody (1:200; Jackson Immuno-Research Laboratories, Inc.).
Statistical Analysis
Quantitative data are expressed as mean ± SD. Means were compared using 1-way ANOVA and the Student t test. P values < 0.05 were considered statistically significant.
RESULTS
Chemistry and Radiochemistry
The synthesis of RGD tetramer was performed through an active ester method by coupling Boc-Glu(OSu)2 with dimeric RGD peptides followed by TFA deprotection. Boc-NH-mini-PEG-COOH was activated with TSTU/DIPEA and then conjugated with the amino group of tetrameric RGD peptide under a slightly basic condition. After TFA deprotection, PRGD4 was obtained as a fluffy white powder. The total synthesis time for 18F-SFB was about 100 min and the decay-corrected yield was 67 ± 11% (n = 10) using the modified GE synthetic module (TRACERlab FXFN). The decay-corrected radiochemical yield of 18F-FPRGD4 based on 18F-SFB was 22.0% ± 0.8% (n = 4). The radiochemical purity of 18F-FPRGD4 was >99% according to analytic HPLC. The specific radioactivity of 18F-FPRGD4 was determined to be about 100–200 TBq/mmol based on the labeling agent 18F-SFB, as the unlabeled PRGD4 was efficiently separated from the product. Starting from 18F-F−, the total synthesis time of 18F-FPRGD4, including the final HPLC purification, was about 180 min, and the overall decay-corrected yield was 15% ± 4%. In comparison, the yield of coupling E{E[c(RGDyK)]2}2 with 18F-SFB was <2% (data not shown). The octanol–water partition coefficient (logP) for 18F-FPRGD4 was −2.67 ± 0.22, which was slightly lower than 18F-FRGD2 (−2.10 ± 0.03) and 18F-FPRGD2 (−2.28 ± 0.05) (26).
In vitro Cell Integrin Receptor–Binding Assay
The receptor-binding affinity of RGD4, PRGD4, and FPRGD4 was determined by performing competitive displacement studies with 125I-echistatin. All peptides inhibited the binding of 125I-echistatin (integrin αvβ3 specific) to U87MG cells in a concentration-dependent manner. The IC50 values for RGD4, PRGD4, and FPRGD4 were 39.1 ± 5.5, 46.5 ± 5.3, and 37.7 ± 7.0 nM, respectively (n = 3) (supplemental Figs. S1–S3 are available online only at http://jnm.snmjournals.org; see supplemental Fig. S1). The comparable IC50 values of all 3 compounds suggest that insertion of the mini-PEG linker and fluorobenzoyl coupling had minimal effect on the receptor-binding affinity.
microPET of 18F-FPRGD4 on Tumor-Bearing Mice
Dynamic microPET scans were performed on the U87MG xenograft model, and selected coronal images at different time points after injection of 18F-FPRGD4 are shown in Figure 2A. The tumor was clearly visible with high contrast to contralateral background as early as 5 min after injection. Quantitation of tumor and major organ activity accumulation in microPET scans was realized by measuring ROIs encompassing the entire organ in the coronal orientation. The U87MG tumor uptake of 18F-FPRGD4 was calculated to be 9.87 ± 0.10, 7.80 ± 0.14, 6.40 ± 0.27, 5.39 ± 0.14, and 4.82 ± 0.22 %ID/g at 5, 30, 60, 120, and 180 min after injection, respectively (n = 3). The averaged time–activity curves for the U87MG tumor, liver, kidneys, heart, lung, and muscle are shown in Figure 3. 18F-FPRGD4 was cleared mainly through the kidneys. Some hepatic clearance was also observed.
FIGURE 2.

(A) Decay-corrected whole-body coronal microPET images of athymic female nude mice bearing U87MG tumor at 5, 15, 30, 60, 120, and 180 min after injection of 18F-FPRGD4 (3.7 MBq [100 μCi]). (B) Decay-corrected whole-body coronal microPET images of c-neu oncomice at 30, 60, and 150min (5-min static image) after intravenous injection of 18F-FPRGD4. (C) Decay-corrected whole-body coronal microPET images of orthotopic MDA-MB-435 tumor-bearing mouse at 30, 60, and 150 min after intravenous injection of 18F-FPRGD4. (D) Decay-corrected whole-body coronal microPET images of DU-145 tumor-bearing mouse (5-min static image) after intravenous injection of 18F-FPRGD4. (E) Coronal microPET images of a U87MG tumor-bearing mouse at 30 and 60 min after coinjection of 18F-FPRGD4 and a blocking dose of c(RGDyK). Arrows indicate tumors in all cases.
FIGURE 3.
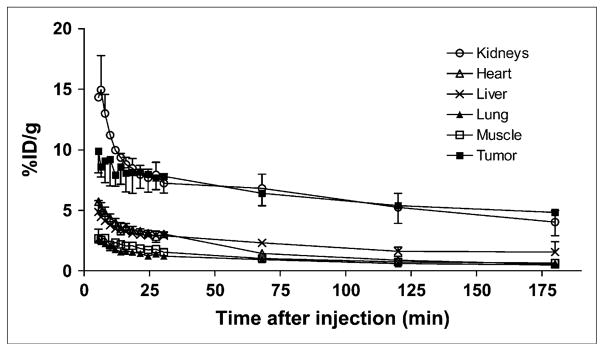
Time–activity curves of major organs after intravenous injection of 18F-FPRGD4. Data were derived from a multiple time-point microPET study. ROIs are shown as the %ID/g ± SD (n = 3).
Representative coronal microPET images of MDA-MB-435 tumor-bearing mice (n = 3) at different times after tracer injection are shown in Figure 2C. As the integrin expression level in MDA-MB-435 tumor is lower than that in U87MG, the tumor uptake of 18F-FPRGD4 in MDA-MB-435 tumor (5.07 ± 0.18, 4.53 ± 0.36, and 3.38 ± 0.48 %ID/g at 30, 60, and 150 min after injection) was also lower than that in U87MG tumor. No significant difference in normal organs and tissues was found between these 2 tumor models.
18F-FPRGD4 was also successful in visualizing a spontaneous murine mammary carcinoma model grown in c-neu oncomice (Fig. 2B) (27–30). The tumor uptakes were 4.22 ± 0.18, 3.56 ± 0.34, and 2.36 ± 0.40 %ID/g at 30, 60, and 150 min, respectively (n = 3). These values are slightly lower than those in MDA-MB-435 human breast cancer tumors grown in nude mice. No significant difference was found in major organs and tissues between the spontaneous tumor model of the BALB/c strain and the xenograft models of the nude mice strain.
We also tested 18F-FPRGD4 in an integrin-negative DU145 tumor model (n = 3). As can be seen in Figure 2D, only slightly higher signal was detected in DU145 tumor (1.44 ± 0.34 and 0.93 ± 0.13 %ID/g at 30 and 60 min after injection) than the contralateral muscle background signal. These values were significantly lower than those in all 3 other integrin-expressing tumor models (P < 0.001). The tumor uptake followed the trend of U87MG > MDA-MB-435 > c-neu > DU145 (supplemental Fig. S2), which is consistent with the integrin αvβ3 expression pattern (quantified by NaDodSO4-polyacrylamide/autoradiography) (19) in these tumor models (data not shown).
The integrin αvβ3 specificity of 18F-FPRGD4 in vivo was also confirmed by a blocking experiment. Representative coronal images of U87MG tumor mice after injection of 18F-FPRGD4 in the presence of a blocking dose of c(RGDyK) (10 mg/kg of mouse body weight) are illustrated in Figure 2E. More than 80% of the uptake in the tumor was inhibited as compared with that in the tumor without blocking (Fig. 2A). Radioactivity accumulation in most other major organs and tissues was also significantly reduced in the presence of nonradioactive RGD peptide.
The tumor uptake and biodistribution of 18F-FPRGD4 derived from quantitative microPET were compared with those of the dimeric analog 18F-FPRGD2 in the same U87MG tumor model (26). As shown in Figure 4, the uptake of 18F-FPRGD4 in U87MG tumor was significantly higher than that of 18F-FPRGD2 at all time points examined (P < 0.001). 18F-FPRGD4 also showed higher uptake than that of 18F-FPRGD2 in the liver and kidneys (P < 0.05). The initial muscle uptake of 18F-FPRGD4 was higher than that of 18F-FPRGD2 (P < 0.05), but the difference was diminished at late time points (P > 0.05).
FIGURE 4.

Comparison between uptake of 18F-FPRGD2 and 18F-FPRGD4 in U87MG tumor, kidneys, liver, and muscle over time. Data were derived from multiple time-point microPET study. ROIs are shown as %ID/g ± SD (n = 3).
Immunofluorescence Staining of c-neu Oncomice
The frozen tumor, liver, kidney, and lung tissue slices harvested from c-neu oncomice were stained for CD31 and mouse β3-integrin. As can be seen in Figure 5, β3-integrin was expressed in both tumor cells and endothelial cells of the murine mammary carcinoma, as most of the CD31-positive vessels were also β3 positive. Integrin β3 was also detected in the liver, lung, and kidneys. In particular, strong staining of integrin β3 was found in the glomerulus, which might be partially responsible for high renal uptake of 18F-FPRGD4. A similar integrin expression pattern was also seen in athymic nude mice (supplemental Fig. S3).
FIGURE 5.

Immunofluorescent staining of β3 and CD31 for tumor, liver, kidney, and lung. For β3 staining, frozen tissue slices (5-μm thick) were stained with a hamster antimouse β3 primary antibody and a Cy3-conjugated goat antihamster secondary antibody. For CD31 staining, frozen tissue slices were stained with a rat antimouse CD31 primary antibody and a FITC-conjugated goat antirat secondary antibody (×200). Arrowheads indicate overlay area in all cases.
DISCUSSION
Various radiolabeled RGD peptides have been evaluated for tumor localization and therapy (9,17,23,31). However, most of the monomeric RGD peptide-based tracers developed so far have fast blood clearance accompanied by relatively low tumor uptake and rapid tumor washout, presumably due to the suboptimal receptor-binding affinity/selectivity and inadequate contact with the binding pocket located in the extracellular segment of integrin αvβ3. The natural functional mode of integrin binding involves multivalent interactions, which could provide not only more effective binding molecules but also systems that could improve the cell targeting and promote cellular uptake. Thus, we and others have applied the polyvalency principle (15,22,31,32) to develop dimeric and multimeric RGD peptides. We have labeled c(RGDyK) and E[c(RGDyK)]2 with 18F using 18F-SFB as a prosthetic group (16,20,32). 18F-FB-RGD (18F-FRGD) had a good tumor-to-muscle ratio but rapid tumor washout and unfavorable hepatobiliary excretion, limiting its potential applications for imaging αv-integrin–positive tumors in the lower abdomen area. In contrast, the dimeric RGD peptide tracer 18F-FRGD2 had a significantly higher tumor uptake and a prolonged tumor retention compared with 18F-FRGD because of the synergistic effect of bivalency and improved pharmacokinetics (20,32). Thus, tetrameric RGD peptide tracer might be superior to the dimeric and monomeric peptide analogs due to the enhanced receptor binding caused by the polyvalency effect. However, the labeling yield of 18F-FRGD4 was not satisfactory, due in part to the bulk of the 4 cyclic penta-peptides and the prosthetic group 18F-SFB. The glutamate α-amine group has a pKa of 9.47, which is also less reactive than the ε-amino group on the lysine side chain (pKa = 8.95) usually used for 18F labeling of peptides.
To overcome the problem of low labeling yield, we wanted to insert a PEG linker between the RGD tetramer and the prosthetic 18F-labeling group. PEG moieties are inert, long-chain amphiphilic molecules produced by linking repeating units of ethylene oxide (33). PEGylation can decrease clearance, retain biologic activity, obtain a stable linkage, and enhance water solubility without significantly altering bioavailability (34). Moreover, PEG spacers are nontoxic and unreactive. PEGylation has been widely used for improving the in vivo kinetics of various pharmaceuticals (27). On the basis of previous studies (16,32), we found that PEGylated (MW 3,400) RGD peptides had lower integrin-binding affinity than non-PEGylated ones. Moreover, long-chain PEGs are mixtures of a broad range of different molecular weight compounds. Polydispersity of PEG complicates the characterization and quality control of the PEGylated compounds. In contrast, a mini-PEG spacer with definite molecular structure has been successfully used to reduce the spatial hindrance and improve the labeling yield for the dimeric RGD peptide (26). This PEGylation had minimal effect on the receptor-binding affinity, imaging quality, tumor uptake, and in vivo kinetics of dimeric RGD peptide E[c(RGDyK)]2. Thus, we decided to use this strategy to make 18F-labeled tetrameric RGD peptide. Indeed, the coupling yield between PRGD4 and 18F-SFB was >20%, whereas the same reaction between RGD4 and 18F-SFB was <2%. PRGD4 and FPRGD4 had an integrin-binding affinity similar to that of RGD4, demonstrating that mini-PEGylation had a minimal effect on the integrin affinity of this RGD tetramer.
The imaging quality of 18F-FPRGD4 was tested in a U87MG human glioblastoma xenograft model, which has been well established to have high integrin expression. Compared with 18F-FPRGD2, the tumor uptake of 18F-FPRGD4 was >50% higher at all time points in the U87MG xenograft model (Fig. 4). No significant difference was observed in the tumor wash-out rate of 18F-FPRGD4 and 18F-FPRGD2. The increased uptake of 18F-FPRGD4 than 18F-FPRGD2 in the liver and kidneys may be due to the increased molecular size and some integrin expression in these organs. Since the tumor-to-nontumor ratios are similar when compared 18F-FPRGD4 with 18F-FPRGD2, further investigation is needed to determine whether the initial high tumor uptake is attributed to the high integrin affinity of 18F-FPRGD4, or due to other factors such as enhanced circulatory half-life of the tetramer, molecular weight, and hydrophilicity. Overall, 18F-FPRGD4 had significantly higher tumor uptake than, and comparable tumor/liver and tumor/muscle ratios (P > 0.1) with 18F-FPRGD2. A similar pattern was also found for 64Cu-labeled RGD peptides (17).
In the blocking experiment, nonradioactive RGD peptide inhibited the uptake of 18F-FPRGD4 not only in U87MG tumor but also in several major organs (Fig. 2E). The biodistribution of 18F-FPRGD4 (Figs. 3 and 4) showed initial rapid clearance of activity in the liver and kidney but then reached a plateau. These phenomena suggest that some normal organs and tissues may also be integrin positive, although to a less extent, as confirmed by immunohistochemistry. Immunohistopathology showed strong positive staining of the endothelial cells of the small glomeruli vessels in the kidneys and weak staining in the branches of the hepatic portal vein. However, other studies have shown that the kidney uptake of multimeric RGD peptides could also be caused by tubular reabsorption (35). Whether the higher renal uptake and retention of 18F-FPRGD4 is integrin αvβ3-mediated is yet to be tested (36).
In this article, we inserted a mini-PEG linker to improve the labeling yield between 18F-SFB and mini-PEGylated RGD tetramer. The coupling yield of slightly higher than 20% based on 18F-SFB is still not satisfactory for routine clinical use. Furthermore, the synthesis of 18F-SFB synthon is quite time consuming. Other 18F-labeling strategies—such as click chemistry (37), reductive amination (38), Michael addition for thiol-specific coupling (20), and oxime formation (39)—may be utilized to simplify the labeling procedure and improve the labeling yield.
Although we have successfully demonstrated the specificity of 18F-FPRGD4 for high (U87MG), medium (MDA-MB-435 and c-neu), and low (DU145) integrin αvβ3–expressing tumors, we did not determine whether the tumor-to-background contrast or the binding potential derived from Logan plot of the dynamic PET scans correlates well with the integrin expression level measured ex vivo by NaDodSO4-PAGE/autoradiography or Western blot. Further characterizations of the metabolites in vivo might also provide more information about the 18F-FPRGD4. Because of the enhanced receptor binding, we found that the tetrameric RGD peptide tracer 18F-FPRGD4 showed significantly higher tumor uptake than its dimeric analog 18F-FPRGD2. However, the ratios of tumor to muscle and tumor to major organ were similar. Thereby, appropriate modification is needed to make it superior to the dimeric peptide analog 18F-FPRGD2 and the monomeric peptide analogs (18F-FRGD or 18F-galacto-RGD). By replacing the mini-PEG linker with other pharmacokinetic modifiers, we may be able to modulate the overall molecular charge, hydrophilicity, and molecular size, thus possibly improving in vivo pharmacokinetics without compromising the tumor-targeting efficacy of the resulting radioconjugates. Moreover, the cost of tetrameric RGD peptides as compared to the dimeric and monomeric analogs cannot be ignored. More careful side-by-side comparisons among 18F-FPRGD4, 18F-FRGD2, and 18F-galacto-RGD in human patients may be needed to assess the dosimetry and tumor-targeting sensitivity/specificity and, eventually, to identify the optimal RGD peptide tracer for PET imaging of integrin expression.
CONCLUSION
A new tetrameric RGD peptide tracer 18F-FPRGD4 was designed and synthesized with good yield. Because of the polyvalency effect, this tracer showed high αvβ3-integrin–binding affinity and specificity in vitro. 18F-FPRGD4 had much higher tumor uptake (6.40 ± 0.27 %ID/g at 60 min after injection) than the monomeric and dimeric RGD peptide analogs (3.80 ± 0.10 %ID/g for 18F-FRGD and 3.40 ± 0.10 %ID/g for 18F-FPRGD2 at 60 min after injection). The microPET studies performed in different tumor models suggest that 18F-FPRGD4 may have great potential as a clinical PET radiopharmaceutical for imaging tumor integrin expression. The mini-PEG spacer (11-amino-3,6,9-trioxaundecanoic acid) is a suitable chemical means to modify the tumor-targeting ability and physiologic behavior of the tetrameric RGD peptide and can improve the radio-labeling yield using 18F-SFB as a prosthetic group.
Acknowledgments
This work was supported by National Institute of Biomedical Imaging and Bioengineering (NIBIB) (grant R21 EB001785), National Cancer Institute (NCI) (grants R21 CA102123, P50 CA114747, U54 CA119367, and R24 CA93862), Department of Defense (DOD) (grants W81XWH-04-1-0697, W81XWH-06-1-0665, W81XWH-06-1-0042, and DAMD17-03-1-0143), a Deans Fellowship from Stanford University and a Benedict Cassen Postdoctoral Fellowship from the Education and Research Foundation of the Society of Nuclear Medicine. We thank Dr. David W. Dick from the cyclotron facility for 18F-F− production.
References
- 1.Castel S, Pagan R, Mitjans F, et al. RGD peptides and monoclonal antibodies, antagonists of αv-integrin, enter the cells by independent endocytic pathways. Lab Invest. 2001;81:1615–1626. doi: 10.1038/labinvest.3780375. [DOI] [PubMed] [Google Scholar]
- 2.Cheng YF, Kramer RH. Human microvascular endothelial cells express integrin-related complexes that mediate adhesion to the extracellular matrix. J Cell Physiol. 1989;139:275–286. doi: 10.1002/jcp.1041390209. [DOI] [PubMed] [Google Scholar]
- 3.Hwang R, Varner J. The role of integrins in tumor angiogenesis. Hematol Oncol Clin North Am. 2004;18:991–1006. doi: 10.1016/j.hoc.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Albelda SM, Mette SA, Elder DE, et al. Integrin distribution in malignant melanoma: association of the β3 subunit with tumor progression. Cancer Res. 1990;50:6757–6764. [PubMed] [Google Scholar]
- 5.Bello L, Francolini M, Marthyn P, et al. αvβ3 and αvβ5 integrin expression in glioma periphery. Neurosurgery. 2001;49:380–389. doi: 10.1097/00006123-200108000-00022. discussion 390. [DOI] [PubMed] [Google Scholar]
- 6.Brooks PC, Stromblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA. Antiintegrin αvβ3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest. 1995;96:1815–1822. doi: 10.1172/JCI118227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai W, Gambhir SS, Chen X. Multimodality tumor imaging targeting integrin αvβ3. Biotechniques. 2005;39:S6–S17. doi: 10.2144/000112091. [DOI] [PubMed] [Google Scholar]
- 8.Chen X. Multimodality imaging of tumor integrin αvβ3 expression. Mini Rev Med Chem. 2006;6:227–234. doi: 10.2174/138955706775475975. [DOI] [PubMed] [Google Scholar]
- 9.Liu S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol Pharm. 2006;3:472–487. doi: 10.1021/mp060049x. [DOI] [PubMed] [Google Scholar]
- 10.Haubner R, Wester HJ, Weber WA, et al. Noninvasive imaging of αvβ3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–1785. [PubMed] [Google Scholar]
- 11.Beer AJ, Haubner R, Goebel M, et al. Biodistribution and pharmacokinetics of the αvβ3-selective tracer 18F-galacto-RGD in cancer patients. J Nucl Med. 2005;46:1333–1341. [PubMed] [Google Scholar]
- 12.Beer AJ, Haubner R, Sarbia M, et al. Positron emission tomography using 18F-galacto-RGD identifies the level of integrin αvβ3 expression in man. Clin Cancer Res. 2006;12:3942–3949. doi: 10.1158/1078-0432.CCR-06-0266. [DOI] [PubMed] [Google Scholar]
- 13.Beer AJ, Haubner R, Wolf I, et al. PET-based human dosimetry of 18F-galacto-RGD, a new radiotracer for imaging αvβ3 expression. J Nucl Med. 2006;47:763–769. [PubMed] [Google Scholar]
- 14.Pichler BJ, Kneilling M, Haubner R, et al. Imaging of delayed-type hypersensitivity reaction by PET and 18F-galacto-RGD. J Nucl Med. 2005;46:184–189. [PubMed] [Google Scholar]
- 15.Thumshirn G, Hersel U, Goodman SL, Kessler H. Multimeric cyclic RGD peptides as potential tools for tumor targeting: solid-phase peptide synthesis and chemoselective oxime ligation. Chemistry (Easton) 2003;9:2717–2725. doi: 10.1002/chem.200204304. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Tohme M, Park R, Hou Y, Bading JR, Conti PS. Micro-PET imaging of αvβ3-integrin expression with 18F-labeled dimeric RGD peptide. Mol Imaging. 2004;3:96–104. doi: 10.1162/15353500200404109. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Zhang X, Xiong Z, et al. microPET imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707–1718. [PubMed] [Google Scholar]
- 18.Chen X, Liu S, Hou Y, et al. MicroPET imaging of breast cancer αv-integrin expression with 64Cu-labeled dimeric RGD peptides. Mol Imaging Biol. 2004;6:350–359. doi: 10.1016/j.mibio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Xiong Z, Wu Y, et al. Quantitative PET imaging of tumor integrin αvβ3 expression with 18F-FRGD2. J Nucl Med. 2006;47:113–121. [PMC free article] [PubMed] [Google Scholar]
- 20.Cai W, Zhang X, Wu Y, Chen X. A thiol-reactive 18F-labeling agent, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide, and synthesis of RGD peptide-based tracer for PET imaging of αvβ3 integrin expression. J Nucl Med. 2006;47:1172–1180. [PMC free article] [PubMed] [Google Scholar]
- 21.Mammen M, Chio S-K, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Z, Wu Y, Xiong Z, Gambhir SS, Chen X. Near-infrared fluorescent RGD peptides for optical imaging of integrin αvβ3 expression in living mice. Bioconjug Chem. 2005;16:1433–1441. doi: 10.1021/bc0501698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Park R, Tohme M, Shahinian AH, Bading JR, Conti PS. MicroPET and autoradiographic imaging of breast cancer αv-integrin expression using 18F- and 64Cu-labeled RGD peptide. Bioconjug Chem. 2004;15:41–49. doi: 10.1021/bc0300403. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Park R, Shahinian AH, et al. 18F-Labeled RGD peptide: initial evaluation for imaging brain tumor angiogenesis. Nucl Med Biol. 2004;31:179–189. doi: 10.1016/j.nucmedbio.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 26.Wu Z, Li Z, Cai W, et al. 18F-Labeled mini-PEG spacered RGD dimer (18F-FPRGD2): synthesis and microPET imaging of αvβ3 integrin expression. Eur J Nucl Med Mol Imaging. 2007 May 5; doi: 10.1007/s00259-007-0427-0. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris TD, Kalogeropoulos S, Nguyen T, et al. Design, synthesis, and evaluation of radiolabeled integrin αvβ3 receptor antagonists for tumor imaging and radiotherapy. Cancer Biother Radiopharm. 2003;18:627–641. doi: 10.1089/108497803322287727. [DOI] [PubMed] [Google Scholar]
- 28.Harris TD, Kalogeropoulos S, Nguyen T, et al. Structure-activity relationships of 111In- and 99mTc-labeled quinolin-4-one peptidomimetics as ligands for the vitronectin receptor: potential tumor imaging agents. Bioconjug Chem. 2006;17:1294–1313. doi: 10.1021/bc060063s. [DOI] [PubMed] [Google Scholar]
- 29.Onthank DC, Liu S, Silva PJ, et al. 90Y and 111In complexes of a DOTA-conjugated integrin αvβ3 receptor antagonist: different but biologically equivalent. Bioconjug Chem. 2004;15:235–241. doi: 10.1021/bc034108q. [DOI] [PubMed] [Google Scholar]
- 30.Mousa SA, Mohamed S. Human αvβ3 integrin potency and specificity of TA138 and its DOTA conjugated form 89Y-TA138. J Cardiovasc Pharmacol. 2005;45:109–113. doi: 10.1097/01.fjc.0000151927.89154.9a. [DOI] [PubMed] [Google Scholar]
- 31.Janssen M, Oyen WJ, Massuger LF, et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm. 2002;17:641–646. doi: 10.1089/108497802320970244. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Hou Y, Tohme M, et al. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor αvβ3-integrin expression. J Nucl Med. 2004;45:1776–1783. [PubMed] [Google Scholar]
- 33.Harris JM, Martin NE, Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40:539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 34.Walsh S, Shah A, Mond J. Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob Agents Chemother. 2003;47:554–558. doi: 10.1128/AAC.47.2.554-558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behr TM, Goldenberg DM, Becker W. Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: present status, future prospects and limitations. Eur J Nucl Med. 1998;25:201–212. doi: 10.1007/s002590050216. [DOI] [PubMed] [Google Scholar]
- 36.Waldner C, Heise G, Meyer-Kirchrath J, Schror K, Grabensee B, Heering P. Selective cyclooxygenase-2 inhibition upregulates renal cortical alpha V integrin expression. Nephron Exp Nephrol. 2003;93:e72–e79. doi: 10.1159/000068519. [DOI] [PubMed] [Google Scholar]
- 37.Marik J, Sutcliffe JL. Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006;47:6881–6884. [Google Scholar]
- 38.Damhaut P, Cantineau R, Lemaire C, Plenevaux A, Christiaens L, Guillaume M. 2- and 4-[18F]fluorotropapride, two specific D2 receptor ligands for positron emission tomography: N.C.A. syntheses and animal studies. Int J Rad Appl Instrum [A] 1992;43:1265–1274. doi: 10.1016/0883-2889(92)90205-s. [DOI] [PubMed] [Google Scholar]
- 39.Poethko T, Schottelius M, Thumshirn G, et al. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J Nucl Med. 2004;45:892–902. [PubMed] [Google Scholar]