

Abstract
Complex functional films containing enzymes and other biomolecules are easily fabricated in nm-scale thicknesses by using layer-by-layer (LbL) methodologies first popularized by Lvov and Decher. In this review, we highlight the high level functional capabilities possible with LbL films of biomolecules based on our own research experiences. We first describe the basics of enzyme film fabrication by LbL alternate electrostatic adsorption, then discuss how to make functional enzyme-polyion films of remarkably high stability. Focusing on cytochrome P450s, we discuss films developed to electrochemically activate the natural catalytic cycle of these key metabolic enzymes. We then describe multifunctional, multicomponent DNA/enzyme/polyion films on arrays and particle surfaces for high throughput metabolic toxicity screening using electrochemiluminescence and LC-MS/MS. Using multicomponent LbL films, complex functionality for bioanalytical and biochemical purposes can be achieved that is difficult or impossible using conventional approaches.
1. Introduction
This review focuses on the fabrication, characterization and use of ultrathin multicomponent films constructed layer-by-layer (LbL) containing enzymes and nucleic acids that are capable of complex functionality. Examples include (1) enzyme films on electrodes and nanoparticles that can be used in biosensors or for chemical syntheses, (2) arrays featuring LbL films of metabolic enzymes and DNA designed for toxicity screening of chemicals, and (3) magnetic beads and nanoparticles coated with enzymes and DNA for metabolic profiling and elucidating chemical pathways of toxicity-related DNA damage.
In the late 1990s, John Schenkman, Yuri Lvov and I were investigating fundamental electrochemical properties and biocatalysis of human cytochrome (cyt) P450s, peroxidases and other heme enzymes in thin films. We developed ultrathin LbL polyion films and these redox enzymes by alternate electrostatic adsorption on electrodes for voltammetric studies and on fused silica for spectroscopy.i–v We also found ways to stabilize enzyme films to enable biocatalysis at high temperatures.vi This research culminated in our development with Sadagopan Krishnan of the first cyt P450 films to enable electrochemical activation of the natural catalytic cycle of this important class of oxidative metabolic enzymes.vii,viii
Once we were able to achieve efficient, functional metabolic reactions in the thin enzyme-polyion films,i,iii–v,ix we targeted molecular-based toxicity screening methodologies in which metabolites could be generated in the thin films and reactivities of the metabolites for DNA damage were monitored. Our aim was to develop devices and methods for metabolic toxicity screening.lii,x We based nearly all of our approaches on multicomponent LbL films of metabolic enzymes, DNA and polyions, and measured DNA damage as an analytical endpoint. Enzymes in these multicomponent LbL films first convert test molecules to their metabolites in a virtual sea of DNA, so that if the metabolites can possibly react with DNA they will do so. When molecules or their metabolites damage DNA, they are usually called genotoxic molecules. Alternative toxicity prediction methods involve novel in vitro bioassays for toxicity assessment,xi–xii,xiii but provide little or no insight into genotoxic chemical pathways.
In the sections below, we describe our research in these endeavors in a tutorial format with an emphasis on fabrication issues and the complex film functionality that can be derived. This report complements recent reviews focused more on bioanalytical aspects of our metabolic toxicity screening approaches.iii–v,xiv Rather than providing a catalog of what has been done in the past, we highlight the functional capabilities of the LbL approach. In the next section, we describe the basics of LbL film fabrication by alternate electrostatic adsorption. This leads to section 3, in which we discuss enzyme-polyion LbL film fabrication, characterization, and stability and provide a few illustrative examples. In section 4, we described multifunctional DNA/enzyme/polyion films on arrays and particle surfaces. We then discuss examples of these films in high throughput metabolic toxicity screening using electrochemical and electrochemiluminescent detection. We also describe high throughput bioreactors using DNA/enzyme films on magnetic beads to produce DNA adducts for LC-MS/MS analysis. In the concluding section, we summarize key features and progress in LbL films, and discuss perspectives for the future.
2. Basic film fabrication methodology
The thin films discussed in this article employ simple but versatile alternate electrostatic layer-by-layer (LbL) film assembly to prepare the necessary multicomponent films of enzymes and other polyions. This method was developed and elaborated by Lvov and Decher,xv–xxi and provides excellent control of film thickness for versatile architectures on the nm scale. The number of components in the film can in principle be the same as the number of individual layers, providing broad opportunities for multicomponent, multifunctional films. Examples include films for sequential enzyme reactions, or for making enzyme products that subsequently react with DNA.
Fabrication by the LbL method begins by adsorbing an initial layer of charged polyion from solution onto an oppositely charged solid surface. Loosely bound polyions are removed by washing with water, then a second layer of polyions of opposite charge to the first is adsorbed. For example, the first layer can be a polycation like poly(diallyldimethylamine) (PDDA, Scheme 1), and the second layer can be the polyanion PSS (Figure 1). The sequence of layer adsorption is repeated as many times and with as many components as desired, changing the sign of the polyion, and consequently the charge on the outer surface, at every successive adsorption step. DNA and enzymes are soluble polyions and are thus perfectly suited for this method. The method is readily amenable for fabricating films containing several different enzymes,xxii or synthetic polyions, DNA and enzymes (Figure 1).i,iii,xxiii Some polyions that have been used to make LbL films are shown in Scheme 1; inorganic nanoparticles can also be used to make film layers.
Scheme 1.

Polyions used to make films by layer-by-layer (LbL) alternate electrostatic adsorption
Figure 1.

Illustration of LbL enzyme-polyion film fabrication on a negatively-charged surface
LbL enzyme film fabrication on a negatively charged electrode is illustrated in Figure 1. The negative charge can be established by oxidation of a carbon surface, by treating a metal oxide surface with base, or by attaching an organothiol layer terminating in sulfonate or carboxylate onto gold. While many conventional LbL approaches use immersion of the surface in the adsorbate solutions, we have mainly used adsorption from single drops of solution on electrodes to conserve valuable biomolecules. Adsorbate solution drops of 1–20 μL can be used, placing the electrode in a humidified chamber to retard evaporation and improve reproducibility. Hydrophobic barriers or wells can also be used to locate the drops, and the resulting LbL film, in specific regions of a flat surface.xxiv
Back to Figure 1, a drop of a 1–3 mg mL−1 positively charged polyion solution is placed onto the electrode. At this concentration, the polycations adsorb on oppositely charged surfaces at steady state in about 15–20 min,xviii,xix effectively reversing the charge on the outer surface. In some cases, better enzyme loading is obtained by using several initial layers of oppositely charged polyions to form a more uniform film.ii After rinsing with water, a drop of 1–3 mg mL−1 negatively charged enzyme is placed on the surface. Again 15–20 min. adsorption times are optimal, unless lower concentrations of the enzyme must be used, in which case adsorption time can be increased to 30–60 min. A negative surface charge on the enzyme is ensured by using a buffer of pH larger than its isoelectric point. After enzyme is adsorbed, the outer surface of the electrode develops a negative charge. This surface charge reversal process should be maintained for each layer of the film adsorbed, with extensive washing to remove loosely bound materials. In this way, the method automatically selects the strongest interactions, usually electrostatic, to form a very stable multilayer film. Alternate polyion and enzyme adsorption steps can now be repeated as many times as desired to obtain the final multilayer film.
When starting with an initial positively charge surface, the procedure would begin with adsorption of an initial polyanion layer, and the enzyme needs to be positively-charged. Several types of experiments have shown that the interior charge of such films is balanced by co-operative electrostatic interactions of the oppositely charged polyions, and the exterior charge depends on the charge of the polyion adsorbed last.xviii,xix
Film fabrication should be monitored at each step using quartz crystal microbalance (QCM) weighing, surface plasmon resonance, spectroscopy, voltammetry, or other suitable methods. Such quality control is essential, especially when designing new films, since in individual cases conditions may need to be fine tuned to achieve successful adsorption at each step. While attractive interactions between the outer surface layers and oppositely charged polyions the polyion in solution are key driving forces for layer formation, these attractive interactions need to be properly balanced. For example, if too large an adsorbate concentration is used, the adsorbate solution can strip off oppositely charged polyions from the film rather than deposit a new layer.
QCM is a valuable quality control tool that can provide mass of each layer deposited, the nominal film thickness, and reveal undesirable events such as layer stripping. QCM resonator frequency decreases with increasing mass on its metal coating with ±1 ng sensitivity, provided that the viscoelasticity of the interface does not change.xxv We typically coat gold QCM electrodes with a monolayer of mercaptopropylsulfonate (MPS), mercaptopropylcarboxylate (MPC), or a mixture of MPC and mercaptopropyl alcohol to provide a uniformed charged underlayer for LbL film fabrication. Estimates of weight of each layer and reproducibility are obtained from QCM films dried in a stream of nitrogen to remove water and minimize bias from interfacial viscoelasticity changes. For 9 MHz quartz resonators, the QCM frequency shift ΔF (Hz) gives the mass per unit area M/A (g cm−2) of a film layer according to
| (1) |
where A is area of the metal disk on the quartz resonator in cm2. Nominal film thickness (d) estimated for a film formed on both sides of the resonator is:
| (2) |
For films formed on only on resonator side, the expression on the right should be multiplied by 2. This expression was verified by comparing films made from a variety of proteins and polyions with cross-sectional scanning electron microscopy images.xxvi
QCM monitoring is illustrated for a multilayer film of the protein myoglobin (Mb) and DNA on an MPS-Au resonator (Figure 2). Frequency decreased linearly and reproducibly throughout multiple adsorption steps, characteristic of formation of a reproducible, stable film. The decrease in ΔF correspond to increases of 840 ng cm−2 for myoglobin (Mb) layers and 370 ng cm−2 for DNA layers,i allowing total amounts of protein and DNA to be estimated for films with specific numbers of bilayers. The nominal thickness of the 18-layer film is 95 nm.
Figure 2.

QCM frequency changes for formation of a DNA-Mb film. Film were dried to constant weight in a stream of nitrogen before each measurement. Reproduced with permission from ref. 1, American Chemical Society, copyright 1998.
The thinnest layers, as thin as 0.5 nm for PDDA and PSS, are achieved using polyion adsorbate solutions in pure water. When polyions in water are used, the first polyion layer of an LbL film generally forms as islands covering a large fraction of the surface, and adsorption of the second and third polyion layers fills in the spaces to produce smooth film surfaces after three layers.ii,xviii,xix As salt content of the polyion solution is increased, e.g. using 0.5–1.0 M NaCl, the polyions tend to coil and thicker, more uniform individual layers can be deposited.xviii,xix,xxvii Although a purely electrostatic approach often leads to the strongest interactions and most stable films, other interactions such as hydrogen bonding have also been used to make LbL films.xix,xx
Polyions such as proteins can often have multiply charged regions on different sides of their 3-dimensional structures. These charged sides tend to orient downward onto oppositely charged layers in the LbL method, but still lead to stable films.ii The general view is that the diffusion adsorption itself is quite fast, on the time scale of seconds, but longer adsorption times (e.g. 15–30 min) are needed to achieve steady state coverage due to additional equilibration and molecular rearrangements of the polyions on the surface. Typically, enzyme-polyion films made by the LbL method are stable in up to 3 M salt solutions, but can be destroyed by detergent solutions or microemulsions. These films are permeable to small molecule reactants and ions as long as they are kept sufficiently thin.xviii,xix
While the LbL method features adsorption of one layer at a time, nearest neighbor protein and polyion layers in the final enzyme-polyion film are intimately mixed (bottom, Figure 1). Neutron reflectivity of polycation/polyanion and PSS/myoglobin (Mb) films on the smooth silicon surfaces confirmed neighbor-layer mixing. These studies showed that up to three adjacent layers can show considerable mixing, whereas polyions three or more layers are not mixed with far distant layers but only with their nearest neighbors.xviii,xix,xxvii Most enzymes retain excellent activity and near native conformations in these films.i,iv,v,xviii–xxii Some polyions that we have used to make layer-by-layer enzyme films are shown in Scheme 1.
3. Films for direct enzyme activation and biosynthesis
Lvov, Ariga, Ichinose, and Kunitake pioneered thin LbL films of proteins, enzymes and polyions on solid surfaces.xxvi This group investigated numerous enzymes in LbL films and even designed multiple enzyme bioreactors based on LbL films capable of sequential enzyme reactions.xxii In nearly all cases, the enzymes retain their catalytic activity, and the films are stable and remain active for up to 3 months when stored a 5 °C.xviii,xix Additional reviews covering this topic have also appeared.iv,v,xx,xxi
After Yuri Lvov joined us in the late 1990s, we reported the first application of LBL films for enzyme electrochemistry using Mb and cyt P450cam.i Successful voltammetry required a monolayer undercoating of MPS on gold to facilitate direct electron exchange with the enzymes. MPS also provides a uniform negative surface to adsorb the first layer of polycation. The FeIII/FeII heme redox couples of cyt P450cam and Mb in these films had chemically reversible CVs.
Below we focus on examples from our own laboratories that illustrate performance and stability of LbL films containing enzymes. First, direct electron transfer for many redox protein and enzyme films on electrodes can be achieved,iv,v,xxviii but using common polyions only a fraction of the enzyme molecules in the film are electroactive. The fraction of electroactive proteins can be measured by comparing total amounts of protein in the film from QCM with electroactive enzyme from voltammetry. Multilayer LbL films containing cyt P450cam or Mb on smooth vapor-deposited gold electrodes had only ~1.5 electroactive layers.i However, the number of electroactive enzyme layers can be greatly increased by using mechanically roughened PG electrodes, which most likely provide disorder-inducing templates that enhance electron transport by “electron hopping” between individual protein redox sites.xxix Examples are shown in Figure 3 for iron heme-protein/polyion films. Figure 3A shows uncorrected cyclic voltammetry (CV) for (cyt P450cam/PEI)n films with different numbers of bilayers (n).xxx Chemically reversible, symmetric CV peaks were found in which all the electrochemically active FeIII cyt P450 is reduced on the forward scan, and FeII cyt P450 is oxidized on the backward scan. Integrating the forward scan CV provides the charge (Q) for one-electron reduction of the FeIII protein. The amount of electroactive protein per unit electrode area (ΓT) is obtained from:iv
Figure 3.
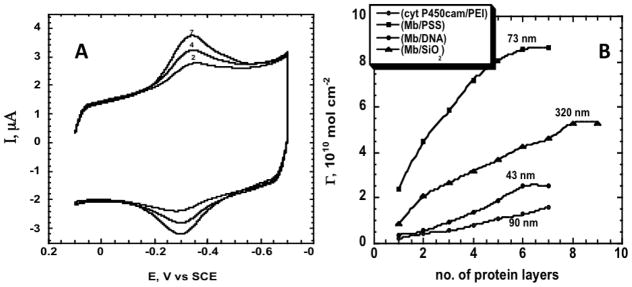
Protein-polyion films on rough PG electrodes in anaerobic pH 5.5 buffer, 50 mM acetate + 100 mM NaCl. (A) Cyclic voltammograms (CV) at 0.3 V s−1 for (cyt P450cam/PEI)n; numbers on curves denote n. (B) Influence of number of protein layers (n) for (Mb/PSS)n (ν), (Mb/SiO2)n (σ), (Mb/DNA)n (λ) and (cyt P450cam/PEI)n (O) films on amount of electroactive protein (Γ) measured by integration of CVs. Numbers near points are film thicknesses estimated from QCM for the given n. Reproduced with permission from ref. 30, ChemPubSoc, Europe, copyright 2003.
| (3) |
where F is Faraday’s constant and A is electrode area. Typical of many enzyme LbL films on rough PG, ΓT increased with the number of protein/polyion bilayers (Figure 3B) with maxima at n = 6 or 7 (Figure 3B). For (Mb/SiO2)n films made with 45 nm silica nanoparticle interlayers, ΓT increased up to n = 8, representing electron transport across 320 nm of the protein-nanoparticle film. For all films, repeated CV scans were reproducible and had good stability for month or more at 4 °C.xxx At maximum ΓT only 30% of cyt P450cam in these films was electroactive, but up to 70% of Mb was electroactive depending on film composition. Using peroxide-initiated catalytic epoxidation of styrene by these enzymes as a test reaction, we found that 12–25 nm films gave the largest catalytic turnover rates, while thicker films exhibit in-film reactant transport limitations.xxx
Nearly complete protein electroactivity can be achieved using a conductive polyion in the film to wire the protein to the electrode.xxxi Stable, fully electroactive films were grown on rough PG with 4 nm underlayers of sulfonated polyaniline (SPAN) covered with a film containing myoglobin (Mb) or horseradish peroxidase (HRP) layers alternated with PSS. Comparisons of films with and without this SPAN underlayer showed that >90% of the protein was electroactive with the SPAN underlayer but only 40% was electroactive when SPAN was absent. The PDDA/SPAN/(enzyme/PSS)3 films exhibited higher sensitivity for electrochemical catalytic reduction of hydrogen peroxide than the films with no SPAN, with an amperometric detection limit of ~3 nM.xxxi
Increasing enzyme stability
Crosslinking protein-polyion films can provide remarkable stability. Cross-linking myoglobin promoted by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide (EDC) after LBL film fabrication greatly improved adhesion to fused silica slides to enable optical spectroscopy measurements. Circular dichroism and visible absorbance spectra confirmed that Mb was in a near-native conformation in polyion films at pH values as low as 2 and up to 11.xxxii At pH 1, Mb in solution is fully unfolded (and partly unfolded at pH 4.5), but it retained considerable characteristic α-helix structure in the crosslinked films at pH 1. Full secondary structure of these acid-treated films was restored by increasing external solution pH to 7.5. The Mb LbL films at pH 1 had better catalytic activity for bioelectronic reduction of trichloroacetic acid than at pH 7.xxxii
Stability of enzyme-polyion films was further improved by covalently linking a poly(L-lysine) (PLL) underlayer to oxidized PG surfaces, then crosslinking subsequent enzyme and PLL layers after deposition using EDC. This approach provided remarkable thermostability and good activity for LbL films containing peroxidases and myoglobin, enabling biocatalysis at 90 °C.xxxiii It allowed enzymes to be used for biocatalysis at temperatures at which they would be rapidly destroyed in solution. Crosslinked LbL films were also prepared on fused silica treated with aminoethyltriethoxysilane for optical spectroscopy. Here, amino groups on the derivatized silica were reacted with polyacrylic acid, and then the PLL underlayer was attached, all using EDC chemistry. Circular dichroism (CD), absorbance spectra, and voltammetry showed that these crosslinked enzyme-PLL films were stable for 9 hr at 90 °C, but enzymes in solution were destroyed completely within 20 min.
CD spectra for Mb, horseradish peroxidase (HRP) and soybean peroxidase (SBP) at 25 °C in neutral buffer feature double minima at 210 and 222 nm characteristic of a high fraction of α-helix in their secondary structures.xxxiv Crosslinked protein-PLL films also show these characteristic minima at 25 °C (Figure 4a,b), and retain these characteristic spectral features after 4 hr at 90 °C, demonstrating remarkable stability.xxxiii Crosslinked films of SBP, a temperature resistant peroxidase, gave similar CD spectra at 90 °C. A control hemin-PLL film gave a featureless CD spectrum characteristic of random coil PLL with no dichroism. For the enzymes dissolved in solution, characteristic minima at 210 and 222 nm collapsed to nearly featureless spectra in <20 min at 90 °C (Figure 4c). These results suggest nearly full retention of enzyme secondary structure in the crosslinked LbL films at 90 °C.
Figure 4.
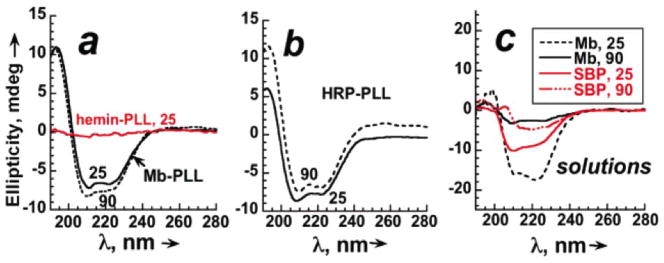
Circular dichroism spectra for crosslinked protein-PLL film on aminosilyl fused silica slides in pH 5.5 acetate buffer + 0.1 M NaCl at 25 °C and after 4 hr at 90 °C for (a) Mb-PLL, and hemin-PLL at 25 °C; (b) HRP-PLL, and (c) 5.5 mM of each protein dissolved in pH 5.5 buffer + 0.1 M NaCl at 25 °C and after 20 min. at 90 °C. Reproduced with permission from ref. 6, American Chemical Society, copyright 2007.
Stability of the enzyme-PLL films linked to PG electrodes at 90 °C was monitored by voltammetric reduction of their FeIIIheme cofactors (Figure 5A). Mb-PLL and SBP-PLL films gave nearly constant peak current for 9 hr, with only a slight decrease at 12 hr. HRP-PLL films gave constant peak current for 5 hr at 90 °C, but fell to 55% of initial in 12 hr. Hemin-PLL had constant peak current for only 3 hr at 90 °C, and peak currents decreased to <50% of initial in 12 hr. Under these same conditions, the enzymes in solution were destroyed in <20 min. Thus, Mb and SBP gave the most stable films, HRP was next, and behavior of hemin (as in CD, Figure 4a) was very different from the enzyme films.
Figure 5.
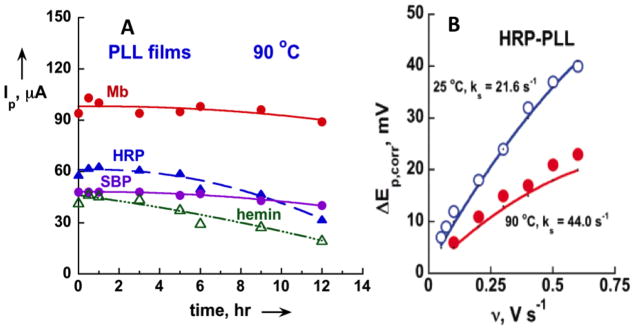
Voltammetric results for covalently crosslinked LbL films of iron heme enzymes and PLL on PG electrodes in anaerobic pH 6.5 buffer + 0.1 M NaCl: (A) Stability at 90 °C monitored by square wave voltammetry peak height; (B) kinetic analysis of corrected CV peak separation vs. scan rate (ν) data at for HRP-PLL films at 25 and 90 °C, pH 6. Points are experimental data; solid lines calculated from Butler-Volmer theory. Reproduced with permission from ref. 6, American Chemical Society, copyright 2007.
The crosslinked enzyme films enabled electron transfer kinetic analysis at high temperature using CV. First, the constant CV reduction-oxidation peak separation (ΔEp) at very low scan rates was subtracted from ΔEp at higher scan rates to correct for non-kinetic influences.xxxv Kinetic analysis depends on observing increases in these corrected ΔEp with scan rate, which were fit to the Butler-Volmer surface electron transfer model.xxxvi These data gave good fits to theory (Figures 5B), providing surface electron transfer rate constants ks for the enzyme FeIII/FeII redox couples that were typically two-fold larger at 90 °C compared to 25 °C.xxxiii The hemin-PLL control was again out of line with the enzyme films, giving ks values 2-fold smaller than the enzymes at both temperatures. In general, all results on control hemin-PLL films differed significantly from those of enzyme-PLL films, indicating that film properties cannot be due to free hemin released from the enzymes into the films, but must be due to the intact enzymes.
Crosslinked enzyme-PLL films were used for biocatalytic reduction of t-butylperoxide (t-BuOOH) using hydrodynamic voltammetry to obtain reaction kinetics. Results showed that horseradish peroxidase (HRP)-PLL was 3-fold more reactive than soybean peroxidase (SBP)-PLL at 25 °C, but SBP-PLL is slightly more reactive at 90 °C. SBP-PLL films were 8-fold more active at 90 °C compared to 25°C.xxxiii
Crosslinked enzyme-PLL films were also stable at 90 °C when prepared on silica nanoparticles. Peroxide driven o-methoxyphenol oxidation by peroxidase-PLL-coated onto 500 nm silica beads gave better yields at 90 °C than 25 °C, suggesting increased catalytic efficiency and selectivity at the higher temperature. These biocolloids were reusable with little loss of activity at 90 °C.xxxiii The crosslinked films were also stable and catalytically active when used in oil-in-water microemulsions,xxxvii whereas without crosslinking LbL films are unstable in surfactant media.
Human cyt P450s
The cytochrome (cyt) P450s are an important human enzyme superfamily controlling the majority of oxidative metabolism in the human liver and other organs (Figure 6). Like Mb and bacterial cyt P450cam, they are iron heme enzymes. Cyt P450s catalyze key oxidations of parent compounds in metabolic processes.xxxviii–xl Such metabolic bioactivation is a major source of toxicity, and can result in metabolites that damage DNA, proteins, and other biomolecules.xli–xliv
Figure 6.
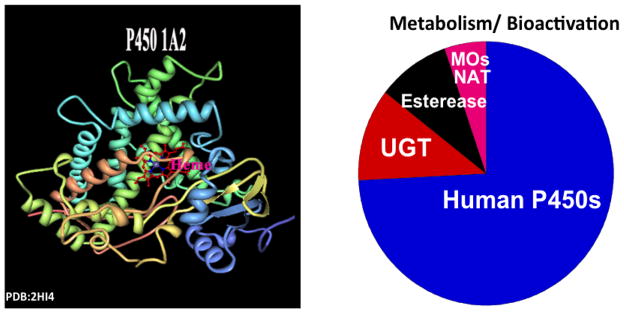
Ribbon structure of human cyt P450 1A2 (left) and the role of the cyt P450 superfamily in human metabolism (right).
We used conventional LbL films of three cyt P450s enzymes to show that heterogeneous electron transfer rate constants (ks) for electrochemical reduction depended on heme iron spin state, varying in the order low spin cytP450cam > mixed spin human cyt P450 2E1 > cyt P450 1A2.xlv Simulation analyses were consistent with faster oxidations than reductions, suggesting a square scheme in which oxidized and reduced forms of the enzymes each participate in conformational equilibria.
In our bodies, cyt P450s utilize oxygen and electrons delivered to the enzyme iron heme cofactor from NADPH by cyt P450 reductase (CPR) to oxidize foreign chemicals.xxxviii–xl However, biocatalysis starting with direct electrochemical reduction of cyt P450s alone in LBL films on electrodes utilizes enzyme-catalyzed reduction of oxygen to hydrogen peroxide that reacts with cyt P450FeIII to form the active ferryloxy species that oxidizes substrates.viii,ix,xxx,xlvi,xlvii Attempts to utilize LbL films of genetically engineered microsomes containing cyt P450s 1A2 and 3A4 and CPR on electrodes showed that electrons entered the films via CPR.xlviii Catalytic turnover of microsomal cyt P450 3A4/CPR for testosterone conversion in films on special hydrophobic electrodes was also reported by Mie et al.xlix They attributed catalysis to electron transfer from electrode to CPR, then to cyt P450 3A4, but a mixed catalytic pathway may interfere as direct electron transfer to cyt P450 3A4 was also found.
The above studies suggested a possible electrochemical pathway to the natural cyt P450 catalytic cycle. We made LbL films using CPR microsomes and pure cyt P450s that achieved a large ratio of cyt P450 to CPR (Figure 7), as exists in the human liver, to mimics the natural biocatalytic pathway of cyt P450s.vii Enzyme activation begins with initial electron injection from the electrode to CPR, which then transfers electrons to cyt P450 to combine with oxygen.viii When cyt P450s were chemically reduced to the FeII form and reacted with CO, cyt P450 FeII-CO spectral bands at 450 nm were found, confirming that native enzymes were present in the films. These microsomal CPR/cyt P450 films had midpoint (formal) potentials from CV near −490 mV vs SCE (−250 mV vs NHE) (Figure 8A). CV potentials did not shift when CO was added, and ks values were near 40 s−1, consistent with the properties of CPR. With only cyt P450s in the films and no CPR, midpoint potentials were near −340 vs SCE (−100 mV vs NHE) (Figure 8B), ~40 mV shifts were found with added CO, and ks was 2–18 s−1.vii These results are all consistent with the pathway electrons→CPR→cyt P450.vii
Figure 7.
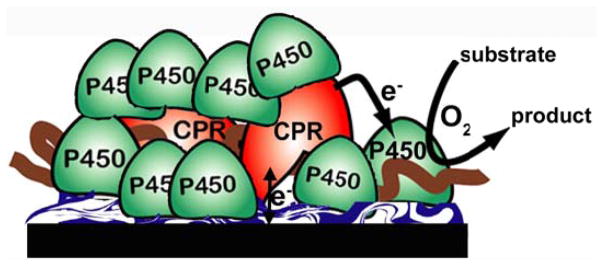
CPR/Cyt P450 films on electrodes. LbL films were assembled on a polyion underlayer on PG disk electrodes and featured six bilayers of purified human cyt P450 (1A2 or 2E1) and microsomal CPR.
Figure 8.

Experimental and simulated CVs for films cyt P450 1A2/CPR on PG electrodes. (A) Background subtracted CVs of {a} CPR films at 0.3 V s−1 with no cyt P450, and cyt P450 1A2/CPR films at scan rates {b} 0.1, {c} 0.2, and {d} 0.3 V s−1. (B) Background subtracted CVs: {a} polyion film, and cyt P450 1A2 film with no CPR at {b} 0.1 and {c} 0.2 V s−1. (C) Digitally simulated theoretical CVs corresponding to {a} reversible electron transfer for only CPR film at 0.3 Vs−1, and {b to d} the ErCEo-model using parameters in Scheme 2 for cyt P450 1A2/CPR films at scan rates {b} 0.1, {c} 0.2, and {d} 0.3 V s−1 showing excellent agreement with experimental CVs in Figure 8A. (D) Influence of scan rate on oxidation (blue circles) and reduction (red diamonds) peak potentials for cyt P450 1A2/CPR films with theoretical peak potentials (lines) simulated using the ErCEo model. Reproduced with permission from ref. 7, American Chemical Society, copyright 2011.
Reduction and oxidation peak currents were much larger for CPR/cyt P450 film CVs (Figure 8A) compared to CPR alone. This is explained by Scheme 2, in which CPR is reduced by the electrode but cyt P450s are not (eq 4). CPRred participates in a redox equilibrium with cyt P450FeIII to give CPRox (eq 5). Electrochemical oxidation of CPRred (eq 6) competes with the equilibrium step. Using this scheme, we accurately simulated CVs (compare Figures 8A&C) and peak potential vs. log (scan rate) plots (Figure 8D) for CPR/cyt P450 films with the best fit parameters shown in Scheme 2.vii Thus, this multicomponent LbL film provides a model system for studies of the complex natural cyt 450 catalysis pathway that can not be achieved using more conventional approaches.
Scheme 2.

ErCEo simulation model for CVs of cyt P450s + CPR.
Catalytic oxidation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) with the CPR/cyt P450 films further confirmed the electrons→CPR→cyt P450 pathway. Catalytic limiting currents were found for oxidation of NNK, which is converted by human cyt P450s 1A2 and 2E1 to 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB).l In constant potential electrolysis using CPR/cyt P450 films, HPB was identified as the major project by LC-MS. Electrode-driven oxidation of NNK by CPR/cyt P450 film was ~1.5x faster than NADPH-driven oxidations using the same films.vii
Catalytic turnover results also support the ErCEo model (Scheme 2). Simulations of film CVs provided a probe of the long postulated equilibriumxxxviii–xl between cyt P450 and CPR (eq 4), which is likely to involve a transient complex in which electron transfer occurs. The best fit kb ≥5kf suggests that in the absence of O2, equilibrium with K = kf/kb≤0.2 lies to in favor of CPRred. vii This explains why CPRox is reduced electrochemically in cyt P450/CPR films preferential to cyt P450. The formal potential difference (ΔE°) between CPR and cyt P450 is ΔE° = [RT/nF] ln(K), where R is the gas constant, T is absolute temperature, and F is Faraday’s constant. Thus, K<0.2 indicates that the cyt P450 formal potential is more negative than that of CPRox in the films ~40 mV, so that CPRox is the more easily reduced species. In catalytic oxidations, O2 reacts with P450-FeII and the equilibrium is drawn to the right to facilitate substrate oxidation.vii
4. Multifunctional enzyme/DNA Films for metabolic toxicity screening
Prediction of the toxicity of commercial chemical candidates to humans is usually based on panels of in vitro bioassays, followed by animal toxicity studies used in a second round of screening. Human clinical trials are initiated if the preceding screens are favorable.xiv,li,lii Despite analytically reliable test protocols, about 30% of drug candidates have toxicity issues that are uncovered only during human clinical testing, or even after the drug is marketed.li,liii Currently, in excess of US$2 billion is needed to develop a new drug. Thus, predicting toxicity at the earliest stages of development is a key to both patient safety and limiting the cost of health care.
There is an intimate relationship between metabolism and toxicity. Most often it is metabolites of an environmental chemical or drug that are the immediate cause of toxic response.xiv These metabolites can react with DNA, proteins and other biomolecules. One deficiency in current in vitro toxicity assessment is that the actual chemistry involved is not clarified by screening bioassays. Armed with knowledge of how make LbL films to facilitate metabolic enzyme reactions, we embarked a decade ago on research to develop toxicity screening approaches that clearly establish chemical details and rates of metabolite-based reactions that may contribute to toxicity.x
DNA damage is a convenient analytical target for toxicity because metabolite-DNA adducts are well-known toxicity biomarkers.liv Damage to DNA from chemicals that produce toxicity in humans is known as genotoxicity. We designed LbL films of metabolic enzymes and DNA on array surfaces or magentic beads to produce potentially reactive metabolites in close proximity to densely-packed DNA.iii,xiv This high DNA concentrations greatly increases the probablility of reaction with metabolites formed by the enzymes in these films.
We used electrochemiluminescence (ECL) to detect relative rates of DNA damage in microfludic arrays for test compounds (Figure 9). LC-MS/MS was used to quantify and establish structures of individual DNA base-metabolite adducts prepared in a high throughput 96-well plate format using the same DNA/enzyme films on magentic beads as bioreactors (or nanoparticles as nanoreators).xiv,lii,lv Both approaches provide DNA-damage rates that are correlated to one another, to cell-based DNA damage assays, and to animal genotoxicity metrics.lvi The arrays can be used to screen metabolites that react with DNA, then biocolloid reactors generate samples that are used to elucidate structures and measure formation rates of individual nucleobase adducts after hydrolyzing the DNA. The devices we have developed are inexpensive, high throughput, and can be employed at early development stages to contribute important toxicity-related reactivity and structure information for chemical product candidates. Our approaches are designed to complement rather than replace toxicity bioassays.
Figure 9.

DNA/enzyme/polyion films used in genotoxicity screening approaches.
We describe here the latest version of our microfluidic genotoxicity screening array using ECL detection and DNA, enzyme, polyion films.xxiv An ideal representation of the LbL films is illustrated in Figure 10. The RuPVP catalyst cation (Scheme 1) is responsible for light generation by ECL when it is electrochemically activated and reacts with guanines in DNA.lvii Films in the toxicity screening array feature DNA (a polyanion), pure enzymes or enzyme sources like microsomes, polycation RuPVP, and polyions such as PDDA or PSS to improve stability. Microsomes are insoluble lipid-enzyme fractions that are negatively charged due to the presence of anionic lipids.
Figure 10.
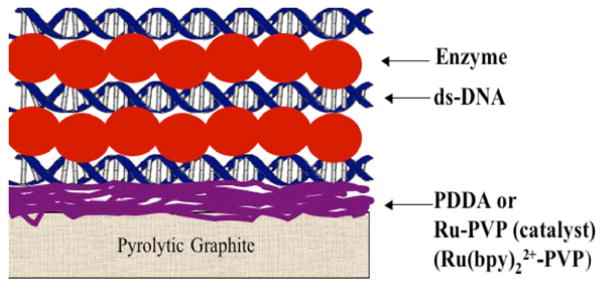
Idealized representation of films in genotoxicity arrays. The enzyme reaction produces metabolites in close proximity to DNA. Damage to DNA is detected by voltammetry or electrochemiluminescence, or by hydrolysis to eject nucleobase adducts, followed by LC-MS.
The heart of the array is a 64-microwell reactor chip housed in a microfluidic chamber (Figure 11). The chip is a conductive pyrolytic graphite (PG) sheet onto which are printed 64 microwells using a computer printed pattern that is heat-transferred onto the PG. The wells hold 1 μL of solution in the wells with hydrophobic sides to facilitate adsorption of film layers.
Figure 11.
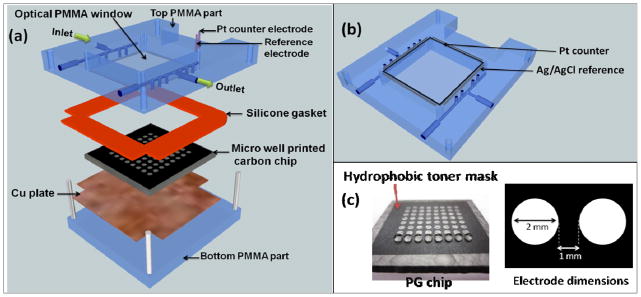
ECL chip and the fluidic reaction chamber: (a) assembly of the flow cell, (b) underside view of reference and counter electrode wires in the top poly(methylmethacrylate) (PMMA) plate, (c) pyrolytic graphite (PG) chip with printed microwells. The first row shows 1 μL water droplets on the wells.
The microfluidic array reactor in Figure 11 was tested using benzo[a]pyrene (B[a]P) as a model compound. Metabolism of B[a]P is well known (Scheme 3), and begins with epoxidation catalyzed by cyt P450s to give B[a]P-7,8-epoxide (eq 7). Hydrolysis of B[a]P-7,8-epoxide catalyzed by the enzyme epoxide hydrolase (EH) then yields B[a]P-7,8-dihydrodiol (eq 8). Subsequent oxidation of this dihydrodiol catalyzed by cyt P450s results in B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE) (Eq 9),lviii the major reactant that forms adducts with guanine and adenine bases in DNA.
Scheme 3.

Metabolic reactions of benzo[a]pyrene leading to DNA-reactive metabolite BPDE
Enzyme/RuPVP/DNA films are fabricated by placing 1 μL droplets of the adsorbate solutions a layer at a time in the microwells at 4 °C. The hydrophilic bottom and hydrophobic walls of the well combine to hold the droplet in place during layer adsorption, which is done in a humidified chamber. Microsomes, partially purified enzyme preparations containing lipids and other proteins, served as major sources of metabolic enzymes. They typically contain many metabolic enzymes, except for supersomes that are genetically engineered to produce a single cyt P450 and its reductase. Sources for the films in this work included human liver microsomes (HLM), human microsomal epoxide hydrolase (EH), pooled human liver S9 enzyme fractions (HS9), supersomes of cyt P450 1B1 and cyt P450 1A2, and rat liver microsomes (RLM). Films had architecture (RuIIPVP/DNA)2/RuPVP/cyt P450 enzyme source/EH or PDDA/DNA. QCM was used to estimate mass, amount of enzyme, and nominal thickness.
The 64-well microfluidic array constitutes a reaction chip in which necessary cofactors, oxygen, and reactants are introduced into the array at a constant flow rate. The chip was loaded with replicate spots including each of the enzyme preparations above, including spots with and without EH to provides the critical epoxide hydrolysis step that ultimately leads to BPDE (Scheme 3). The microsomal cyt P450s are activated electrochemically, as described above.vii,viii Once the reactions proceed for a desired time, the array is washed with buffer and 1.25 V vs. Ag/AgCl reference electrode is applied for 180 s in a dark box to generate ECL light that is detected by a CCD camera.xxiv Electrochemical oxidation of RuII→ RuIII in Ru-PVP initiates a multistep electron transfer pathway involving guanines in DNA to generated electronically excited RuII* sites that decay to the ground state with the emission of visible ECL light.lvii,lix
After the enzyme reactions are stopped, ECL is measure from array spots. Spots with cyt P450s increased in ECL intensity with enzyme reaction time (Figure 12a,b). ECL spot intensity was extracted with software to generate plots of % ECL increase vs. enzyme reaction time (Figure 12c,d), and the slopes of these linear plots directly monitor relative rates of DNA damage. ECL increases results mainly from formation of covalent nucleobase adducts of metabolites that disorder the DNA double helix and make guanines in the films more accessible to the RuIII sites in the LbL films.xiv,lii Slightly larger relative rates of DNA damage were found when EH was included in the spots with cyt P450 enzymes, suggesting formation of the major damage agent BPDE when both cyt P450 and EH are present, leading to more DNA damage. LC-MS/MS analysis of products of magnetic bead reactors coated with the same enzyme/DNA films as in the arrays resulted in product ion spectra of dG-BPDE and dA-BPDE that confirmed formation of these adducts when cyt P450 and EH were on the beads.xxiv
Figure 12.
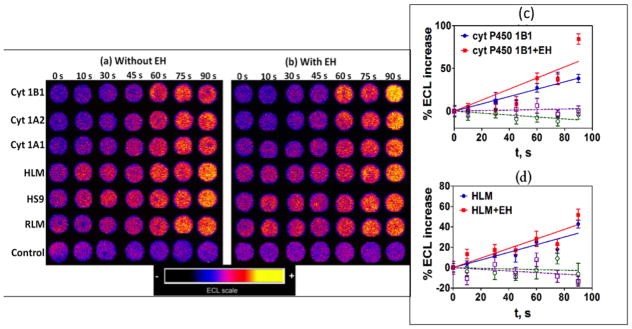
Results from genotoxicity array study of oxygenated 25 μM B[a]P in pH 7.4 phosphate buffer with electrochemical activation of cyt P450s at −0.65 V vs. Ag/AgCl (0.14 M KCl): (a and b) Reconstructed ECL array data from RuIIPVP/enzyme/DNA spots for reaction times 0–90 s. Control spots contained cyt P450 1B1 without (a) and with (b) EH subjected to the same reaction conditions as above without activation of cyt P450s. (c and d) Influence of enzyme reaction time on ECL increase for (c) cyt P450 1B1, and (d) human liver microsomes (HML). Controls for cyt P450s (green) and cyt P50s + EH (purple) behaved equivalently without substrate or with substrate but no activation of cyt P450s. Reproduced from ref. 24 with permission of the Royal Society of Chemistry.
The arrays were also used to study inhibition of cyt P450 enzymes by measuring the decrease in DNA damage in the presence of known inhibitors.xxiv Results were consistent with reported enzyme activities and inhibition.
5. Summary and outlook for the future
Discussion and examples above illustrate the utility of thin functional polyion films focused mainly on enzymes and enzyme-catalyzed reactions. The common thread of all these systems is alternate electrostatic adsorption LbL film fabrication designed to provide the desired functions. The presentation is in the order of increasing complexity. First, we addressed films of enzymes on electrodes, and their characterization. In most cases, enzyme activity in LbL films is equivalent or better than that of the enzyme in solution, and stability is greatly improved.iv,viii,xviii,xix Next, we discussed simple modifications that result in remarkably stable films on electrodes or colloidal particles that can be used for biocatalysis in acidic solutions or at high temperatures where function and secondary structures of the free enzymes would be completely destroyed. These chemical modifications remove and stability limitations of the LbL films under extreme conditions and allow enzymes to be used in acid, basic, detergent-based media, and high temperatures. The most complex enzyme film combined cyt P450s with redox partner cyt P450 reductase to obtain the natural electrode-driven pathway cyt P450 catalysis at high activity. The mixed interlayer structure of LbL films is advantageous for such applications since the enzymes involved in these multistep reactions need to interact in ordered to achieve high-level catalytic function.
Our final example involved films of DNA, enzymes and a redox metallopolymer that provide complex functionality enabling screening and more detailed investigations of genotoxicity-related chemistry. These latter films combine metabolic enzyme catalysis and reactions of the metabolites with DNA, with detection by ECL or LC-MS/MS. These complex LbL films clearly enable such applications, since formation of DNA adducts and their measurement is quite difficult in other ways. For example, in our hands, reactions catalyzed by microsomal enzymes to make metabolites that react with DNA with all components in the same solution may run for several days, and still not lead to products detectable by LC-MS/MS. In the best cases, at least 8 hr reactions are required, resulting reaction mixture are quite complicated, and require considerable fractionation or purification before analysis. Using DNA/enzyme LbL films on arrays or magnetic beads, enzyme reaction times as small as 1 min yield viable results (cf. Figure 12). This is essentially because the films provide very large enzyme and DNA concentrations and short diffusion lengths to facilitate metabolite-DNA reactions. Of course, reaction times may be somewhat longer when enzymes of low activity are used, or when a detailed LC-MS/MS analysis of all DNA adducts is needed, but a detailed analysis is often not possible at all using conventional solution-based reactions that are too slow to generate sufficient products for the analysis. While not presented specifically herein, the bead bioreactor/LC-MS/MS approach has provided detailed information on genotoxicity chemistry of tamoxifen in rats vs. humans,lx DNA adducts of unusual polyaromatic hydrocarbons,lxi and metabolite profiling and enzyme inhibition.lxii,lxiii
Clearly, LbL films are now moving beyond the realm of interesting and novel applications, and can be used to enable practical alternatives for rapid, high throughput bioanalytical and biochemical studies. Using multicomponent films, complex functionality can be readily developed that is difficult or impossible to achieve using conventional methodology. We expect that LbL films will feature in many future applications in which high stability or multiple biomolecule-related operations are required.
Supplementary Material
Acknowledgments
This work was supported financially by US PHS grant No. ES03154 from the National Institute of Environmental Health Sciences (NIEHS), NIH, USA. The authors thank coworkers and collaborators named in joint publications for their many excellent contributions to this research, without which progress would not have been possible.
Acronyms and Abbreviations
- LbL
layer-by-layer
- PDDA
poly(diallyldimethylamine)
- PSS
polystyrene sulfonate
- QCM
quartz crystal microbalance
- MPS
mercaptopropylsulfonate
- MPC
mercaptopropylcarboxylate
- Mb
myoglobin
- CV
cyclic voltammetry
- PG
pyrolytic graphite
- SPAN
sulfonated polyaniline
- HRP
horseradish peroxidase
- PLL
poly(L-lysine)
- EDC
1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide
- CD
Circular dichroism
- SBP
soybean peroxidase
- cyt P450s
cytochrome P450 enzymes
- CPR
cyt P450 reductase
- NADPH
nicotinamide adenine dinucleotide
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- ErCEo model
electrochemical reduction step, chemical step, electrochemical oxidation step
- ECL
electrochemiluminescence
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- RuPVP
[Ru(bpy)2(PVP)10]2+ {(PVP, poly(4-vinylpyridine)} (see Scheme 1 for structure)
- B[a]P
benzo[a]pyrene
- BPDE
B[a]P-7,8-dihydrodiol-9,10-epoxide
- HLM
human liver microsomes
- EH
epoxide hydrolase
- HS9
pooled human liver S9 enzyme fractions
- RLM
rat liver microsomes
- CCD
charge coupled device
References
- i.Lvov YM, Lu Z, Schenkman JB, Zu X, Rusling JF. J Am Chem Soc. 1998;120:4073–4080. [Google Scholar]
- ii.Schenkman JB, Jansson I, Lvov YM, Rusling JF, Boussaad S, Tao NJ. Arch Biochem Biophys. 2001;385:78–87. doi: 10.1006/abbi.2000.2140. [DOI] [PubMed] [Google Scholar]
- iii.Rusling JF, Hvastkovs EG, Hull Dominic O, Schenkman JB. Chem Commun. 2008:141–154. doi: 10.1039/b709121b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iv.Rusling JF, Zhang Z. In: Biomolecular Films: Design, Function and Applications. Rusling JF, editor. Marcel Dekker; N. Y: 2003. pp. 1–64. [Google Scholar]
- v.Rusling JF, Wang B, Yun S. In: Bioelectrochemistry. Bartlett PN, editor. John Wiley; N. Y: 2008. pp. 39–86. [Google Scholar]
- vi.Guto PM, Kumar CV, Rusling JF. J Phys Chem B. 2007;111:9125–9131. doi: 10.1021/jp071525h. [DOI] [PubMed] [Google Scholar]
- vii.Krishnan S, Wasalathanthri D, Zhao L, Schenkman JB, Rusling JF. J Am Chem Soc. 2011;133:1459–1465. doi: 10.1021/ja108637s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.Krishnan S, Schenkman JB, Rusling JF. J Phys Chem B. 2011;115:8371–8380. doi: 10.1021/jp201235m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ix.Zu X, Lu Z, Zhang Z, Schenkman JB, Rusling JF. Langmuir. 1999;15:7372–7377. [Google Scholar]
- x.Rusling JF. Biosens Bioelectron. 2004;20:1022–1028. doi: 10.1016/j.bios.2004.06.033. [DOI] [PubMed] [Google Scholar]
- xi.Lynch AM, Sasaki JC, Elespuru R, et al. New and emerging technologies for genetic toxicity testing. Environ Mol Mutagen. 2011;52:205–223. doi: 10.1002/em.20614. [DOI] [PubMed] [Google Scholar]
- xii.Kramer JA, Sagartz JE, Morris DL. Nat Rev Drug Discov. 2007;6:636–649. doi: 10.1038/nrd2378. [DOI] [PubMed] [Google Scholar]
- xiii.Mayne JT, Ku WW, Kennedy SP. Curr Opin Drug Discov Dev. 2006;9:75–83. [PubMed] [Google Scholar]
- xiv.Hvastkovs EG, Schenkman JB, Rusling JF. Annu Rev Anal Chem. 2012;5:79–105. doi: 10.1146/annurev.anchem.111808.073659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xv.Decher G, Hong JD. Ber Bunsen-Ges Phys Chem. 1991;95:1430. [Google Scholar]
- xvi.Decher G, Hong JD, Schmitt J. Thin Solid Films. 1992;210:831. [Google Scholar]
- xvii.Lvov Y, Decher G, Möhwald H. Langmuir. 1993;9:481. [Google Scholar]
- xviii.Lvov Y. In: Protein Architecture: Interfacing Molecular Assemblies and Immobilization Biotechnology. Lvov Y, Möhwald H, editors. Marcel Dekker; New York: 2000. pp. 125–167. [Google Scholar]
- xix.Lvov Y. Handbook Of Surfaces And Interfaces Of Materials. In: Nalwa RW, editor. Nanostructured Materials, Micelles and Colloids. Vol. 3. Academic Press; San Diego: 2001. pp. 170–189. [Google Scholar]
- xx.Zhang X, Chen H, Zhang H. Chem Commun. 2007:1395–1405. doi: 10.1039/b615590a. [DOI] [PubMed] [Google Scholar]
- xxi.Ariga K, Hill JP, Ji Q. Phys Chem Chem Phys. 2007;9:2319–2340. doi: 10.1039/b700410a. [DOI] [PubMed] [Google Scholar]
- xxii.Ariga K, Kunitake T. In: Protein Architecture: Interfacing Molecular Assemblies and Immobilization Biotechnology. Lvov Y, Möhwald H, editors. Marcel Dekker; New York: 2000. pp. 169–192. [Google Scholar]
- xxiii.Zhou L, Yang J, Estavillo C, Stuart JD, Schenkman JB, Rusling JF. J Am Chem Soc. 2003;125:1431–1436. doi: 10.1021/ja0290274. [DOI] [PubMed] [Google Scholar]
- xxiv.Wasalathanthri DP, Malla Spundana, Bist Itti, Tang CK, Faria RC, Rusling JF. High- Lab on a Chip. 2013;13:4554–4562. doi: 10.1039/c3lc50698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxv.Buttry DA, Ward MD. Chem Rev. 1992;92:1355–1379. [Google Scholar]
- xxvi.Lvov Y, Ariga K, Ichinose I, Kunitake T. J Amer Chem Soc. 1995;117:6117–6123. [Google Scholar]
- xxvii.Decher G. Science. 1997;277:1231–1237. [Google Scholar]
- xxviii.Rusling JF. In: Protein Architecture: Interfacing Molecular Assemblies and Immobilization Biotechnology. Lvov Y, Mohwald H, editors. Marcel Dekker; N. Y: 2000. pp. 337–354. [Google Scholar]
- xxix.Ma H, Hu N, Rusling JF. Langmuir. 2000;16:4969–4975. [Google Scholar]
- xxx.Munge B, Estavillo C, Schenkman JB, Rusling JF. ChemBiochem. 2003;4:82–89. doi: 10.1002/cbic.200390018. [DOI] [PubMed] [Google Scholar]
- xxxi.Yu X, Sotzing GA, Papadimitrakopoulos F, Rusling JF. Anal Chem. 2003;75:4565–4571. doi: 10.1021/ac034188r. [DOI] [PubMed] [Google Scholar]
- xxxii.Panchagnula V, Kumar CV, Rusling JF. J Am Chem Soc. 2002;124:12515–12525. doi: 10.1021/ja020683d. [DOI] [PubMed] [Google Scholar]
- xxxiii.Guto PM, Kumar CV, Rusling JF. J Phys Chem B. 2007;111:9125–9131. doi: 10.1021/jp071525h. [DOI] [PubMed] [Google Scholar]
- xxxiv.Holzwarth G, Doty P. J Am Chem Soc. 1965;87:218–228. doi: 10.1021/ja01080a015. [DOI] [PubMed] [Google Scholar]
- xxxv.Hirst J, Armstrong FA. Anal Chem. 1998;70:5062–5071. doi: 10.1021/ac980557l. [DOI] [PubMed] [Google Scholar]
- xxxvi.Laviron E. J Electroanal Chem. 1979;101:19–28. [Google Scholar]
- xxxvii.Guto PM, Kumar CV, Rusling JF. Langmuir. 2008;24:10365–10370. doi: 10.1021/la801644e. [DOI] [PubMed] [Google Scholar]
- xxxviii.Schenkman JB, Greim H, editors. Cytochrome P450. Springer-Verlag; Berlin: 1993. [Google Scholar]
- xxxix.Ortiz de Montellano PR, editor. Cytochrome P450. 3. Kluwer/Plenum; New York: 2005. [Google Scholar]
- xl.Guengerich FP. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- xli.Singer B, Grunberger D. Molecular Biology of Mutagens and Carcinogens. Plenum Press; New York: 1983. [Google Scholar]
- xlii.Jacoby WB, editor. Enzymatic Basis of Detoxification. I and II. Academic; New York: 1980. [Google Scholar]
- xliii.Friedberg EC. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- xliv.Scharer OD. Angew Chem Int Ed. 2003;42:2946–2974. doi: 10.1002/anie.200200523. [DOI] [PubMed] [Google Scholar]
- xlv.Krishnan S, Abeykoon A, Schenkman JB, Rusling JF. J Am Chem Soc. 2009;131:16215–16224. doi: 10.1021/ja9065317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xlvi.Bistolas N, Wollenberger U, Jung C, Scheller FW. Biosens Bioelec. 2005;20:2408–2423. doi: 10.1016/j.bios.2004.11.023. [DOI] [PubMed] [Google Scholar]
- xlvii.Fleming BD, Johnson DL, Bond AM, Martin LL. Expert Opin Drug Metab Toxicol. 2006;2:581–589. doi: 10.1517/17425255.2.4.581. [DOI] [PubMed] [Google Scholar]
- xlviii.Sultana N, Schenkman JB, Rusling JF. J Am Chem Soc. 2005;127:13460–13461. doi: 10.1021/ja0538334. [DOI] [PubMed] [Google Scholar]
- xlix.Mie Y, Suzuki M, Komatsu Y. J Am Chem Soc. 2009;131:6646–6647. doi: 10.1021/ja809364r. [DOI] [PubMed] [Google Scholar]
- l.Hecht SS. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- li.Caldwell GW, Yan L. Curr Opin Drug Discov Dev. 2006;9:47–50. [PubMed] [Google Scholar]
- lii.Rusling JF, Hvastkovs EG, Schenkman JB. In: Drug Metabolism Handbook. Nassar A, Hollenburg PF, Scatina JJ, editors. Trenton, NJ: Wiley; 2009. pp. 307–340. [Google Scholar]
- liii.Nasser AEF, Kamel AM, Clarimont C. Drug Dev Today. 2004;9:1055–1064. doi: 10.1016/S1359-6446(04)03297-0. [DOI] [PubMed] [Google Scholar]
- liv.Farmer PB, Brown K, Tompkins E, et al. Toxicol Appl Pharmacol. 2005;207:S293–301. doi: 10.1016/j.taap.2004.12.020. [DOI] [PubMed] [Google Scholar]
- lv.Zhao L, Schenkman JB, Rusling JF. Anal Chem. 2010;82:10172–10178. doi: 10.1021/ac102317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lvi.Pan S, Zhao L, Schenkman JB, Rusling JF. Anal Chem. 2011;83:2754–2760. doi: 10.1021/ac200050n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lvii.Dennany L, Forster RJ, Rusling JF. J Am Chem Soc. 2003;125:5213–5218. doi: 10.1021/ja0296529. [DOI] [PubMed] [Google Scholar]
- lviii.Benigni R, Bossa C. Chem Rev. 2011;111:2507–2536. doi: 10.1021/cr100222q. [DOI] [PubMed] [Google Scholar]
- lix.Forster RJ, Bertoncello P, Keyes TE. Annu Rev Anal Chem. 2009;2:359–85. doi: 10.1146/annurev-anchem-060908-155305. [DOI] [PubMed] [Google Scholar]
- lx.Zhao L, Krishnan S, Zhang Y, Schenkman JB, Rusling JF. Chem Res Toxicology. 2009;22:341–347. doi: 10.1021/tx8004295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxi.Pan S, Li D, Zhao L, Schenkman JB, Rusling JF. Chem Res Toxicology. 2013;26:1229–1239. doi: 10.1021/tx400147c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxii.Bajrami B, Zhao L, Schenkman JB, Rusling JF. Anal Chem. 2009;81:9921–9929. doi: 10.1021/ac9015853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxiii.Bajrami B, Krishnan S, Rusling JF. Drug Metabolism Lett. 2008;2:158–162. doi: 10.2174/187231208785425854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.