

Background: Arachidonic acid and its metabolites regulate pancreatic glucose-stimulated insulin secretion (GSIS) through multiple mechanisms.
Results: Group X secretory phospholipase A2 (GX sPLA2) suppresses GSIS; suppression was abolished when COX-2 activity or PGE2-EP3 receptor signaling were inhibited.
Conclusion: GX sPLA2 inhibits GSIS by augmenting PGE2 production.
Significance: GX sPLA2 may be targeted for ameliorating beta cell dysfunction in type 2 diabetes.
Keywords: Cyclooxygenase (COX), Insulin Secretion, Pancreatic Islet, Prostaglandin, Type 2 Diabetes
Abstract
Group X secretory phospholipase A2 (GX sPLA2) potently hydrolyzes membrane phospholipids to release arachidonic acid (AA). While AA is an activator of glucose-stimulated insulin secretion (GSIS), its metabolite prostaglandin E2 (PGE2) is a known inhibitor. In this study, we determined that GX sPLA2 is expressed in insulin-producing cells of mouse pancreatic islets and investigated its role in beta cell function. GSIS was measured in vivo in wild-type (WT) and GX sPLA2-deficient (GX KO) mice and ex vivo using pancreatic islets isolated from WT and GX KO mice. GSIS was also assessed in vitro using mouse MIN6 pancreatic beta cells with or without GX sPLA2 overexpression or exogenous addition. GSIS was significantly higher in islets isolated from GX KO mice compared with islets from WT mice. Conversely, GSIS was lower in MIN6 cells overexpressing GX sPLA2 (MIN6-GX) compared with control (MIN6-C) cells. PGE2 production was significantly higher in MIN6-GX cells compared with MIN6-C cells and this was associated with significantly reduced cellular cAMP. The effect of GX sPLA2 on GSIS was abolished when cells were treated with NS398 (a COX-2 inhibitor) or L-798,106 (a PGE2-EP3 receptor antagonist). Consistent with enhanced beta cell function, GX KO mice showed significantly increased plasma insulin levels following glucose challenge and were protected from age-related reductions in GSIS and glucose tolerance compared with WT mice. We conclude that GX sPLA2 plays a previously unrecognized role in negatively regulating pancreatic insulin secretion by augmenting COX-2-dependent PGE2 production.
Introduction
Type 2 diabetes (T2D)2 occurs when pancreatic beta cells are unable to secrete sufficient insulin to meet the metabolic requirements associated with insulin resistance (1). Pancreatic dysfunction is characterized by a reduction in mass as well as function of islets (2). While the molecular mechanisms of beta cell failure are poorly understood, arachidonic acid (AA) and its metabolites are thought to play important roles in pancreatic islet function. Islets have the highest AA-containing phospholipid (PL) content of any known tissue, constituting more than 30% of the total esterified fatty acyl mass of glycerolipids in rodent and human islets (3). Acute AA treatment promotes glucose-stimulated insulin secretion (GSIS) (4, 5). However, chronic treatment with fatty acids including AA suppresses insulin secretion by beta islet cells (6). Thus, the role of AA in GSIS likely depends on its metabolic fate in beta cells. While free AA is known to be an activator of GSIS in general (7, 8), prostaglandin E2 (PGE2), a product of AA metabolism and the major prostaglandin (PG) produced by islets, is considered to be an inhibitor of GSIS (9–13). PGE2 exerts its effects by interacting with one or more of its four PGE2 (EP) receptors, EP1, EP2, EP3, and EP4 (14). EP3 is the most abundant PGE2 receptor expressed in islets (13, 15). Upon binding to the EP3 receptor subtype, PGE2 decreases adenylyl cyclase activity with a subsequent reduction in cAMP (16), a known potentiator of GSIS (17). Cyclooxygenase (COX) enzymes catalyze the key step in the synthesis of PGE2 from AA (18). Unlike most cell types, COX-2 rather than COX-1 is the predominant, constitutively expressed COX in pancreatic islet cells (19). Inhibition of COX-2 enhances GSIS in C57BL/6 mice with a parallel reduction in PGE2 production, consistent with a role for PGE2 in suppressing GSIS (20). The increase in diabetes susceptibility in the BTBR mouse strain has recently been attributed to elevated PGE2 production and EP3 receptor expression in pancreatic beta cells (13). Islets from T2D humans produce significantly more PGE2 compared with islets from non-diabetic donors (13). Furthermore, L-798,106, a specific EP3 receptor antagonist significantly enhanced GSIS only in islets from T2D donors and not non-diabetic donors, suggesting that the PGE2/EP3 axis contributes to beta cell dysfunction in humans (13). Hyperglycemic conditions increase COX-2 expression and hence PGE2 production in beta cells, suggesting a mechanism by which hyperglycemia contributes to beta cell dysfunction (21).
AA is produced in cells through the action of phospholipase A2, which hydrolyzes PL at the sn-2 position to generate free fatty acids (in particular AA) and lysophospholipids (22). Thus, an important question to address is the identity of the cellular PLA2(s) that provide AA for PGE2 production in beta cells. Cytosolic phospholipase A2 (cPLA2) is considered to be the major source of AA for eicosanoid production in many cell types and is reportedly expressed by pancreatic islet cells (23). However, inhibition of cPLA2 does not enhance GSIS (24, 25), indicating it is not involved in PGE2-mediated suppression of insulin secretion. The role of Ca2+-independent PLA2 (iPLA2) in pancreatic islet function has undergone extensive investigation; the enzyme has been shown to stimulate GSIS (26, 27). Taken together, these results suggest that the major intracellular PLA2's, cPLA2 and iPLA2, do not generate PGE2 that leads to suppressed GSIS.
Comprehensive studies investigating the other major class of PLA2s, the secretory PLA2s (sPLA2s), in beta cell function are lacking. In mammals, ten enzymatically active sPLA2 isoforms have been identified, which differ in tissue distribution and substrate specificity (28). Among the members of the sPLA2 family, Group X sPLA2 (GX sPLA2) is the most potent in hydrolyzing phosphatidylcholine to release AA for COX1/2-dependent eicosanoid formation. In the current study, we demonstrate the expression of GX sPLA2 in insulin-producing islet cells and provide evidence that it negatively regulates GSIS through a COX-2/PGE2-dependent mechanism.
EXPERIMENTAL PROCEDURES
Biochemical Reagents and Assays
Assays for Insulin (Crystal Chem Inc), PGE2 metabolites (Cayman) and cAMP (ENZO Life Sciences) were performed according to the manufacturers' instructions. Phospholipase activity in conditioned media was measured using a colorimetric assay as we previously described (29) with 1-palmitoyl-2-oleoylphosphatidylglycerol (POPG; Matreya LLC) as a substrate. Briefly, mixed micelles were prepared by warming 7 mg of POPG to 37 °C in a 0.2 ml mixture of 4.0% (w/v) Nonidet-40 and 2.0% sodium deoxycholate, and then adding 1.8 ml warm assay buffer (0.12 mol/liter Tris-HCl, pH 8, 12 mmol/liter CaCl2, 0.1 mmol/liter EDTA). For enzyme assays, 10 μl of conditioned media was added to 40 μl of substrate solution. After incubating at 37 °C, the amount of free fatty acids (FFA) released was quantified using a NEFA-C kit (Wako Chemicals); 1 unit of activity corresponds to 1 nmol of FFA released in 20 min per mg cell protein. NS-398 was purchased from Cayman Chemicals. L-798,106 was from Santa Cruz Biotechnology.
Animals
We previously described the generation of C57BL/6 mice with targeted deletion of GX sPLA2 (gene PLA2G10) (30). Heterozygous GX sPLA2+/− mice were bred to produce GX sPLA2+/+ (WT) and GX sPLA2−/− (GX KO) mice for the experiments. Mice (3–15-month-old) were maintained on a 10-h light/14-h dark cycle and received standard mouse chow and water ad libitum. Male mice were used throughout the study. All procedures were in accordance with the guidelines of the Lexington Veterans Affairs and the University of Kentucky Institutional Animal Care and Use Committees.
Islet Isolation
Mouse islets were isolated via intraductal collagenase (Roche) digestion and ficoll gradient centrifugation by a method adopted by the Islet Procurement and Analysis Core, Diabetes Research and Training Center, Vanderbilt University Medical Center (Nashville, TN). A detailed method will be provided on request. Following isolation, islets were hand-picked and maintained in RPMI containing 5 mm glucose, 10% (v/v) FBS, and penicillin and streptomycin.
Immunohistochemistry
Pancreata from WT and GX KO mice were embedded in paraffin and 5 μm-thick sections were mounted on glass slides. After antigen retrieval with sodium citrate-based reagent (Dako) for 15 min at 95 °C, sections were blocked with 2% horse serum. Following blocking, the sections were immunostained using rabbit anti-mouse GX sPLA2 (gift from Dr. M. Gelb, University of Washington) and goat anti-mouse insulin (Santa Cruz Biotechnology), both at a dilution of 1:100. For fluorescent images, Alexa Fluor-conjugated secondary antibodies were used (Invitrogen).
Metabolic Experiments
To assess GSIS in vivo, mice were fasted for 16 h, and then plasma samples were collected from the retro-orbital sinus before and 15 min after intraperitoneal glucose injection (3 g/kg). For glucose tolerance tests, mice were fasted for 6 h prior to i.p. glucose injection (2 g/kg).
Real-time RT-PCR
Total RNA was prepared from 150–200 islets isolated from WT and GX KO mice or transfected MIN6 cells using the RNeasy Mini kit (Promega). Quantification was performed in duplicate using the standard curve method and normalized to 18S. The primer sequence used for various genes will be provided on request.
Cell Culture and Transfections
MIN6 cells were cultured in DMEM supplemented with 15% heat-inactivated fetal bovine serum (FBS), 2 mmol/liter l-glutamine, 45 mmol/liter β-mercaptoethanol, 100 units/ml penicillin, and 100 μg/ml streptomycin. The GX sPLA2 and control expression constructs and procedure for transient transfections have been described (31). When assayed 48 h after transfection, phospholipase activity secreted by transfected MIN6-GX cells was increased 1.5–2.5-fold compared with control MIN6-C cells.
In Vitro GSIS Assays
After isolation and handpicking, islets from WT and GX KO mice were cultured overnight in RPMI media. Islets were then selected and transferred to culture inserts (Greiner bio-one) in 12-well plates (25 islets per well) washed and equilibrated for 1 h in Buffer 1 (DMEM supplemented with 38 mm sodium bicarbonate, 4 mm l-glutamine, 1 mm sodium pyruvate, 4.65 mm HEPES, and 1 g/liter BSA) containing 5 mm glucose, and then incubated successively for 40 min in Buffer 1 containing 5 mm glucose (low glucose) followed by Buffer 1 containing 20 mm glucose (high glucose). At the end of the incubations, insulin content in the low glucose and high glucose buffer was assayed and normalized to total cellular insulin content, which was determined after lysing islets in acid-ethanol (75% ethanol, 0.2 mol/liter HCl). For GSIS in MIN6 cells, cells in 24-well plates were washed once with Kreb's Ringer buffer containing 0.2% BSA (KRB-BSA) and 5 mm glucose and then equilibrated in the same buffer for 1 h. The media was replaced with fresh KRB-BSA supplemented with either 5 mm (low) glucose or 20 mm (high) glucose for 40 min. Insulin concentrations in conditioned media were normalized to total cell protein.
Statistics
Data are expressed as mean ± S.E. Results were analyzed by t test or by 1-way ANOVA followed by a Bonferroni post-test. p < 0.05 was considered statistically significant. All statistical analyses were carried out using GraphPad Prism 4.
RESULTS
GX sPLA2 Is Expressed in Pancreatic Islets and MIN6 Cells
GX sPLA2 mRNA was detected by RT-PCR in mouse pancreata and was relatively enriched in isolated mouse islets (Fig. 1A). MIN6 cells (a mouse pancreatic beta cell line) also expressed GX sPLA2 mRNA (Fig. 1A). To assess the distribution of GX sPLA2 protein in mouse pancreas, we performed double immunofluorescence staining of pancreatic sections from C57BL/6 mice for GX sPLA2 (Fig. 1B; green) and insulin (red). GX sPLA2 immunoreactivity co-localized with insulin-secreting cells (yellow on merged image, Fig. 1B) indicating expression of GX sPLA2 in beta cells. As expected, GX sPLA2 was not detected in pancreatic sections from GX KO mice. There were no apparent morphological differences in the pancreas of WT and GX KO mice, and the density and size distribution of islets in the two strains appeared to be similar (not shown).
FIGURE 1.
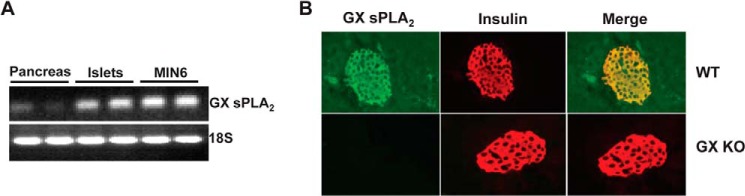
GX sPLA2 is expressed in pancreatic islet cells and MIN6 cells. A, total RNA was isolated from mouse pancreas, islets isolated from mouse pancreas, and MIN6 cells, a mouse pancreatic beta-cell line. RT-PCR was performed using 0.5 μg of RNA and primers for GX sPLA2 and 18 S. B, pancreatic sections from WT and GX KO mice were stained by indirect immunofluorescence for GX sPLA2 (green) and insulin (red) and visualized (20× magnification) by fluorescence microscopy. A merged image shows co-expression of GX sPLA2 in insulin-producing cells of WT pancreas (yellow).
GX sPLA2 Deficiency Is Associated with Enhanced Glucose-stimulated Insulin Secretion in Mice and in Isolated Islets
Prompted by our novel finding that GX sPLA2 is expressed by pancreatic islet cells, we next investigated whether the enzyme plays any role in beta islet function. Glucose-stimulated insulin secretion (GSIS) was assessed in WT and GX KO mice by measuring plasma insulin levels before and 15 min following i.p. injection of glucose (3 mg/kg body weight). Fasting insulin levels were similar in WT mice (0.34 ± 0.04 ng/ml) compared with GX KO mice (0.31 ± 0.03 ng/ml). Plasma insulin levels increased significantly in both WT and GX KO mice 15 min following glucose injection (Fig. 2A). Interestingly, insulin levels were significantly higher in GX KO mice (0.95 ± 0.1 ng/ml) compared with WT mice (0.72 ± 0.04 ng/ml) following glucose challenge (Fig. 2A). The increased plasma insulin levels were associated with a modest (∼22%) but not statistically significant decrease in blood glucose concentrations in GX KO mice compared with WT mice (Fig. 2B). To investigate whether endogenous GX sPLA2 expressed in islets modulates GSIS, insulin secretion by isolated islet cells from 4-month-old WT and GX KO mice was assessed ex vivo. While there was no significant difference in basal insulin secretion (5 mm glucose) for the two strains, insulin secretion in response to 20 mm glucose was significantly higher for islets isolated from GX KO mice compared with WT mice (Fig. 2C). Thus, GX sPLA2 negatively regulates GSIS in mice, which appears to be mediated through a direct effect on islet beta cells.
FIGURE 2.
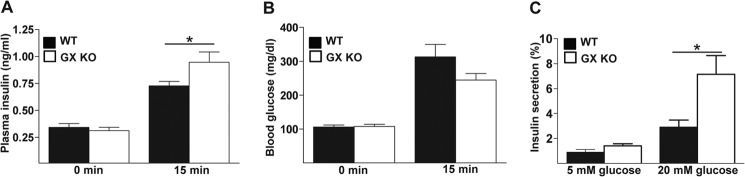
Deficiency of GX sPLA2 enhances glucose-stimulated insulin secretion. Plasma (A) insulin and (B) glucose concentrations were determined before and 15 min after intraperitoneal glucose injection (3 g/kg; n = 10 per group) in 4-month-old WT and GX KO mice. C, islets (25 islets per mouse) were isolated from 4-month-old WT and GX KO mice (n = 4), cultured overnight, washed, and equilibrated in assay buffer containing 5 mm glucose, and then incubated for 40 min in either 5 or 20 mm glucose as indicated. Insulin in the assay media was determined and normalized to total insulin in the corresponding islets. Data are presented as mean ± S.E.; *, p < 0.05.
GX sPLA2 Enhances PGE2 Production and Suppresses GSIS and cAMP Levels in MIN6 Cells
As a gain-of-function strategy, we analyzed GSIS in MIN6 cells transiently overexpressing GX sPLA2 protein. As expected, MIN6 cells overexpressing GX sPLA2 (MIN6-GX) demonstrated a significant increase in phospholipase activity in the culture medium compared with cells transfected with vector control (MIN6-C) (Fig. 3A). Consistent with the results from primary islets (Fig. 2C), the increase in sPLA2 activity in MIN-GX cells was associated with a significant ∼32% reduction in GSIS compared with MIN6-C cells (Fig. 3B).
FIGURE 3.
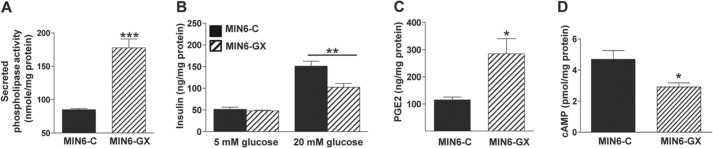
GX sPLA2 regulates GSIS, PGE2 and cAMP levels in MIN6 cells. MIN6 cells were transfected with empty expression vector (MIN6-C) or vector encoding mouse GX sPLA2 (MIN6-GX). A, phospholipase activity in 20 h conditioned media from MIN6-C and MIN6-GX cell cultures 48 h after transfection was determined and normalized to cell protein. B, 48 h after transfection, MIN6-C and MIN6-GX cells were incubated in buffer containing either 5 or 20 mm glucose for 40 min. Insulin concentrations in cell media were determined and normalized to total cell protein (n = 4). C, PGE2 levels in culture media from MIN6-C and MIN6-GX cells 48 h after transfection, normalized to total cell protein (n = 4; independent transfections per construct). D, cellular cAMP content in MIN6-C and MIN6-GX cells was determined 48 h after transfection and normalized to total cell protein (n = 5). Data are presented as mean ± S.E.; *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
We and others have reported that GX sPLA2 potently releases arachidonic acid (AA) and thus enhances PGE2 production in multiple cell types (30, 32, 33). Here we show that increased GX sPLA2 activity leads to enhanced PGE2 production in beta cells. MIN6-GX cells produced a significant 2.5-fold increase in PGE2 secretion compared with MIN6-C cells (Fig. 3C). PGE2 binding to the EP3 receptor, the most abundant EP receptor expressed in islets, results in decreased adenylate cyclase activity and consequently, reduced cellular cAMP (34). Increased intracellular cAMP enhances insulin secretion, whereas decreased cAMP is known to at least partially mediate PGE2-induced beta cell dysfunction (34). In accordance with these concepts, increased PGE2 production by MIN6-GX cells (Fig. 3C) was associated with significantly reduced cAMP levels compared with MIN6-C cells (Fig. 3D). Taken together, our data supports the conclusion that GX sPLA2 negatively regulates GSIS, possibly by enhancing the generation of PGE2 in islet cells, leading to reduced cAMP.
GX sPLA2-mediated Suppression of GSIS Involves COX-2 and the PGE2-EP3 Receptor in MIN6 Cells
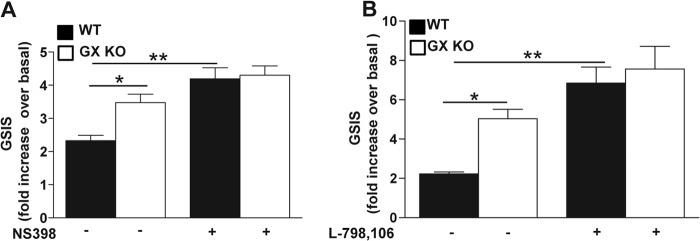
To investigate the possibility that GX sPLA2 inhibits GSIS through COX-2-dependent PGE2 production, we assessed the effect of a specific COX-2 inhibitor, NS398, on GSIS in MIN6-GX and MIN6-C cells. Under basal conditions, insulin secretion was not significantly altered by NS398 treatment in either MIN6-C or MIN6-GX cells (Fig. 4A). However, insulin secretion under high glucose conditions was significantly increased in MIN6-C cells treated with NS-398, consistent with published studies (5, 15). Interestingly, after treatment with NS398 there was no significant difference in GSIS between MIN6-C and MIN6-GX cells, indicating that COX-2 is required for GX sPLA2's suppressive effect (Fig. 4A). To define the role of EP3 receptor signaling, we next assessed GSIS in MIN6-C and MIN6-GX cells in the presence and absence of the specific EP3 antagonist L-798,106 (13). Treatment with L-798,106 caused a significant 2.9-fold increase in GSIS in MIN6-C cells (Fig. 4B) in accordance with previous reports (13, 15). In the absence of L-798,106 MIN6-GX cells demonstrated a significant (∼30%) reduction in GSIS compared with MIN6-C cells, as shown above (Figs. 3B and 4A). Treatment with L-798,106 significantly enhanced GSIS in both MIN6-C and MIN6-GX cells, and completely abolished the difference in GSIS between the two cell types (Fig. 4B). Findings in transfected cells were corroborated in an additional series of experiments whereby untransfected MIN6 cells were incubated for 16 h with 0.1 μg/ml recombinant GX sPLA2 (rGX). Consistent with the overexpression results, exogenous treatment with rGX resulted in significantly enhanced PGE2 production (3.4-fold; Fig. 4C) and significantly reduced GSIS (first two bars, Fig. 4D). Importantly, the ability of rGX to reduce GSIS was abolished in cells treated with 10 μm NS398 (Fig. 4D). We conclude that the suppressive effect of GX sPLA2 on GSIS in MIN6 cells is mediated by COX-2-dependent PGE2 production and EP3 receptor signaling.
FIGURE 4.

Treatment with COX-2 inhibitor or EP3 receptor antagonist abolishes GX sPLA2-mediated suppression of GSIS in MIN6 cells. A, 24 h after transfection, MIN6-C and MIN6-GX cells were treated with the COX-2 inhibitor, NS398 (10 μm) or an equivalent volume of vehicle (DMSO) in regular culture medium containing 1% FBS for 20 h (n = 4) prior to assaying GSIS as described under “Experimental Procedures.” B, GSIS assay was performed in the presence of either EP3 antagonist, L-798,106 (10 μm) or an equivalent volume of vehicle (DMSO) in MIN6-C and MIN6-GX cells 48 h after transfection. C, MIN6 cells were treated with or without 0.1 μg/ml recombinant human GX sPLA2 (+rGX) for 16 h (n = 4), and PGE2 levels in the medium was estimated. D, MIN6 cells were treated with or without 0.1 μg/ml rGX in regular culture medium containing 1% FBS either with 10 μm NS398 or vehicle (DMSO) for 20 h prior to assaying GSIS as described under “Experimental Procedures.” Basal insulin secretion in 5 mm glucose was not altered by NS398 treatment in either group of cells (data not shown). Data are presented as mean ± S.E.; *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Treatment with COX-2 Inhibitor or EP3 Receptor Antagonist Completely Abolishes the Effect of Endogenous GX sPLA2 on GSIS in Primary Islet Cells
Primary islets isolated from WT and GX KO mice were treated with either NS398 or the corresponding volume of vehicle (DMSO) for 20 h prior to assessing GSIS. Another set of islets were treated with L-798,106 or DMSO during the GSIS assay. Basal insulin secretion was not altered by either NS398 or L-798,106 in either WT or GX KO islets (data not shown). As expected based on our previous findings (Fig. 2C), control-treated islets from GX KO mice demonstrated a significant increase in GSIS compared with islets from control-treated WT mice (Fig. 5, A and B). NS398 and L-798,106 caused a significant, 1.8-fold and 3.1-fold increase in GSIS in WT islets, respectively (black bars, Fig. 5, A and B). In contrast, there was no statistically significant increase in GSIS in islets from GX KO mice when treated with either NS398 or L-798,106 (p = 0.29 and 0.21, respectively). Interestingly, the difference in GSIS observed between islets from WT and GX KO mice was completely abolished in the presence of either inhibitor, consistent with the conclusion that endogenous GX sPLA2 suppresses insulin secretion through the COX-2, EP3 receptor pathway.
FIGURE 5.
COX-2 inhibitor and EP3 receptor antagonist enhance GSIS in primary mouse islets isolated from WT but not GX sPLA2-deficient mice. A, 25 islets isolated from 3-month-old WT and GX KO mice (n = 4 mice per group) were treated with the COX-2 inhibitor, NS398 (10 μm) or an equivalent volume of vehicle (DMSO) in regular culture medium containing 1% FBS for 20 h (n = 4) prior to assaying GSIS as described under “Experimental Procedures.” B, GSIS assay was performed as described under “Experimental Procedures” in 25 primary islets isolated from WT and GX KO mice (n = 4) with either the EP3 antagonist, L-798,106 (10 μm) or an equivalent volume of vehicle in the assay buffer. NS398 and L-798,106 treatment did not alter insulin secretion from either WT or GX KO islets under basal conditions. Data are presented as mean ± S.E.; *, p < 0.05; **, p < 0.01.
GX KO Mice Are Protected from the Development of Age-related Glucose Intolerance and GSIS Impairment
Given our data that GX sPLA2 modulates GSIS, we investigated whether GX sPLA2 influences glucose homeostasis in mice. For these studies we carried out i.p. glucose tolerance tests in WT and GX KO mice at 3 months (Fig. 6A) and 15 months (Fig. 6B) of age. The glucose challenge was kept constant for the two age groups (2 mg/g) to allow for direct comparison of results. For both age groups, fasting glucose levels were not significantly different between the two strains. The 15-month-old WT mice exhibited a severe impairment in glucose tolerance, as evidenced by a marked increase in plasma glucose concentrations at 60–180 min after glucose challenge compared with younger WT mice. Interestingly, GX KO mice appeared to be protected from this age-related glucose intolerance. To investigate whether differences in glucose tolerance could be at least partly attributed to differences in beta cell function, we assessed GSIS in islets isolated from young and old WT and GX KO mice (Fig. 6C). For both age groups, basal insulin secretion in islets from WT and GX KO mice were similar. Interestingly, GSIS was significantly higher in islets from GX KO mice compared with WT mice at both ages. Islets from older WT mice demonstrated significantly reduced GSIS compared with younger WT mice, reflecting a decline in beta cell function with age in the WT mice. Although there was a trend for decreased GSIS in islets from older GX KO mice compared with younger GX KO mice, GSIS in islets from 14-month-old GX KO mice was nevertheless comparable to that of islets from 2-month-old WT mice (Fig. 6C). Thus, the absence of GX sPLA2 activity during the course of aging partially ameliorates the development of beta cell dysfunction.
FIGURE 6.
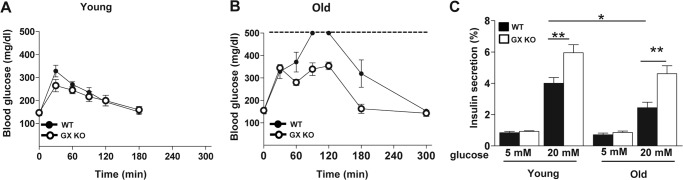
GX sPLA2 deficiency enhances GSIS in vivo and protects mice from age-related glucose intolerance and beta-cell dysfunction. A, young (3-month-old) and (B) old (15-month-old) WT and GX KO mice were fasted for 6 h prior to intraperitoneal injection of 2 mg glucose/g bodyweight, and glucose concentrations were determined at the indicated time after injection (n = 4–5). Glucose values in 15-month-old WT mice measured 90 and 120 min after glucose injection exceeded the limit of the glucose meter (dotted line). C, Islets (25 islets per mouse) were isolated from 2- and 14-month-old WT and GX KO mice (n = 4–5), cultured overnight, washed, and equilibrated in assay buffer containing 5 mm glucose, incubated for 40 min in either 5 or 20 mm glucose as indicated. Insulin in the assay media was determined and normalized to total insulin in the corresponding islets. For both groups, values are expressed as percentage of total cellular insulin. Data are presented as mean ± S.E.; *, p < 0.05; **, p < 0.01.
DISCUSSION
Insulin secretion by pancreatic beta cells is a complex process that is regulated by a number of autocrine, paracrine, and endocrine regulators (1, 35). Pancreatic beta cell dysfunction leading to defective insulin secretion is thought to be one of the major contributors to the development of T2D (2). Thus, there is a pressing need to understand the factors and pathways that regulate insulin secretion by beta cells and their respective roles in beta cell dysfunction. In the present study, we report the following novel findings: 1) mouse pancreatic islet cells express GX sPLA2, a member of the secretory phospholipase A2 family known to potently hydrolyze membrane phospholipids to release AA; 2) increased expression or exogenous addition of GX sPLA2 suppresses GSIS in MIN6 cells, a mouse beta cell line; 3) islets isolated from GX KO mice exhibit enhanced GSIS ex vivo compared with islets from WT mice; and 4) GX KO mice exhibit significantly enhanced GSIS and appear to be protected from age-related glucose intolerance and beta cell dysfunction. In addition, our in vitro studies support the conclusion that GX sPLA2 suppresses GSIS through a COX-2-dependent, EP3 receptor signaling pathway (Fig. 7).
FIGURE 7.
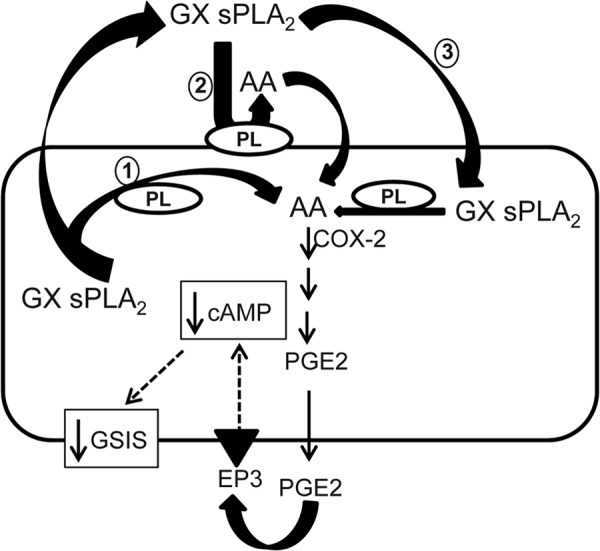
GX sPLA2-mediated regulation of GSIS. GX sPLA2 hydrolysis of cellular membrane phospholipids generates arachidonic acid (AA), which is converted to PGE2 in a COX2-dependent manner. PGE2 released from the cell binds the EP3 receptor on the cell surface, leading to reduced cellular cAMP content and suppressed GSIS. The cellular membrane(s) hydrolyzed by GX sPLA2 to release AA have not been delineated. GX sPLA2 may generate AA during (1) or after (2) its secretion from cells, or subsequent to re-uptake (3).
Recently, Kimple et al. reported that EP3 receptor expression and PGE2 production are significantly elevated in islets from diabetic mice (40–200-fold) and human T2D donors (∼7-fold) compared with non-diabetic controls. Interestingly, treatments with an EP3 receptor antagonist augmented GSIS only in islets from diabetic mouse or human donors and not non-diabetic donors. Furthermore, activation of the EP3 receptor suppressed the effects of glucagon-like peptide-1 on GSIS in this same study (13). Despite extensive evidence establishing the role of PGE2/EP3 axis in suppressing GSIS, the prerequisite PLA2 that generates AA for PGE2 production in beta cells has not been identified. Our results from experiments using a selective COX-2 inhibitor (NS398) and an EP3 receptor antagonist (L-798,106) clearly show that GX sPLA2 suppresses GSIS through a mechanism that is at least partially mediated through the COX2/PGE2/cAMP pathway.
Of the 10 members of the sPLA2 family known to be expressed by mammalian cells, to date only group IB and group IIA sPLA2 have been shown to be expressed in rodent pancreatic islets (36, 37). However, group IB sPLA2 deficiency does not lead to alterations in basal or glucose-stimulated insulin secretion in mice (38) and group IIA sPLA2 is naturally deficient in the inbred C57BL/6 mouse strain (39), ruling out a role for these two isozymes in the current study. We determined by real time RT-PCR that GV and GIID sPLA2 mRNAs are expressed by MIN6 cells, whereas group III, group XIIA, and group XIIB mRNAs were not detected (data not shown). Our studies indicate that expression of GIID sPLA2 is confined to non-insulin producing cells in pancreatic islets (data not shown). Among the known sPLA2s, GX- and GV sPLA2s are reported to be potent in releasing AA leading to eicosanoid generation in a variety of cells both in vitro and in vivo (40). Our unpublished results indicate that GV sPLA2 does not contribute to PGE2 production in islets and in contrast to GX sPLA2 activates GSIS. Further studies are needed to understand how these two closely related enzymes exert opposing effects in beta cells. Furthermore, the cellular membrane(s) hydrolyzed by GX sPLA2 to release AA for PG production has not yet been clearly delineated. While it is generally believed that GX sPLA2 hydrolyzes PLs in the outer leaflet of the plasma membrane, there is also evidence indicating that AA is released prior to secretion of GX sPLA2 (41). Alternatively, externalized GX sPLA2 may be taken up by the cells, where it is coupled to intracellular cyclooxygenases for the production of AA metabolites (Fig. 7). We previously reported that GX sPLA2 modulates cellular functions by negatively regulating LXR activity in macrophages, adrenal cells, and fat cells (30, 31, 43). The LXR target genes ABCA1 and ABCG1 play critical roles in maintaining beta cell function by regulating cellular cholesterol homeostasis. Indeed, cellular cholesterol accumulation is an emerging mechanism leading to beta cell dysfunction in T2D (44). Deficiency of either ABCA1 or ABCG1 in beta cells leads to alterations in cellular cholesterol distribution and impaired insulin secretion (45). We assessed whether GX sPLA2 impacts LXR target gene expression or cholesterol content in primary mouse islets and/or MIN6 cells (data not shown). While GX sPLA2 suppresses LXR target gene expression in beta cells, this effect did not result in significant alterations in total cholesterol content. WT and GX KO islets exhibit similar GSIS when treated with an EP3 receptor antagonist (Fig. 5B), supporting our conclusion that GX sPLA2 suppresses GSIS primarily through PGE2 signaling, and not through altered cholesterol homeostasis. An interesting possibility is that under certain conditions, such as in the setting of hyperlipidemia or in aging, GX sPLA2 contributes to disruptions in cholesterol homeostasis and beta cell dysfunction. Our observation that 15-month-old GX KO mice, unlike WT mice, do not develop glucose intolerance may be related to a cumulative detrimental effect of GX sPLA2 on LXR activation, cholesterol homeostasis, and function in beta cells during aging.
In summary, our results demonstrate that GX sPLA2 negatively regulates GSIS in pancreatic islet cells possibly by generating AA substrate for PGE2 production and consequent reduction of intracellular cAMP. Thus, GX sPLA2 may act as a “molecular brake” to modulate insulin secretion. An important question to address is the regulation of GX sPLA2 in beta cells. GX sPLA2 is synthesized as an inactive zymogen that requires proteolytic cleavage for hydrolytic activity (46). The identity of the protease(s) involved and the factor(s) that regulate their action are not yet known, although it has been suggested that members of the furin-like proprotein convertases may play a role in the processing of GX sPLA2 (47). Interestingly, two proprotein convertases, PC1 and PC2, are highly expressed in pancreatic beta cells, where they mediate the cleavage of proinsulin to form insulin (48, 49), raising the possibility that proteolytic activation of proinsulin and GX sPLA2 may be coordinately regulated. Further studies are needed to address whether aberrant activation of GX sPLA2 exacerbates beta cell dysfunction under pathological conditions, possibly including chronic inflammation (42). Targeting GX sPLA2 may be an effective therapeutic option in enhancing beta cell function in the treatment of diabetes.
Acknowledgments
We thank Dr. Sabire Ozcan, University of Kentucky for providing MIN6 cells and Dr. M.Gelb, University of Washington for supply of recombinant GX sPLA2 enzyme. We thank Dr. Deneys R. van der Westhuyzen for helpful discussion and review of the manuscript. We also thank Dr. Wendy Katz for helping us with paraffin embedding and tissue sectioning. This article is the result of work supported with resources and the use of facilities at the Lexington Veterans Affairs Medical Center, Lexington, KY.
This work was supported in whole or in part by National Institutes of Health Grant R01 DK082419 (to N. R. W.) and the NIGMS Grant 8 P20 GM103527-05 (to P. S.).
- T2D
- type 2 diabetes
- AA
- arachidonic acid
- GSIS
- glucose-stimulated insulin secretion
- GX
- group X
- PLA
- phospholipase A
- COX
- cyclooxygenase
- PGE
- prostaglandin E
- WT
- wild-type
- POPG
- 1-palmitoyl-2-oleoylphosphatidylglycerol
- LXR
- liver X receptor
- ABCA1
- ATP binding cassette transporter A1
- ABCG1
- ATP binding cassette transporter G1
- cAMP
- cyclic AMP.
REFERENCES
- 1. Kahn S. E., Hull R. L., Utzschneider K. M. (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 [DOI] [PubMed] [Google Scholar]
- 2. Prentki M., Nolan C. J. (2006) Islet β cell failure in type 2 diabetes. J. Clin. Invest. 116, 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramanadham S., Bohrer A., Mueller M., Jett P., Gross R. W., Turk J. (1993) Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: Prominence of plasmenylethanolamine molecular species. Biochemistry 32, 5339–5351 [DOI] [PubMed] [Google Scholar]
- 4. Jones P. M., Persaud S. J. (1993) Arachidonic acid as a second messenger in glucose-induced insulin secretion from pancreatic β-cells. J. Endocrinol. 137, 7–14 [DOI] [PubMed] [Google Scholar]
- 5. Persaud S. J., Muller D., Belin V. D., Kitsou-Mylona I., Asare-Anane H., Papadimitriou A., Burns C. J., Huang G. C., Amiel S. A., Jones P. M. (2007) The role of arachidonic acid and its metabolites in insulin secretion from human islets of langerhans. Diabetes 56, 197–203 [DOI] [PubMed] [Google Scholar]
- 6. Keane D., Newsholme P. (2008) Saturated and unsaturated (including arachidonic acid) non-esterified fatty acid modulation of insulin secretion from pancreatic beta-cells. Biochem. Soc. Trans. 36, 955–958 [DOI] [PubMed] [Google Scholar]
- 7. Landt M., Easom R. A., Colca J. R., Wolf B. A., Turk J., Mills L. A., McDaniel M. L. (1992) Parallel effects of arachidonic acid on insulin secretion, calmodulin-dependent protein kinase activity and protein kinase C activity in pancreatic islets. Cell calcium 13, 163–172 [DOI] [PubMed] [Google Scholar]
- 8. Metz S. A. (1988) Exogenous arachidonic acid promotes insulin release from intact or permeabilized rat islets by dual mechanisms. Putative activation of Ca2+ mobilization and protein kinase C. Diabetes 37, 1453–1469 [DOI] [PubMed] [Google Scholar]
- 9. Meng Z. X., Sun J. X., Ling J. J., Lv J. H., Zhu D. Y., Chen Q., Sun Y. J., Han X. (2006) Prostaglandin E2 regulates Foxo activity via the Akt pathway: implications for pancreatic islet beta cell dysfunction. Diabetologia 49, 2959–2968 [DOI] [PubMed] [Google Scholar]
- 10. Meng Z., Lv J., Luo Y., Lin Y., Zhu Y., Nie J., Yang T., Sun Y., Han X. (2009) Forkhead box O1/pancreatic and duodenal homeobox 1 intracellular translocation is regulated by c-Jun N-terminal kinase and involved in prostaglandin E2-induced pancreatic β-cell dysfunction. Endocrinology 150, 5284–5293 [DOI] [PubMed] [Google Scholar]
- 11. Tran P. O. T., Gleason C. E., Poitout V., Robertson R. P. (1999) Prostaglandin E2 mediates inhibition of insulin secretion by interleukin-1β. J. Biol. Chem. 274, 31245–31248 [DOI] [PubMed] [Google Scholar]
- 12. Luo P., Wang M.-H. (2011) Eicosanoids, β-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 95, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kimple M. E., Keller M. P., Rabaglia M. R., Pasker R. L., Truchan N. A., Neuman J. C., Brar H. K., Attie A. D. (2013) The prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes 62, 1904–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coleman R. A., Smith W. L., Narumiya S. (1994) International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 46, 205–229 [PubMed] [Google Scholar]
- 15. Tran P. O. T., Gleason C. E., Robertson R. P. (2002) Inhibition of interleukin-1β-induced COX-2 and EP3 gene expression by sodium salicylate enhances pancreatic islet β-cell function. Diabetes 51, 1772–1778 [DOI] [PubMed] [Google Scholar]
- 16. Smyth E. M., Grosser T., Wang M., Yu Y., FitzGerald G. A. (2009) Prostanoids in health and disease. J. Lipid Res. 50, S423–S428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yajima H., Komatsu M., Schermerhorn T., Aizawa T., Kaneko T., Nagai M., Sharp G. W., Hashizume K. (1999) cAMP enhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes 48, 1006–1012 [DOI] [PubMed] [Google Scholar]
- 18. Dubois R. N., Abramson S. B., Crofford L., Gupta R. A., Simon L. S., Van De Putte L. B., Lipsky P. E. (1998) Cyclooxygenase in biology and disease. FASEB J. 12, 1063–1073 [PubMed] [Google Scholar]
- 19. Sorli C. H., Zhang H.-J., Armstrong M. B., Rajotte R. V., Maclouf J., Robertson R. P. (1998) Basal expression of cyclooxygenase-2 and nuclear factor–interleukin 6 are dominant and coordinately regulated by interleukin 1 in the pancreatic islet. Proc. Natl. Acad. Sci. U.S.A. 95, 1788–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujita H., Kakei M., Fujishima H., Morii T., Yamada Y., Qi Z., Breyer M. D. (2007) Effect of selective cyclooxygenase-2 (COX-2) inhibitor treatment on glucose-stimulated insulin secretion in C57BL/6 mice. Biochem. Biophys. Res. Commun. 363, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Persaud S. J., Burns C. J., Belin V. D., Jones P. M. (2004) Glucose-induced regulation of COX-2 expression in human islets of Langerhans. Diabetes 53, S190–S192 [DOI] [PubMed] [Google Scholar]
- 22. Dennis E. A., Cao J., Hsu Y.-H., Magrioti V., Kokotos G. (2011) Phospholipase A2 Enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chemical Reviews 111, 6130–6185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen M., Yang Z., Naji A., Wolf B. A. (1996) Identification of calcium-dependent phospholipase A2 isoforms in human and rat pancreatic islets and insulin secreting beta-cell lines. Endocrinology 137, 2901–2909 [DOI] [PubMed] [Google Scholar]
- 24. Juhl K., Høy M., Olsen H. L., Bokvist K., Efanov A. M., Hoffmann E. K., Gromada J. (2003) cPLA2α-evoked formation of arachidonic acid and lysophospholipids is required for exocytosis in mouse pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 285, E73–E81 [DOI] [PubMed] [Google Scholar]
- 25. Jones P. M., Burns C. J., Belin V. D., Roderigo-Milne H. M., Persaud S. J. (2004) The role of cytosolic phospholipase A2 in insulin secretion. Diabetes 53, S172–S178 [DOI] [PubMed] [Google Scholar]
- 26. Bao S., Song H., Wohltmann M., Ramanadham S., Jin W., Bohrer A., Turk J. (2006) Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express group VIA Phospholipase A2 and effects of metabolic stress on glucose homeostasis. J. Biol. Chem. 281, 20958–20973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song K., Zhang X., Zhao C., Ang N. T., Ma Z. A. (2005) Inhibition of Ca2+-independent phospholipase A2 results in insufficient insulin secretion and impaired glucose tolerance. Mol. Endocrinol. 19, 504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lambeau G., Gelb M. H. (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77, 495–520 [DOI] [PubMed] [Google Scholar]
- 29. Wooton-Kee C. R., Boyanovsky B. B., Nasser M. S., de Villiers W. J. S., Webb N. R. (2004) Group V sPLA2 Hydrolysis of Low-Density Lipoprotein Results in Spontaneous Particle Aggregation and Promotes Macrophage Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 24, 762–767 [DOI] [PubMed] [Google Scholar]
- 30. Shridas P., Bailey W. M., Gizard F., Oslund R. C., Gelb M. H., Bruemmer D., Webb N. R. (2010) Group X secretory phospholipase A2 negatively regulates ABCA1 and ABCG1 expression and cholesterol efflux in macrophages. Arterioscler. Thromb. Vasc. Biol. 30, 2014–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shridas P., Bailey W. M., Boyanovsky B. B., Oslund R. C., Gelb M. H., Webb N. R. (2010) Group X secretory phospholipase A2 regulates the expression of steroidogenic acute regulatory protein (StAR) in mouse adrenals. J. Biol. Chem. 285, 20031–20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Degousee N., Ghomashchi F., Stefanski E., Singer A., Smart B. P., Borregaard N., Reithmeier R., Lindsay T. F., Lichtenberger C., Reinisch W., Lambeau G., Arm J., Tischfield J., Gelb M. H., Rubin B. B. (2002) Groups IV, V, and X phospholipases A2s in human neutrophils: role in eicosanoid production and gram-negative bacterial phospholipid hydrolysis. J. Biol. Chem. 277, 5061–5073 [DOI] [PubMed] [Google Scholar]
- 33. Morioka Y., Ikeda M., Saiga A., Fujii N., Ishimoto Y., Arita H., Hanasaki K. (2000) Potential role of group X secretory phospholipase A(2) in cyclooxygenase-2-dependent PGE(2) formation during colon tumorigenesis. FEBS Lett. 487, 262–266 [DOI] [PubMed] [Google Scholar]
- 34. Robertson R. P., Tsai P., Little S. A., Zhang H. J., Walseth T. F. (1987) Receptor-mediated adenylate cyclase-coupled mechanism for PGE2 inhibition of insulin secretion in HIT cells. Diabetes 36, 1047–1053 [DOI] [PubMed] [Google Scholar]
- 35. Newgard C. B., McGarry J. D. (1995) Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu. Rev. Biochem. 64, 689–719 [DOI] [PubMed] [Google Scholar]
- 36. Ramanadham S., Ma Z., Arita H., Zhang S., Turk J. (1998) Type IB secretory phospholipase A2 is contained in insulin secretory granules of pancreatic islet beta-cells and is co-secreted with insulin from glucose-stimulated islets. Biochim. Biophys. Acta 1390, 301–312 [DOI] [PubMed] [Google Scholar]
- 37. Ishida-Oku M., Iwase M., Sonoki K., Sasaki N., Imoto H., Uchizono Y. (2010) Expression of secretory phospholipase A2 in insulitis of human transplanted pancreas and its insulinotropic effect on isolated rat islets. Islets 2, 274–277 [DOI] [PubMed] [Google Scholar]
- 38. Labonté E. D., Kirby R. J., Schildmeyer N. M., Cannon A. M., Huggins K. W., Hui D. Y. (2006) Group 1B phospholipase A2-mediated lysophospholipid absorption directly contributes to postprandial hyperglycemia. Diabetes 55, 935–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kennedy B. P., Payette P., Mudgett J., Vadas P., Pruzanski W., Kwan M., Tang C., Rancourt D. E., Cromlish W. A. (1995) A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J. Biol. Chem. 270, 22378–22385 [DOI] [PubMed] [Google Scholar]
- 40. Murakami M., Taketomi Y., Miki Y., Sato H., Hirabayashi T., Yamamoto K. (2011) Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog. Lipid Res. 50, 152–192 [DOI] [PubMed] [Google Scholar]
- 41. Mounier C. M., Ghomashchi F., Lindsay M. R., James S., Singer A. G., Parton R. G., Gelb M. H. (2004) Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A(2) occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A(2)-α. J. Biol. Chem. 279, 25024–25038 [DOI] [PubMed] [Google Scholar]
- 42. Ohtsuki M., Taketomi Y., Arata S., Masuda S., Ishikawa Y., Ishii T., Takanezawa Y., Aoki J., Arai H., Yamamoto K., Kudo I., Murakami M. (2006) Transgenic expression of group V, but not group X, secreted phospholipase A2 in mice leads to neonatal lethality because of lung dysfunction. J. Biol. Chem. 281, 36420–36433 [DOI] [PubMed] [Google Scholar]
- 43. Li X., Shridas P., Forrest K., Bailey W., Webb N. R. (2010) Group X secretory phospholipase A2 negatively regulates adipogenesis in murine models. FASEB J. 24, 4313–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fryirs M., Barter P. J., Rye K. A. (2009) Cholesterol metabolism and pancreatic beta-cell function. Curr. Opin. Lipidol. 20, 159–164 [DOI] [PubMed] [Google Scholar]
- 45. Kruit J. K., Wijesekara N., Westwell-Roper C., Vanmierlo T., de Haan W., Bhattacharjee A., Tang R., Wellington C. L., LütJohann D., Johnson J. D., Brunham L. R., Verchere C. B., Hayden M. R. (2012) Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function. Diabetes 61, 659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cupillard L., Koumanov K., Mattéi M.-G., Lazdunski M., Lambeau G. (1997) Cloning, chromosomal mapping, and expression of a novel human secretory phospholipase A2. J. Biol. Chem. 272, 15745–15752 [DOI] [PubMed] [Google Scholar]
- 47. Jemel I., Ii H., Oslund R. C., Payré C., Dabert-Gay A.-S., Douguet D., Chargui K., Scarzello S., Gelb M. H., Lambeau G. (2011) Group X secreted phospholipase A2 proenzyme is matured by a furin-like proprotein convertase and releases arachidonic acid inside of human HEK293 cells. J. Biol. Chem. 286, 36509–36521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smeekens S. P., Montag A. G., Thomas G., Albiges-Rizo C., Carroll R., Benig M., Phillips L. A., Martin S., Ohagi S., Gardner P. (1992) Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proc. Natl. Acad. Sci. U.S.A. 89, 8822–8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bennett D. L., Bailyes E. M., Nielsen E., Guest P. C., Rutherford N. G., Arden S. D., Hutton J. C. (1992) Identification of the type 2 proinsulin processing endopeptidase as PC2, a member of the eukaryote subtilisin family. J. Biol. Chem. 267, 15229–15236 [PubMed] [Google Scholar]