

Background: Point mutations in two genes encoding chaperonin subunits have been implicated in neuropathies.
Results: CCT4 and CCT5 proteins carrying these mutations were expressed in bacteria and investigated for their biochemical defects.
Conclusion: H147R CCT5 is faulty in chaperoning function, whereas C450Y CCT4 may be defective in protein stability.
Significance: These biochemical defects may be the source of these neuropathies in patients.
Keywords: Actin, Crystallin, Molecular Chaperone, Protein Folding, Protein Stability, CCT, TRiC, Chaperonin, Complex Assembly, Neuropathy
Abstract
Hereditary sensory neuropathies are a class of disorders marked by degeneration of the nerve fibers in the sensory periphery neurons. Recently, two mutations were identified in the subunits of the eukaryotic cytosolic chaperonin TRiC, a protein machine responsible for folding actin and tubulin in the cell. C450Y CCT4 was identified in a stock of Sprague-Dawley rats, whereas H147R CCT5 was found in a human Moroccan family. As with many genetically identified mutations associated with neuropathies, the underlying molecular basis of the mutants was not defined. We investigated the biochemical properties of these mutants using an expression system in Escherichia coli that produces homo-oligomeric rings of CCT4 and CCT5. Full-length versions of both mutant protein chains were expressed in E. coli at levels approaching that of the WT chains. Sucrose gradient centrifugation revealed chaperonin-sized complexes of both WT and mutant chaperonins, but with reduced recovery of C450Y CCT4 soluble subunits. Electron microscopy of negatively stained samples of C450Y CCT4 revealed few ring-shaped species, whereas WT CCT4, H147R CCT5, and WT CCT5 revealed similar ring structures. CCT5 complexes were assayed for their ability to suppress aggregation of and refold the model substrate γd-crystallin, suppress aggregation of mutant huntingtin, and refold the physiological substrate β-actin in vitro. H147R CCT5 was not as efficient in chaperoning these substrates as WT CCT5. The subtle effects of these mutations are consistent with the homozygous disease phenotype, in which most functions are carried out during development and adulthood, but some selective function is lost or reduced.
Introduction
Sensory neurons are nerve cells that convert external stimuli from the environment into internal stimuli. A rare group of disorders, hereditary sensory neuropathies (HSNs),2 affect sensory neurons, resulting in a range of clinical symptoms (1). These disorders are marked by the degeneration of the myelinated nerve fibers in the peripheral sensory neurons and the autonomic neurons that control the involuntary nervous system (1, 2). These defects may manifest as ulceration of the feet, inability to feel pain (especially in the lower limbs), and severe pain in the distal limbs (1, 2). Genetic screening of many neuropathy families has led to the discovery of several mutated genes associated with HSNs and other related neuropathy diseases. These neuropathies may be inherited through autosomal dominant or autosomal recessive forms and are heterogeneous in their pathological and behavioral symptoms (2–4). Although age of onset is variable, severe instances of this disease can result in both onset and death in childhood (2).
Point mutations in three chaperonin genes have been implicated in this class of neuropathies (Table 1) (5–7). Although only two are true HSNs, the other, hereditary spastic paraplegia, has some important phenotypic overlaps with HSNs (8). One of these HSNs is actually characterized as being an HSN with spastic paraplegia, even further showing the phenotypic heterogeneity of these disorders (5). Two of these have been found in human populations, making their study potentially valuable for understanding and eventually treating human neuropathy diseases. How these mutations lead to the disease phenotypes is still unknown (1).
TABLE 1.
Mutations in chaperonin genes leading to neuropathy diseases
Chaperonins are ATP-dependent chaperones that assist in folding substrate proteins inside a cavity. They are made of back-to-back rings of seven to nine subunits each (9). Chaperonins are divided into two classes: type I, found in bacteria, chloroplasts, and mitochondria; and type II, found in archaeal and eukaryotic cytosol (9). Although there are structural and functional differences between the two classes, they share the same domain architecture: an equatorial domain making subunit-subunit contacts and forming the ATP-binding and hydrolysis site, an apical domain recognizing substrate to be brought into the cavity, and an intermediate domain acting as a hinge-like region between the other two domains (9). The eukaryotic cytosolic chaperonin TRiC (TCP-1 ring complex) is involved in the folding and assembly of dozens of essential eukaryotic proteins (9, 10). The most important proteins it folds are tubulin and actin, which are especially crucial in neurons (11). Unlike most of the type I and some of the archaeal type II chaperonins, which contain identical subunits in both rings, TRiC contains eight different subunits (termed CCT1–8, chaperonin containing TCP-1–8) in each of its two rings (10).
Two of the identified HSNs have mutations in two of the CCT genes: CCT4 and CCT5. A point mutation in the CCT5 gene (A492G) has been associated with human HSN in a Moroccan family (5). These patients are homozygous recessive for this mutation in exon 4 of the CCT5 gene, which translates to H147R in the protein (5). HSN has also been identified in a Sprague-Dawley rat strain, associated with a single-point mutation in the CCT4 gene (G1349A) (7). The affected rats are homozygous recessive for this mutation in CCT4, resulting in the C450Y mutant in the protein (7). Both His147 in CCT5 and Cys450 in CCT4 are well conserved in a variety of species (5, 7). Both mutant amino acid replacements are in the equatorial domain of the CCT subunit, therefore possibly affecting intra- or inter-ring formation in the chaperonin complex or ATP hydrolysis activity. However, the actual molecular basis has not been investigated.
The other chaperonin mutation leading to neuropathy is V98I in mitochondrial Hsp60 (HSPD1 gene), identified in a French family with hereditary spastic paraplegia (6). Although this is in a type I chaperonin, unlike the type II chaperonin CCT mutations, the two chaperonins have similar functions and may therefore share a molecular defect to manifest similar disease phenotypes. This mutant protein was studied biochemically and within bacterial cells. In vitro studies showed that this substitution affected both ATP hydrolysis and chaperoning (aggregation suppression and refolding) ability as a homo-oligomer (12). In vivo studies showed that the ATP hydrolysis defect was ameliorated when only a few of the mutant subunits were in the chaperonin rings. However, the chaperoning defect, although slight, was enough to cause problems with protein folding (12). Having a subtle defect in these diseases is not too surprising because these patients do live to adulthood, so the chaperonins have to be functional, albeit slightly suppressed, through their lifetimes.
Human TRiC expressed in HeLa cells is assembled from eight different protein subunits (13) and has not been amenable to efficient genetic manipulation. However, CCT4 and CCT5 subunits form homo-oligomeric TRiC-like rings when expressed in Escherichia coli (14). These rings have eight subunits per ring and are active in hydrolyzing ATP, suppressing aggregation, and refolding a variety of substrates (14). Therefore, we have used expression of the single CCT4 and CCT5 subunits as an experimental system to study the biochemical basis of the CCT4 and CCT5 mutants associated with HSNs.
EXPERIMENTAL PROCEDURES
Mutagenesis and Expression
Wild-type plasmids were previously constructed by modifying the pET21b vector to contain a tobacco etch virus (TEV) protease cleavage site between the end of the inserted gene (CCT4 or CCT5) and the C-terminal His6 tag (14). Site-directed mutagenesis was used to introduce the neuropathy mutations (G1349A to make C450Y in CCT4, and A440G to make H147R in CCT5) into the plasmids. Mutations were confirmed by sequencing (GENEWIZ). Plasmids were transformed into E. coli BL21(DE3)-RIL cells. Proteins were expressed as described previously (14). Briefly, the cells were grown in Super Broth to A = 5.0 at 37 °C and then shifted to 18 °C and induced with 0.5 mm isopropyl β-d-thiogalactopyranoside. After overnight induction, cultures were pelleted by centrifugation for 15 min, and cells were resuspended in CCT-A buffer (20 mm HEPES/KOH (pH 7.4), 300 mm NaCl, 10 mm MgCl2, 10% glycerol, 1 mm DTT, and 1 mm ATP) with the addition of one EDTA-free Complete protease inhibitor tablet (Roche Applied Science) per liter of culture.
Long-term Lysate Supernatant/Pellet Separation
E. coli cells expressing WT and mutants CCT4 and CCT5 were grown and expressed as described above but without the addition of protease inhibitors. The cells were lysed via a French press and incubated at 4 °C. At specified time points (0, 4, 7, 11, and 14 days), 200-μl aliquots were taken from the lysates and spun down at 11,500 × g for 30 min. The supernatants were extracted, and the pellets were resuspended in CCT-A buffer. SDS-PAGE loading dye (see below) was added to both the supernatant and pellet, and samples were boiled for 10 min and then frozen at −20 °C until all samples were collected.
Sucrose Gradient Sedimentation
Using CCT-A buffer, 5–40% sucrose gradients were prepared using the Gradient Master (BioComp Instruments, Inc.). Lysates (100 μl) were added carefully to the top, and gradients were ultracentrifuged at 4 °C for 18 h at 37,000 rpm using a Beckman SW41 rotor. Twenty fractions were collected using the Gradient Fractionator (BioComp Instruments, Inc.), and one bottom fraction was collected from the remaining gradient.
SDS-PAGE and Immunoblotting
Proteins were separated by SDS-PAGE (10, 12, or 14%) at 165 V for 1 h after boiling in reducing buffer (60 mm Tris (pH 6.8), 2% SDS, 5% β-mercaptoethanol, 10% glycerol, and bromphenol blue for color) for 5 min. The gels were stained with Coomassie Blue. Transfer was conducted for 1.5 h at 300 mA in transfer buffer (10% methanol, 25 mm Tris, and 192 mm glycine) onto 0.45-μm PVDF membranes (Millipore). The anti-CCT4 (sc-48865) and anti-CCT5 (sc-13886) primary antibodies for the CCT subunits were from Santa Cruz Biotechnology. The secondary antibodies were alkaline phosphatase-conjugated (Millipore), and the membranes were visualized using an alkaline phosphatase-conjugate substrate kit (Bio-Rad). Band quantification was done using ImageJ.
CCT Subunit Purification
Purification was carried out as described previously (14) with a few slight differences outlined below. Briefly, after lysis via a French press, the lysate was centrifuged, and the supernatant was removed by pipetting. The supernatant was passed through a 0.45-μm filter and loaded on a cobalt-nitrilotriacetic acid (Co-NTA) column (Pierce). After loading, the column was first washed with 100% CCT-A buffer and then with 5% CCT-B buffer (CCT-A buffer with 250 mm imidazole), and the CCT subunit was eluted from the column in a linear gradient from 5 to 100% CCT-B buffer. The CCT4 protein was washed with more column volumes of 5% CCT-B buffer than the CCT5 protein due to the presence of a 53-kDa fragment that could be decreased by more thorough washing at that percentage of imidazole. The fractions containing the CCT subunit were combined, concentrated, and diluted with CCT-A buffer down to 25 mm imidazole. The His tag was cleaved by TEV protease, and the protein was applied again to the Co-NTA column, to which it no longer bound. The fractions containing the CCT subunit were combined, further concentrated, and passed over a Superose 6 10/300 GL size exclusion column (GE Healthcare). CCT subunits were eluted from the size exclusion column with CCT-A buffer but with 5% glycerol and no ATP. These fractions were pooled and concentrated, and the protein concentration was measured using the Bradford assay (Bio-Rad) with BSA as the standard.
Electron Microscopy and Circular Dichroism
Negative stain transmission electron microscopy was carried out as described previously (14). The secondary structure of the chaperonins was assayed by far-UV CD at 100 μg/ml protein in filtered and degassed 10 mm Tris and 20 mm KCl. Spectra from 260 to 195 nm were obtained for each chaperonin and the buffer using an Aviv Model 202 CD spectrophotometer. Thermal denaturation was carried out by increasing the temperature in 5 °C increments from 25 to 100 °C, with a 5-min incubation before each spectrum was measured. Mean molar ellipticity at 227 nm was used as the metric for percentage of protein folding. Points were fit to a two-state denaturation curve in Prism (GraphPad).
Native Gel Electrophoresis
CCT5 and its mutant were diluted to 0.5 mg/ml and mixed 2:1 with Bio-Rad native sample buffer. Samples were loaded on Criterion XT 3–8% Tris acetate gels (Bio-Rad) with 100 mm Tricine and 100 mm Tris base running buffer (the cathode buffer contained 0.02% Coomassie Blue G 250). Gels were run at 4 °C either for 3 h at 150 V or overnight at 10 mA and stained with Coomassie Blue.
ATP Hydrolysis and Human γd-Crystallin Refolding Assays
The ATP hydrolysis assay was first described by Reissmann et al. (15) and repeated with slight modifications by Sergeeva et al. (14). The human γd-crystallin aggregation suppression assay is described in detail by Acosta-Sampson and King (16) and Knee et al. (17) and was modified by Sergeeva et al. (14) to the conditions used in this study. Refolding percentages were calculated as described by Sergeeva et al. (18) with the same mutant human γd-crystallin protein (Y92A/Y97A) purification outlined there.
Mutant Huntingtin Aggregation Suppression Assay
The mutant huntingtin (mHtt) aggregation suppression assay was modified from that described by Tam et al. (19). Briefly, GST-, His-, and S·Tag-tagged exon 1 of Htt with 51 polyglutamines and containing a TEV protease cleavage site between the GST tag and the rest of construct was purified using a Co-NTA column, followed by a glutathione-agarose column (Pierce). To initiate an aggregation suppression reaction, 5 μm mHtt protein in buffer (20 mm Tris, 50 mm KCl, 5 mm MgCl2, 5 mm DTT, and 1 mm ATP) containing various concentrations of chaperonin was cleaved with 0.1 mm TEV protease. This reaction was left at 30 °C for 16 h. The reaction was stopped by equal volume addition of 4% SDS, boiled for 10 min, and filtered through 0.22-μm cellulose acetate membrane (GE Healthcare). The membrane was washed and blocked using 5% milk in TBS. An alkaline phosphatase-conjugated antibody against the S·Tag (EMD Millipore) was used to detect the amount of mHtt trapped in the membrane. Ovalbumin was used as a control, and the concentration of CCT5 was calculated as described for the human γD-crystallin assay. Quantification was performed using ImageJ, and suppression was calculated as the decrease from the ovalbumin control.
Actin Refolding Assay
The actin refolding assay was modified from Machida et al. (20). Briefly, pET28a containing T7- and His-tagged β-actin was translated using the PURExpress in vitro synthesis kit (New England Biolabs) for 2 h at 37 °C. The translated actin was diluted by half into an equal mixture of buffer (100 mm HEPES/KOH (pH 7.5), 300 mm KCl, 10 mm MgCl2, and 1 mm ATP) and 4 mg/ml chaperonin or BSA, and actin was allowed to refold for 2 h at 37 °C. Ionic strength (changing the KCl concentration to 100 and 500 mm) and concentration (changing the chaperonin concentration to 1 and 2 mg/ml) were also varied. Trypsin was added to a final concentration of 20 ng/μl for 15 min at 32 °C to degrade all non-native actin. SDS-PAGE loading dye (see above) was added to the samples, and samples were boiled for 10 min. Samples were run on 12% SDS-polyacrylamide gel, transferred to PVDF, and probed with anti-T7 antibody (Novagen). Quantification was performed using ImageJ, with ratios taken for each in vitro actin experiment and normalized to 1000 for WT CCT5.
RESULTS
Mutant Protein Expression and Stability
Each neuropathy mutation was introduced into the plasmid constructs containing the WT CCT4 or CCT5 sequences using site-directed mutagenesis. Both full-length mutant proteins were expressed in E. coli at sufficiently high levels to be directly identifiable using Coomassie Blue stain (Fig. 1). The mutant expression level was divided by the WT expression levels for each dilution to quantify how much less of the mutant was expressed. For C450Y CCT4, the levels monitored by Coomassie Blue stain were reduced to ∼80% compared with WT CCT4 in both the supernatant and pellet. By Coomassie Blue stain, CCT5 expression levels were comparable for both the WT and H147R mutant in the supernatant. In the pellet, the levels of the mutant were slightly lower than those of the WT, at ∼80% of the WT accumulation.
FIGURE 1.

Expression levels of CCT4, CCT5, and their neuropathy mutants. The supernatant (left) and pellet (right) of E. coli cells expressing CCT4 or CCT5 were diluted by half from 1:25 to 1:800. Solid arrows point to the full-length CCT protein, and dashed arrows point to the CCT4 fragment (Frag.) of 53 kDa. The expression levels were quantified using ImageJ and calculated for the mutants as mutant level divided by WT level for each dilution. For the Coomassie Blue-stained gels (upper), the expression levels were almost the same in the WT and mutant. The immunoblots (against CCT4 and CCT5, respectively; lower) show a decreased recovery of antigenic C450Y CCT4 compared with WT CCT4 in the supernatant.
To increase the sensitivity of detection of the truncated chains, the same gels were probed with anti-CCT4 and anti-CCT5 antibodies. With the increased sensitivity of immunoblotting, a shorter fragment of 53 kDa was clearly detected for both WT and C450Y CCT4 chains. This truncated product was previously shown to be missing the first 60 amino acids of the protein due to either a delayed translation start or a specific protease in the E. coli lysate (14). The shortened mutant chain was present at higher levels in the pellet than in the supernatant. This suggests association into an inclusion body, common for misfolded or incomplete polypeptide chains.
A more significant difference was seen in the recovery of C450Y CCT4 compared with WT CCT4. The mutant chains accumulated to ∼30% of the WT level in the supernatant and 60% of the WT level in the pellet. This presumably represents reduced efficiency in the partial refolding of the chains during the transfer out of SDS to the membrane in the immunoblot procedure. This is consistent with increased fractionation of the mutant chains into the pellet fractions. In the immunoblot assays using the anti-CCT5 antibody, H147R CCT5 was not significantly decreased compared with WT CCT5 in either the supernatant or pellet. Therefore, the expression and recovery of the H147R CCT5 mutant in E. coli did not differ from that of WT CCT5.
To understand the fate of both WT and mutant chains, we incubated the lysate at 4 °C without protease inhibitors for up to 2 weeks, taking samples for pellet/supernatant separations every 3–4 days. Over time, both WT and mutant proteins accumulated in the pellet, suggesting that they became aggregated rather than becoming susceptible to proteases and being degraded in the lysates (Fig. 2). This was especially true for C450Y CCT4, which was mostly in the pellet fraction by about day 7, as shown by both the Coomassie Blue-stained gel and immunoblotting. WT CCT4 had a much higher level in the pellet initially compared with C450Y CCT4 as seen by immunoblotting, especially for the 53-kDa fragment. However, by looking at the Coomassie Blue-stained gel, we see that WT CCT4 also accumulated in the pellet over time, but more slowly compared with C450Y CCT4. As seen on the Coomassie Blue-stained gel and immunoblot of CCT5, both WT and mutant levels in the pellet increased from days 0 to 11, suggesting that a fraction of the chains aggregated. However, overall, the amount of CCT5 in the pellet and supernatant changed much less over time compared with that of CCT4. In general, CCT5 was more stable than CCT4 in the lysate over the period assayed, with C450Y CCT4 being the least stable subunit of the four tested. The loss of soluble chains seems to be due to aggregation rather than proteolysis for all four proteins.
FIGURE 2.

Long-term lysate incubation of CCT4, CCT5, and their neuropathy mutants. Lysates of CCT4 (A) and CCT5 (B) and their neuropathy mutants (Mut) were incubated for 0, 4, 7, or 11 days and then subjected to pellet (P)/supernatant (S) separations. Both Coomassie Blue-stained gels and immunoblots are shown with full-length CCT4 or CCT5 between the dotted lines, respectively. Two E. coli fragments (asterisks) and two CCT4 fragments (+) that accumulated in the pellet are indicated. The full-length proteins are quantified to the right of each gel, with the WT in blue and the mutant in magenta. The pellet is indicated by circles with solid lines, and the supernatant is indicated by squares with dashed lines. Both CCT4 and CCT5 and their neuropathy mutants (especially C450Y CCT4) accumulated in the pellet over time, suggesting aggregation of the full-length species. AU, arbitrary units.
Mutant Protein Sedimentation
The supernatants of the E. coli lysates expressing both WT and mutant chaperonins were assayed by sucrose gradient ultracentrifugation to identify whether they were organized into high molecular weight complexes (Fig. 3). The sedimentation patterns for both WT and H147R CCT5 were similar, with a distinct species in the 18 S complex region and some presence of soluble subunits at the top of the gradient. WT CCT4 exhibited a distinct 22 S complex species composed of both full-length and truncated CCT4 chains. For the C450Y CCT4 lysate, recovery of unassembled subunits was sharply reduced compared with the WT control. The majority of C450Y subunits recovered sedimented at the 22 S region of the gradient, but the mutant species seemed to sediment slightly faster and more broadly than the WT species. The rapidly sedimenting chains to the right of the 22 S peak may represent aggregated chains, corresponding to the increased recovery in the pellets from Fig. 1. The mutant fragments behaved similarly to the full-length mutant chains. This overall pattern is consistent with misfolding and loss of mutant soluble subunits: either through degradation or inclusion body formation, but with some successful assembly of the remaining subunits. The two CCT oligomer species, identified here as 18 S and 22 S, sedimented similarly to the sedimentation at 20 S seen for endogenous WT TRiC isolated from HeLa cells (13).
FIGURE 3.
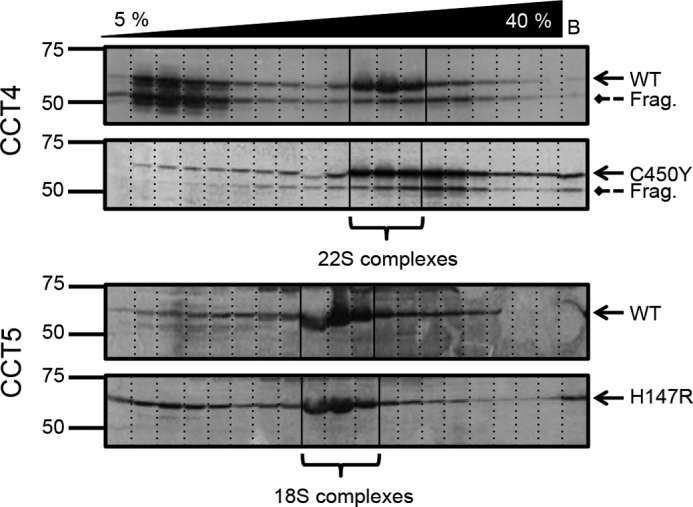
Sucrose ultracentrifugation gradients of CCT4, CCT5, and their neuropathy mutants. Centrifuged lysates were immunoblotted for CCT4 (upper) and CCT5 (lower). Solid arrows point to the full-length CCT protein, and dashed arrows point to the CCT4 fragment (Frag.) of 53 kDa. C450Y CCT4 showed a distinctly different sedimentation pattern (no soluble monomer species and a more broad 22 S species, possibly sedimenting slightly faster) compared with WT CCT4. WT and H147R CCT5 had very similar sedimentation patterns. The WT sedimentation patterns shown here are consistent with those published by Sergeeva et al. (14) but have been more specifically labeled as 22 S and 18 S for CCT4 and CCT5, respectively.
Mutant Protein Purification
The CCT chaperonins and their neuropathy mutants were purified from the lysates by cobalt affinity chromatography (Fig. 4). The elution profiles of WT and H147R CCT5 were similar, with both proteins eluting from the cobalt affinity column in approximately the same amounts. For CCT4, the pattern of elution of full-length WT chains differed from that of the fragment, suggesting that they were not in a complex with each other under these conditions. Note that the fragments also bound to the cobalt column, indicating that they carry the C-terminal His tags. To decrease the amount of CCT4 fragment eluting with full-length CCT4 from the cobalt column, a longer 5% CCT-B buffer wash was used. Therefore, the WT CCT4 protein partitioned between weakly bound chains eluting at low imidazole and tightly bound chains eluting at higher concentrations. C450Y CCT4 (both full-length and the fragment) was recovered from the column at significantly lower levels compared with WT CCT4. This suggested that the conformation and stability of the mutant CCT4 subunits were altered, so the protein was either aggregating or it no longer efficiently bound to the cobalt column. This may be because the His tag was buried or otherwise inaccessible for binding.
FIGURE 4.
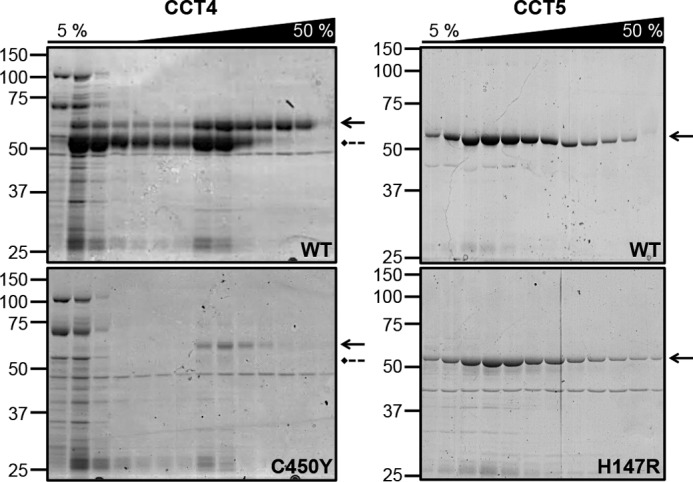
CCT4 and CCT5 purification from a Co-NTA column. Fractions of CCT4 (left: WT, upper, and C450Y, lower) and CCT5 (right: WT, upper, and H147R, lower) from the 5% wash (5%) and elution (50%) from the Co-NTA column were run on 10% Coomassie Blue-stained SDS-polyacrylamide gel. Solid arrows point to the full-length CCT protein, and dashed arrows point to the CCT4 fragment of 53 kDa. WT CCT4 had significantly more protein eluting from the column compared with C450Y CCT4, even with the difference in expression levels taken into account. There was no significant difference between the elution of WT and H147R CCT5.
Both WT and mutant proteins were further purified by TEV protease cleavage to remove the His tag, followed by size exclusion chromatography. Due to the low concentration eluted from the cobalt column, the C450Y CCT4 mutant protein was much less pure and at a significantly lower yield compared with WT CCT4. However, it did elute from the size exclusion column at the same place as WT CCT4, suggesting that some of the ring-like complexes were assembled, but they were not stable or sufficient enough for a large sample to be purified. This limited our ability to assay the properties of C450Y CCT4 compared with WT CCT4. In contrast, the neuropathy mutant of CCT5 was successfully purified with the His tag removed to levels similar to those of WT CCT5.
Mutant Protein Structure
The final purified samples eluted from the size exclusion column were examined by negative stain transmission electron microscopy. WT CCT4, WT CCT5, and H147R CCT5 all had similar morphology (Fig. 5), appearing as well formed rings oriented along the beam axis. C450Y CCT4 had few-to-no rings and, for the most part, appeared as aggregated species by negative stain transmission electron microscopy. The lack of ring species at the end of the mutant CCT4 purification suggests that the mutant CCT4 protein may be unstable, even in the multimeric state. Fractions of mutant CCT4 eluted from the cobalt column did show a few rings by negative stain EM, and the size exclusion elution volume and lysate sucrose ultracentrifugation gradients did suggest a chaperonin-sized species. Although C450Y CCT4 may be capable of forming rings, they did not persist throughout the purification, possibly succumbing to aggregation or dissociation. Taken together, the experiments in Figs. 1–5 indicate that the defect in C450Y is one of subunit folding and stability.
FIGURE 5.
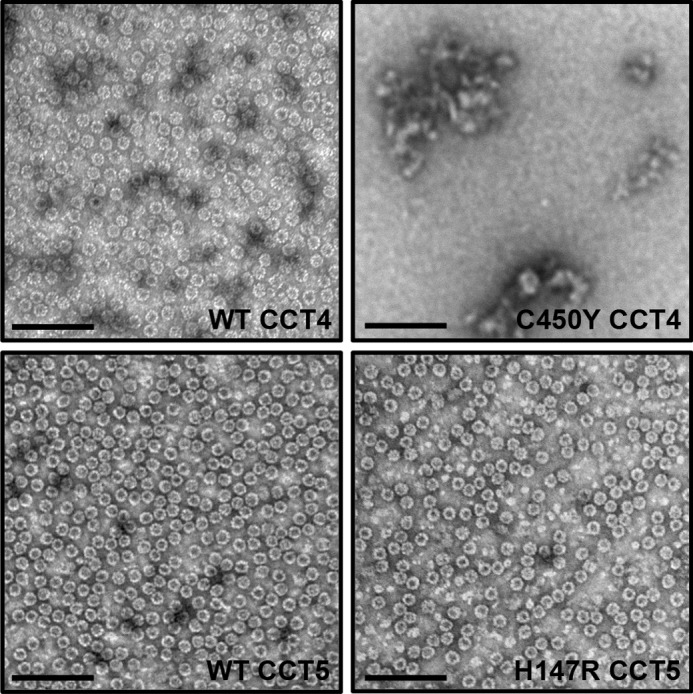
Negative stain transmission electron micrographs of CCT4, CCT5, and their neuropathy mutants. WT CCT4 (upper left), WT CCT5 (lower left), and H147R CCT5 (lower right) formed TRiC-like rings of approximately the same size. They are visualized here after a full purification and elution from the size exclusion column. At the end of the purification, C450Y CCT4 (upper right) contained more aggregates and did not display rings by transmission electron microscopy. Scale bars = 100 nm. WT CCT4 and WT CCT5 rings are consistent with those published by Sergeeva et al. (14).
Purified samples could be obtained for WT and H147R CCT5 and were subjected to native gel electrophoresis. H147R CCT5 repeatedly ran slightly slower than WT CCT5, suggesting that its charge difference was on the surface of the protein, therefore altering its running properties on a native gel (Fig. 6). Additionally, both WT and mutant CCT5 were well formed complexes of ∼1 MDa, with no smear of degraded subunits or monomer subunits. This assay also verified that the protein purified was indeed mutant CCT5.
FIGURE 6.
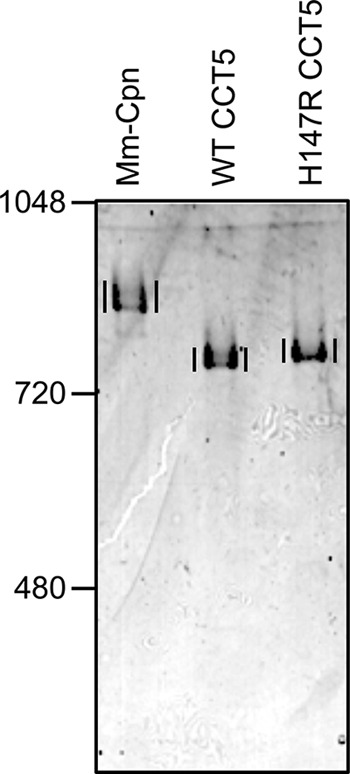
Native gel electrophoresis of CCT5 and its neuropathy mutant. M. marapaludis chaperonin (Mm-Cpn, control), WT CCT5, and H147R CCT5 were subjected to native gel electrophoresis. Vertical lines are for visual comparison and indicate the chaperonin complexes in each lane. The H147R CCT5 mutant ran slightly slower than WT CCT5, suggesting that the mutation alters the outer charge of the mutant chaperonin.
To assess the conformation of the mutant CCT5 subunits, far-UV CD scans of WT and H147R CCT5 were obtained, along with thermal melts of both proteins as tracked by CD. They exhibited very similar spectra with minima at 227 nm and a very similar thermal denaturation midpoint of ∼60 °C (Fig. 7A). The denaturation of the mutant was less cooperative than the denaturation of the WT, possibly pointing to some difference in subunit contacts within or between the rings (Fig. 7B). However, in general, the H147R mutation in CCT5 did not disrupt subunit structure or complex assembly.
FIGURE 7.
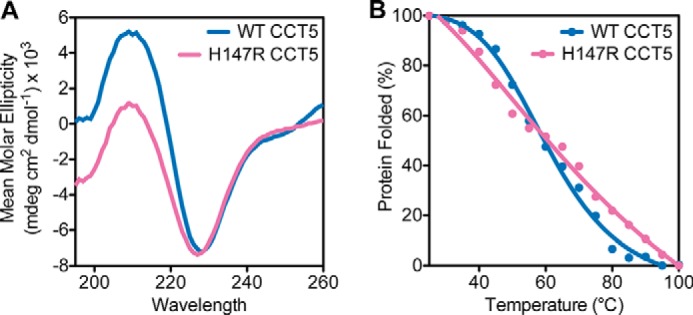
Far-UV CD scans and thermal denaturation of CCT5 and its neuropathy mutant. A, CD scans of WT CCT5 (blue) and H147R CCT5 (magenta) showed similar spectra from 260 to 195 nm, with minima at ∼227 nm. B, thermal denaturation of WT CCT5 (blue) and H147R CCT5 (magenta) by CD had approximately the same midpoint of 60 °C, although the profiles were slightly different in terms of cooperativity. The mean molar ellipticity at 227 was used as the proxy for the percentage of protein folding. The WT CCT5 scan and thermal melt are consistent with those published by Sergeeva et al. (14).
CCT5 Mutant Activity
To investigate how the mutation may lead to neuropathy, chaperonin activity assays were performed. Due to the position of the mutation in the equatorial domain, one likely defect might be in ATP hydrolysis of the mutant chaperonin. The purified WT and H147R CCT5 complexes were therefore assayed for their ability to hydrolyze ATP. As shown in Fig. 8, the hydrolysis rates were very similar between WT and mutant CCT5.
FIGURE 8.
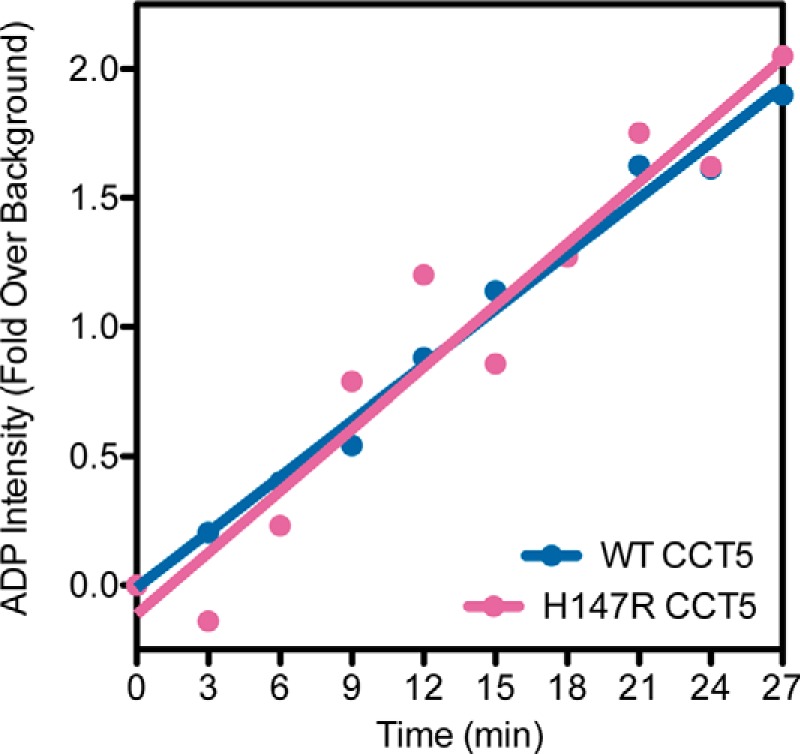
ATP hydrolysis of CCT5 and its neuropathy mutant. WT CCT5 (blue) and H147R CCT5 (magenta) showed similar rates of ATP hydrolysis as measured by quantified generation of [α-32P]ADP over time. The values shown for WT CCT5 were published previously by Sergeeva et al. (14).
The critical functions of group II chaperonins are believed to be suppressing the intracellular aggregation of partially folded intermediates and assisting the folding to the native state. We therefore assayed WT and H147R CCT5 for suppression of off-pathway aggregation and refolding in vitro to the native state. One substrate used in these experiments was human γD-crystallin, whose off-pathway aggregation and productive refolding have been systematically studied (16, 21–28). Endogenous human TRiC purified from HeLa cells and WT CCT4 and CCT5 homo-oligomers are active in both assays (13, 14).
As shown in Fig. 9, the human γD-crystallin chains aggregated to high molecular weight complexes after dilution out of the denaturant (27). When WT CCT5 was added, the aggregation of WT human γD-crystallin was suppressed. This is consistent with what was seen previously for CCT5 suppression of WT human γD-crystallin aggregation (14). H147R CCT5 was able to suppress mutant aggregation at first, but showed an increase in turbidity that was similar to WT human γD-crystallin alone at the end of the reaction (Fig. 9A). Therefore, the mutant protein appears to have an altered reaction with the substrate in this reaction compared with WT CCT5. A potentially more stringent substrate was also assayed with WT and H147R CCT5. In this case, the aggregating protein was human γD-crystallin carrying a double-alanine substitution of tyrosine residues, Y92A/Y97A (18, 25). Suppression of aggregation by WT CCT5 was similar to that found with WT human γD-crystallin (Fig. 9B). The H147R CCT5 protein had an altered interaction compared with WT CCT5, mimicking the results seen for WT human γD-crystallin. For both human γD-crystallin substrates, H147R CCT5 suppressed aggregation less efficiently compared with WT CCT5.
FIGURE 9.
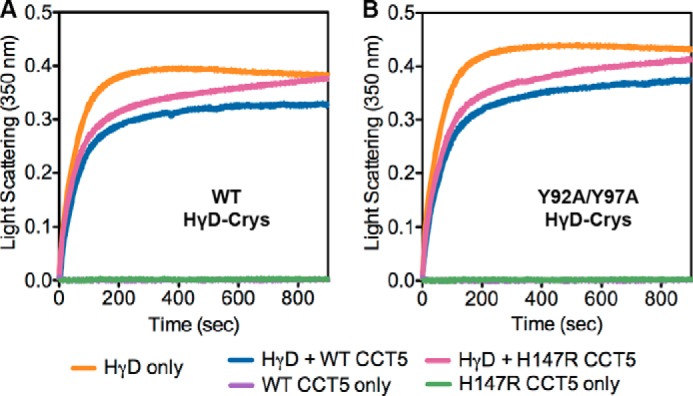
Aggregation suppression of human γD-crystallin by CCT5 and its neuropathy mutant. Aggregation of WT (A, orange) or Y92A/Y97A (B, orange) human γD-crystallin (HγD-Crys, HγD) was suppressed more efficiently by WT CCT5 (blue) than by H147R CCT5 (magenta). Without human γD-crystallin, WT CCT5 (purple) and H147R CCT5 (green) did not show any self-polymerization. The curves are representative; the assays were repeated three to five times and showed the same trends.
We showed that CCT5 had an increase in turbidity throughout the assay, which we attributed to self-polymerization. However, when WT and H147R CCT5 were added to the assay without human γD-crystallin (Fig. 9), we did not see an increase in turbidity, suggesting that it was not self-polymerization but rather polymerization or aggregation of the complex between CCT5 and human γD-crystallin that was causing the increase in turbidity throughout the assay. We cannot exclude that the decrease in aggregation suppression of human γD-crystallin by H147R CCT5 may be due to increased aggregation of the complex between H147R CCT5 and human γD-crystallin.
Along with aggregation suppression, we also assayed the amount of human γD-crystallin refolded by the chaperonin. Residual background refolding was present but was significantly less than the amount of human γD-crystallin actively refolded by the chaperonins (Fig. 10A). When we quantified the amount of WT and Y92A/Y97A human γD-crystallin refolded by WT and H147R CCT5, we observed a significant decrease in the amount refolded by H147R CCT5 compared with WT CCT5 (Fig. 10B). This decrease was ∼30% for WT human γD-crystallin and ∼20% for Y92A/Y97A human γD-crystallin, but the amount of human γD-crystallin refolded by H147R CCT5 was not significantly different from background refolding in both cases. In general, Y92A/Y97A human γD-crystallin was refolded to lower levels compared with WT human γD-crystallin, contrary to what was seen for the archaeal Methanococcus marapaludis chaperonin previously (18).
FIGURE 10.
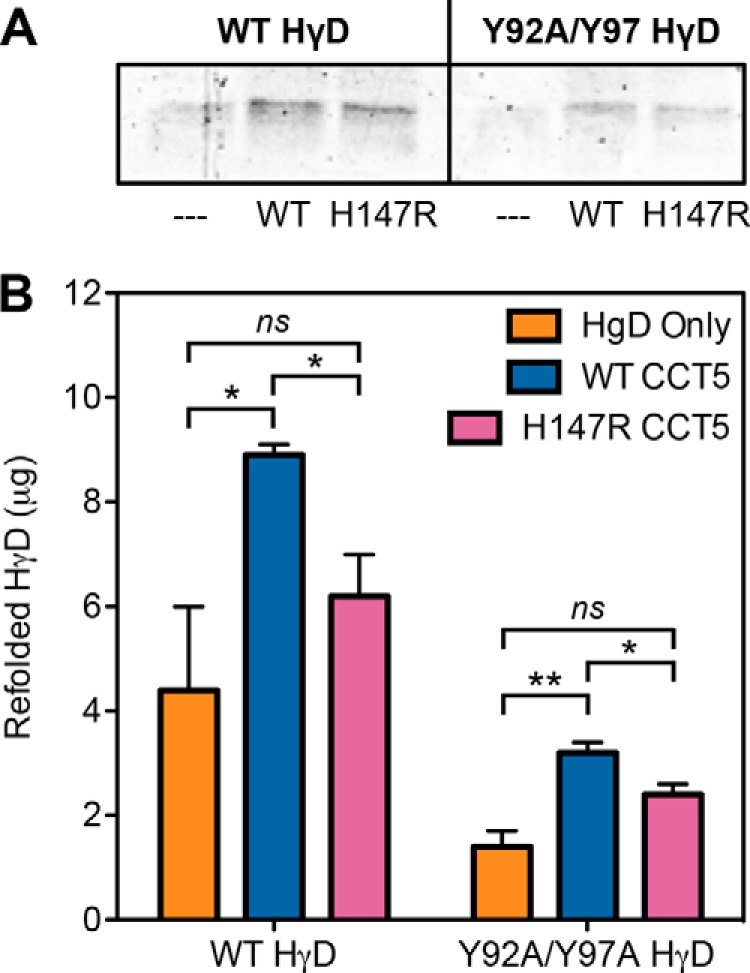
SDS-PAGE and quantification of human γD-crystallin refolded by CCT5 and its neuropathy mutant. A, 14% Coomassie Blue-stained SDS-polyacrylamide gel of either WT (left) or Y92A/Y97A (right) refolded human γD-crystallin (HγD) alone (– – –), with WT CCT5, or with H147R CCT5 is shown. Some residual background refolding can be seen, but there was significantly more refolding by the chaperonins. B, WT CCT5 (blue) refolded significantly more WT (left) or Y92A/Y97A (right) human γD-crystallin than H147R CCT5 (magenta). Both chaperonins refolded more WT than Y92A/Y97A human γD-crystallin. Error bars are S.E. from three independent quantifications. *, significance at p < 0.05 by t test; **, significance at p < 0.01 by t test; ns, not significant.
Although human γD-crystallin is an authentic substrate of TRiC in the periphery of the eye lens, its value is limited when surveying how H147R CCT5 may lead to neuropathy. Therefore, we also challenged mutant CCT5 with two other human substrates associated with the brain. The first was Htt, a very large, 3144-amino acid (348 kDa), soluble cytoplasmic protein. Although it is ubiquitously expressed, it is found at high levels in the central nervous system and testes (29). WT Htt has various functions in cells, such as acting as a scaffold protein and playing a role in neuronal gene transcription and in axonal and vesicular transport (30). Htt in its pathological form contains an expanded repeat of CAG, resulting in 36+ polyglutamines (31). Aggregates of mHtt have been found in patient brains, consistent with the idea that aggregation of the pathological protein is part of the disease (32, 33). These aggregates contain fragments of the mHtt protein, the shortest of which includes only the first exon of Htt, wherein the polyglutamine region is located (29). Previous studies have shown that TRiC interacts with mHtt and decreases its aggregation (19, 34–36). We assayed both WT and H147R CCT5 for their ability to suppress mHtt. For this assay, we used an mHtt protein that was GST-tagged and contained a TEV protease site. When we added TEV protease to the reaction containing mHtt and either WT or H147R CCT5, mHtt aggregated. We were able to see how much aggregation was suppressed by the chaperonins by using a filter trap assay and probing with an antibody against the mHtt construct. Although both were able to suppress mHtt, WT CCT5 was more efficient in at least one concentration compared with H147R CCT5 (Fig. 11).
FIGURE 11.
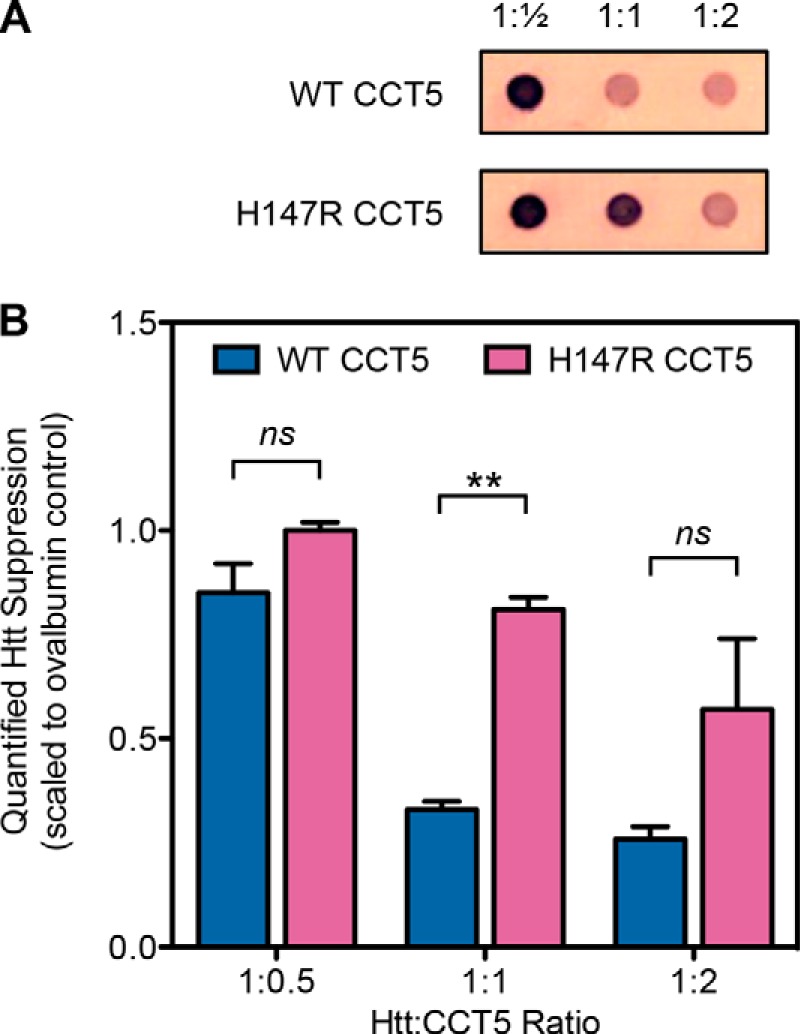
mHtt aggregation suppression by CCT5 and its neuropathy mutant. A, representative filter trap samples probed with an antibody to mHtt. Ratios are mHtt to CCT5. B, quantifications of multiple experiments as in A. WT CCT5 suppressed mHtt more efficiently than H147R CCT5 at all ratios, but only significantly at a ratio of 1:1. Data were normalized to the ovalbumin control (1.0). Error bars are S.E. from two independent quantifications. **, significance at p < 0.01 by t test; ns, not significant.
The second more neuropathy-related substrate we assayed is highly expressed in neurons and is one of the major substrates of TRiC: β-actin (11). For this assay, we synthesized T7-tagged β-actin in vitro and allowed WT or H147R CCT5 to fold it to the native state (with BSA as the control). The samples were cleaved with trypsin so only the native β-actin persisted, run on SDS-polyacrylamide gel, transferred to an immunoblot, and probed with anti-T7 antibody. We found that H147R CCT5 folded significantly less β-actin than WT CCT5 (Fig. 12). However, in this assay, unlike the human γD-crystallin refolding assay, the background folding of β-actin as seen by the BSA negative control was minimal, so the amount folded by H147R CCT5 was still significant. To further investigate the actin-refolding properties, we varied both the concentration of chaperonin and the ionic strength of the buffer (Fig. 13). For each of these conditions, the CCT5 mutant did not refold as much as WT CCT5. Interestingly, although we saw a concentration dependence when we varied the concentration, we were able to confirm that the concentration of KCl in the buffer we used above was the optimal concentration for actin refolding.
FIGURE 12.
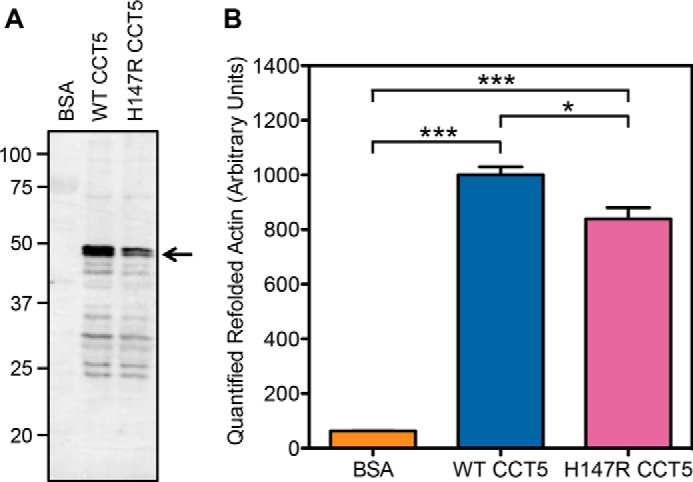
Quantification of β-actin refolded by CCT5 and its neuropathy mutant. A, representative 12% SDS-PAGE immunoblot of refolded actin probed with anti-T7 antibody in the presence of BSA, WT CCT5, or H147R CCT5. The arrow points to β-actin. B, quantification of multiple experiments as in A. H147R CCT5 refolded significantly less actin than WT CCT5. WT CCT5 refolded intensity was normalized to 1000. Error bars are S.E. from four independent quantifications. *, significance at p < 0.05 by t test; ***, significance at p < 0.001 by t test.
FIGURE 13.
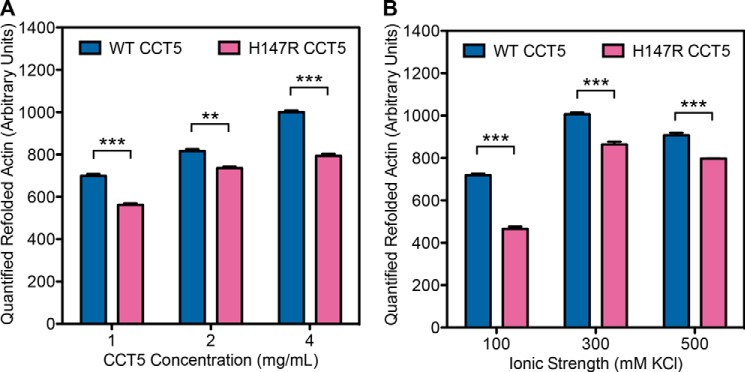
Variations in protein concentration and ionic strength of β-actin refolded by CCT5 and its neuropathy mutant. The same assay as described in the legend to Fig. 12 was performed but with variations in protein concentration (A) and ionic strength of the buffer (B). H147R CCT5 refolded significantly less actin than WT CCT5 in all variations. There was a protein concentration dependence (A), and the optimal ionic strength was 300 mm KCl (B). The conditions used for Fig. 12 were normalized to 1000. Error bars are S.E. from three independent quantifications. **, significance at p < 0.01 by t test; ***, significance at p < 0.001 by t test.
Overall, WT CCT5 was more efficient in suppressing human γD-crystallin aggregation, refolding human γD-crystallin (by ∼30%), suppressing mHtt aggregation (by ∼40%), and folding β-actin compared with H147R CCT5 (by ∼20%). This suggests that the defect in H147R CCT5 is that of chaperonin function.
DISCUSSION
Human CCT4 and CCT5 subunits of the TRiC group II chaperonins assemble into double-barrel TRiC-like rings in the absence of the other seven CCT subunits (14). We have used this homo-oligomerization of CCT4 and CCT5 subunits to investigate two neuropathy mutations identified in these chaperonin subunits. Based on the in vitro work on V98I Hsp60 and the fact that these patients survive to adulthood, we were expecting only subtle differences in the function of these mutant subunits (12).
The H147R CCT5 mutant subunits assembled into oligomeric rings with similar efficiency as WT CCT5 subunits. The melting temperature for the mutant rings was similar to that for WT CCT5, indicating that the H147R substitution did not cause a major defect in chaperonin structure. These chaperonin-like complexes hydrolyzed ATP with similar efficiency as WT CCT5 complexes. However, when assayed for the ability to suppress in vitro aggregation of human γD-crystallin, their efficiency was reduced. The ability of the mutant complexes to chaperone the refolding of human γD-crystallin back to the native-like state was also significantly reduced. Additionally, H147R CCT5 did not suppress mHtt aggregation as efficiently as WT CCT5 and folded significantly less β-actin than WT CCT5. Note, however, that in most of these assays, the mutant complexes exhibited substantial levels of activity with respect to WT CCT5 and negative controls. Our experiments do not distinguish a reduction in the initial efficiency of recognizing and binding partially folded substrates from an actual alteration of the chaperoning reaction that proceeds within the lumen of the complex.
The human γD-crystallin aggregation suppression and refolding assay used in this study has been used for many other chaperonins (13, 14, 17, 18). Interestingly, the crystallin mutant used herein, Y92A/Y97A, was refolded to higher levels by the archaeal M. marapaludis chaperonin (18). Here, both WT and H147R CCT5 refolded the mutant substrate chains to levels of about one-third of those of WT human γD-crystallin chains. H147R CCT5 refolded less of both WT and Y92A/Y97A human γD-crystallin compared with WT CCT5, showing that the refolded amount is even lower than when mutant substrates are chaperoned by this mutant chaperonin. The β-actin assay was used before in various iterations (20, 37, 38), but its use is novel for these homo-oligomeric complexes. Seeing a significant difference between our WT and mutant CCT5 with this stringent substrate bolstered the theory that this mutant is responsible for neuropathy due to its decreased chaperoning ability.
Another discrepancy worth noting is that of mHtt and CCT5. It was shown previously that only CCT1 and CCT4 can suppress aggregation of mHtt (19). Here, we have shown that CCT5 is also capable of suppressing mHtt aggregation. In the previous study, CCT subunits were co-overexpressed in yeast with mHtt constructs. The conformation of the CCT subunits in these overexpressed cells is unknown, so they could have been misfolded or aggregated, therefore not showing any efficacy. These latest results of CCT5 being able to suppress mHtt aggregation may allow the study of CCT subunits other than CCT1 in modulating mHtt aggregation.
The H147R substitution introduces a charge change into the CCT5 subunits. The guanido group of arginine is generally found at the surface of soluble proteins. Direct evidence of this change was seen by native gel electrophoresis. Such increased charge density might reduce chaperonin activity in both suppressing aggregation and refolding due to electrostatic effects.
The defect in C450Y CCT4 was in the folding and stability of the mutant subunit itself, which may affect the complex formation ability. Compared with WT CCT4 chains, a larger percentage of the mutant protein aggregated in the pellet fraction of cells, accumulation of mutant soluble subunits by C450Y CCT4 was reduced, and formation of organized rings by C450Y CCT4 was sharply reduced.
Due to the homo-oligomeric nature of our system, it was hard to assess whether a normal TRiC ring with seven other CCT subunits would be equally disrupted. That may depend on how TRiC is assembled in the cell. If CCT4 is one of the last subunits added, even an unstable CCT4 subunit may be incorporated and function sufficiently as part of the full ring. However, if CCT4 needs to form homo-oligomeric rings on the way to the mature TRiC complex, the mutation may result in a more defective phenotype. In either case, if the C450Y CCT4 mutant folds less efficiently in the cytoplasm or is subject to increased aggregation, it could reduce the levels of functional TRiC, thus affecting folding of any of the numerous TRiC substrates. These are likely to have differential importance in different cell types.
Another feature of the C450Y CCT4 mutant subunit is that the amino acid change itself is from a cysteine to a tyrosine, which may be easily post-translationally modified by a kinase. Either the loss of the cysteine or the gain of the tyrosine could affect post-translational modifications for downstream signaling (39). If C450Y CCT4 does incorporate itself as part of TRiC, it may be modified with respect to WT subunits. Unfortunately, very little is known about the control of chaperonin activity by post-translational modifications of TRiC.
It was encouraging to see a similar chaperoning defect in H147R CCT5 as seen for V98I Hsp60 (12). Although in our system the defect was exaggerated due to the homo-oligomeric nature of the chaperonins, any decrease in the protein-folding function of TRiC will negatively affect many essential substrate proteins, including tubulin and actin. Because neurons contain a high abundance of microtubules, tubulin is a good candidate for a substrate that may be most affected (11). There have been reports of sensory neuropathy induced by taxanes (paclitaxel and docetaxel; anti-cancer drugs used in chemotherapy), and it has been postulated that the taxanes promote microtubule aggregation, specifically in neurons (40). Therefore, the H147R CCT5 HSN may very well be working through the same mechanism.
By studying purified human C450Y CCT4 and H147R CCT5 expressed in E. coli, we have found very subtle biochemical defects in these neuropathy-associated mutants compared with the WT. Whether these defects are exactly the issues contributing to neuropathy within the Moroccan family or the Sprague-Dawley rat strain remains to be determined. To further investigate the chaperonin activity of the CCT5 neuropathy mutant, it will be crucial to use more physiological neuronal substrates in these aggregation and refolding assays. β-Actin is a good first candidate, but others will need to be tested. However, sorting out which substrates are predominantly affected by the CCT mutant substitutions will require characterizing the substrates associated with TRiC within human neuronal cells expressing the neuropathy mutations. The use of patient or rat cell lines of these neuropathies would be ideal for investigating these mutants in the disease context.
Acknowledgments
We thank Dr. Chengbiao Wu for helpful discussions, Koning Shen and Dr. Judith Frydman for the mHtt plasmid, and Drs. Emily Sontag and Leslie Thompson for mHtt protocol suggestions. We thank the Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (supported by National Science Foundation Grant NSF-007031).
This work was supported, in whole in in part, by National Institutes of Health Grants PN2EY016525 and R01EY015834.
- HSN
- hereditary sensory neuropathy
- TEV
- tobacco etch virus
- Co-NTA
- cobalt-nitrilotriacetic acid
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- mHtt
- mutant huntingtin.
REFERENCES
- 1. Auer-Grumbach M. (2008) Hereditary sensory neuropathy type I. Orphanet J. Rare Dis. 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas P. K., Misra V. P., King R. H. M., Muddle J. R., Wroe S., Bhatia K. P., Anderson M., Cabello A., Vilchez J., Wadia N. H. (1994) Autosomal recessive hereditary sensory neuropathy with spastic paraplegia. Brain 117, 651–659 [DOI] [PubMed] [Google Scholar]
- 3. Cavanagh N. P. C., Eames R. A., Galvin R. J., Brett E. M., Kelly R. E. (1979) Hereditary sensory neuropathy with spastic paraplegia. Brain 102, 79–94 [DOI] [PubMed] [Google Scholar]
- 4. Rotthier A., Baets J., De Vriendt E., Jacobs A., Auer-Grumbach M., Lévy N., Bonello-Palot N., Kilic S. S., Weis J., Nascimento A., Swinkels M., Kruyt M. C., Jordanova A., De Jonghe P., Timmerman V. (2009) Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain 132, 2699–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bouhouche A., Benomar A., Bouslam N., Chkili T., Yahyaoui M. (2006) Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 43, 441–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hansen J. J., Dürr A., Cournu-Rebeix I., Georgopoulos C., Ang D., Nielsen M. N., Davoine C.-S., Brice A., Fontaine B., Gregersen N., Bross P. (2002) Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 70, 1328–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee M.-J., Stephenson D. A., Groves M. J., Sweeney M. G., Davis M. B., An S.-F., Houlden H., Salih M. A. M., Timmerman V., de Jonghe P., Auer-Grumbach M., Di Maria E., Scaravilli F., Wood N. W., Reilly M. M. (2003) Hereditary sensory neuropathy is caused by a mutation in the delta subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct4) gene. Hum. Mol. Genet. 12, 1917–1925 [DOI] [PubMed] [Google Scholar]
- 8. Timmerman V., Clowes V. E., Reid E. (2013) Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp. Neurol. 246, 14–25 [DOI] [PubMed] [Google Scholar]
- 9. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 10. Frydman J. (2001) Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu. Rev. Biochem. 70, 603–647 [DOI] [PubMed] [Google Scholar]
- 11. Lundin V. F., Leroux M. R., Stirling P. C. (2010) Quality control of cytoskeletal proteins and human disease. Trends Biochem. Sci. 35, 288–297 [DOI] [PubMed] [Google Scholar]
- 12. Bross P., Naundrup S., Hansen J., Nielsen M. N., Christensen J. H., Kruhøffer M., Palmfeldt J., Corydon T. J., Gregersen N., Ang D., Georgopoulos C., Nielsen K. L. (2008) The Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J. Biol. Chem. 283, 15694–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Knee K. M., Sergeeva O. A., King J. A. (2013) Human TRiC complex purified from HeLa cells contains all eight CCT subunits and is active in vitro. Cell Stress Chaperones 18, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sergeeva O. A., Chen B., Haase-Pettingell C., Ludtke S. J., Chiu W., King J. A. (2013) Human CCT4 And CCT5 chaperonin subunits expressed in Escherichia coli form biologically active homo-oligomers. J. Biol. Chem. 288, 17734–17744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reissmann S., Parnot C., Booth C. R., Chiu W., Frydman J. (2007) Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat. Struct. Mol. Biol. 14, 432–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Acosta-Sampson L., King J. (2010) Partially folded aggregation intermediates of human γD-, γC-, and γS-crystallin are recognized and bound by human αB-crystallin chaperone. J. Mol. Biol. 401, 134–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Knee K. M., Goulet D. R., Zhang J., Chen B., Chiu W., King J. A. (2011) The group II chaperonin Mm-Cpn binds and refolds human γD crystallin. Protein Sci. 20, 30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sergeeva O. A., Yang J., King J. A., Knee K. M. (2014) Group II archaeal chaperonin recognition of partially folded human γD-crystallin mutants. Protein Sci. 23, 693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tam S., Geller R., Spiess C., Frydman J. (2006) The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 8, 1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Machida K., Masutani M., Kobayashi T., Mikami S., Nishino Y., Miyazawa A., Imataka H. (2012) Reconstitution of the human chaperonin CCT by co-expression of the eight distinct subunits in mammalian cells. Protein Expr. Purif. 82, 61–69 [DOI] [PubMed] [Google Scholar]
- 21. Chen J., Flaugh S. L., Callis P. R., King J. (2006) Mechanism of the highly efficient quenching of tryptophan fluorescence in human γD-crystallin. Biochemistry 45, 11552–11563 [DOI] [PubMed] [Google Scholar]
- 22. Flaugh S. L., Kosinski-Collins M. S., King J. (2005) Contributions of hydrophobic domain interface interactions to the folding and stability of human γD-crystallin. Protein Sci. 14, 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flaugh S. L., Kosinski-Collins M. S., King J. (2005) Interdomain side-chain interactions in human γD crystallin influencing folding and stability. Protein Sci. 14, 2030–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flaugh S. L., Mills I. A., King J. (2006) Glutamine deamidation destabilizes human γD-crystallin and lowers the kinetic barrier to unfolding. J. Biol. Chem. 281, 30782–30793 [DOI] [PubMed] [Google Scholar]
- 25. Kong F., King J. (2011) Contributions of aromatic pairs to the folding and stability of long-lived human γD-crystallin. Protein Sci. 20, 513–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kosinski-Collins M. S., Flaugh S. L., King J. (2004) Probing folding and fluorescence quenching in human γD crystallin Greek key domains using triple tryptophan mutant proteins. Protein Sci. 13, 2223–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kosinski-Collins M. S., King J. (2003) In vitro unfolding, refolding, and polymerization of human γD crystallin, a protein involved in cataract formation. Protein Sci. 12, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moreau K. L., King J. (2009) Hydrophobic core mutations associated with cataract development in mice destabilize human γD-crystallin. J. Biol. Chem. 284, 33285–33295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wetzel R. (2012) Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J. Mol. Biol. 421, 466–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bates G. P. (2005) History of genetic disease: the molecular genetics of Huntington disease–a history. Nat. Rev. Genet. 6, 766–773 [DOI] [PubMed] [Google Scholar]
- 31. Walker F. O. (2007) Huntington's disease. Lancet 369, 218–228 [DOI] [PubMed] [Google Scholar]
- 32. Arrasate M., Finkbeiner S. (2012) Protein aggregates in Huntington's disease. Exp. Neurol. 238, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clabough E. B. D. (2013) Huntington's disease: the past, present, and future search for disease modifiers. Yale J. Biol. Med. 86, 217–233 [PMC free article] [PubMed] [Google Scholar]
- 34. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M. A., Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sontag E. M., Joachimiak L. A., Tan Z., Tomlinson A., Housman D. E., Glabe C. G., Potkin S. G., Frydman J., Thompson L. M. (2013) Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc. Natl. Acad. Sci. U.S.A. 110, 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shahmoradian S. H., Galaz-Montoya J. G., Schmid M. F., Cong Y., Ma B., Spiess C., Frydman J., Ludtke S. J., Chiu W. (2013) TRiC's tricks inhibit huntingtin aggregation. eLife 2, e00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pappenberger G., McCormack E. A., Willison K. R. (2006) Quantitative actin folding reactions using yeast CCT purified via an internal tag in the CCT3/γ subunit. J. Mol. Biol. 360, 484–496 [DOI] [PubMed] [Google Scholar]
- 38. Llorca O., McCormack E. A., Hynes G., Grantham J., Cordell J., Carrascosa J. L., Willison K. R., Fernandez J. J., Valpuesta J. M. (1999) Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature 402, 693–696 [DOI] [PubMed] [Google Scholar]
- 39. Abe Y., Yoon S.-O., Kubota K., Mendoza M. C., Gygi S. P., Blenis J. (2009) p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J. Biol. Chem. 284, 14939–14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hagiwara H., Sunada Y. (2004) Mechanism of taxane neurotoxicity. Breast Cancer 11, 82–85 [DOI] [PubMed] [Google Scholar]