

Background: The nature of endoplasmic reticulum (ER) stress in chronic pancreatitis (CP) is not known.
Results: ER stress is activated early, independently of trypsinogen activation, and remains sustained in CP.
Conclusion: Pathologic ER stress activation may be a novel pathogenic mechanism of CP.
Significance: ER stress is a key event independent of the traditional central event of pancreatitis-trypsinogen activation.
Keywords: Endoplasmic Reticulum (ER), ER Stress, NF-kappa B (NF-KB), Pancreas, Unfolded Protein Response (UPR), Chronic Pancreatitis, Trypsinogen Activation
Abstract
The pathogenesis of chronic pancreatitis (CP) is poorly understood. Endoplasmic reticulum (ER) stress has now been recognized as a pathogenic event in many chronic diseases. However, ER stress has not been studied in CP, although pancreatic acinar cells seem to be especially vulnerable to ER dysfunction because of their dependence on high ER volume and functionality. Here, we aim to investigate ER stress in CP, study its pathogenesis in relation to trypsinogen activation (widely regarded as the key event of pancreatitis), and explore its mechanism, time course, and downstream consequences during pancreatic injury. CP was induced in mice by repeated episodes of acute pancreatitis (AP) based on caerulein hyperstimulation. ER stress leads to activation of unfolded protein response components that were measured in CP and AP. We show sustained up-regulation of unfolded protein response components ATF4, CHOP, GRP78, and XBP1 in CP. Overexpression of GRP78 and ATF4 in human CP confirmed the experimental findings. We used novel trypsinogen-7 knock-out mice (T−/−), which lack intra-acinar trypsinogen activation, to clarify the relationship of ER stress to intra-acinar trypsinogen activation in pancreatic injury. Comparable activation of ER stress was seen in wild type and T−/− mice. Induction of ER stress occurred through pathologic calcium signaling very early in the course of pancreatic injury. Our results establish that ER stress is chronically activated in CP and is induced early in pancreatic injury through pathologic calcium signaling independent of trypsinogen activation. ER stress may be an important pathogenic mechanism in pancreatitis that needs to be explored in future studies.
Introduction
Chronic pancreatitis (CP)3 is a fibro-inflammatory disease of the pancreas with significant morbidity and mortality (1). Its pathogenesis remains poorly understood with the current theories heavily based on extrapolations from acute pancreatitis (2, 3). However, the relationship between acute and chronic pancreatitis has not been well established either (3, 4). Pathologic intra-acinar trypsinogen activation has been regarded as the key event of pancreatitis for more than a century. Recent data (5, 6) using trypsinogen-7 knock-out mouse lacking pathologic intra-acinar trypsinogen activation (T−/−) to study pancreatitis suggest the possible existence of other trypsinogen-independent pathogenic mechanisms. Currently, little is known about alternate pathogenic mechanisms in pancreatic injury.
In this study, we wish to explore the possibility of pathologic endoplasmic reticulum (ER) stress in chronic pancreatitis. ER is a multifunctional organelle involved in synthesis, folding, and quality control of proteins (7). Our understanding of ER homeostasis and its mechanisms has vastly increased in the last two decades (8–10). ER stress activates a network of cell signaling machinery called unfolded protein response (UPR) (summarized in Fig. 1 for ease of reference in subsequent discussions) aimed to restore homeostasis or lead to pathologic cellular events if homeostasis fails (8). Pathologic downstream responses of ER stress have now been implicated in the pathogenesis of several chronic diseases (11–19). Pancreatic acinar cells, which are specialized in synthesis, storage, and secretion of digestive enzymes, have the highest rate of protein synthesis among human tissues (20) and possess a characteristically rich volume of ER. Thus, because of the dependence on high ER volume and functionality, pancreatic acinar cells might be especially susceptible to perturbations in ER homeostasis. Indeed, ER stress has been previously described in pancreatic acinar cells during in vitro hyperstimulation with caerulein (21) and in vivo l-arginine-induced experimental acute pancreatitis (22). However, the nature of ER stress in chronic pancreatitis and its induction, mechanism, natural, course and role in pancreatic injury have never been investigated so far.
FIGURE 1.
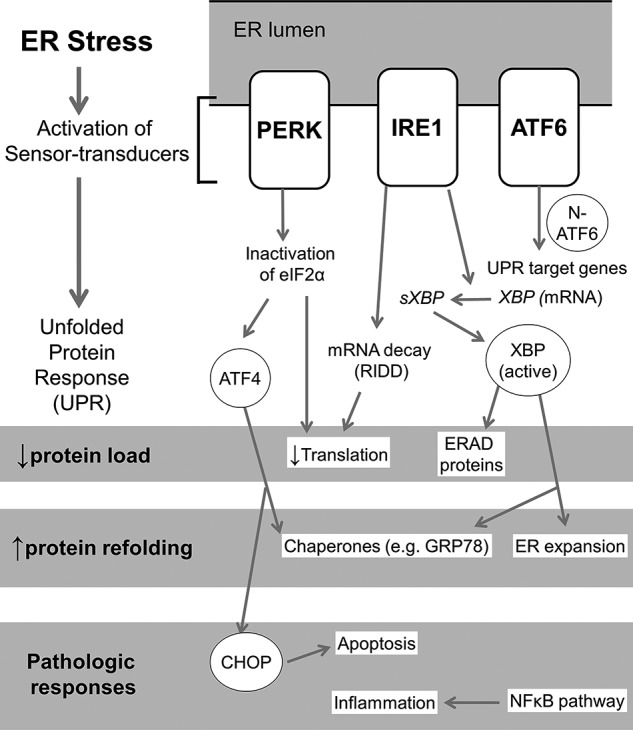
ER stress and UPR pathways. ER stress sensing triggers a) auto-phosphorylation of PERK; b) oligomerization, auto-phosphorylation, and activation of ribonuclease activity of IRE-1; and c) migration of ATF6 to Golgi resulting in its cleavage and liberation of the N-terminal cytosolic fragment, which translocates to the nucleus (8). Reduction in translation occurs due to inactivation of EIF2α after undergoing phosphorylation by pPERK, as well as due to degradation of any mRNA in the vicinity of activated IRE-1 by its nonspecific ribonuclease activity (termed regulated IRE1-dependent degradation (RIDD)) (8). IRE-1 has a specific ribonuclease activity resulting in targeted splicing of XBP-1 transcript (sXBP1), which is translated into active transcription factor XBP-1 (8, 29). XBP-1 leads to synthesis of a) chaperones such as GRP78; b) refolding proteins such as protein disulfide isomerase; c) endoplasmic reticulum-associated degradation (ERAD) products; and d) lipids for the expansion of ER membrane (8). Transcription of XBP-1 is induced by nuclear N-terminal fragment of ATF6 (29). Selective translation of ATF-4 occurs in low EIF2α abundance, which induces transcription of CHOP and GADD34. The initial UPR responses are aimed at restoring ER homeostasis. Pathogenic responses result when homeostasis fails due to unabated ER stress. Significant redundancies and overlaps occur in each of the three pathways; their relative roles may be specific to cell type and context. ER stress- and UPR-related downstream transcription factors are shown in circles.
Although ER stress and UPR activation are universal cellular responses to several kinds of stress, the nature of the stressors seem to be specific to particular pathologies. For example, the stressor is excessive demand of insulin production in type 2 diabetes (11) and expression of mutant protein in inherited diseases such as retinitis pigmentosa (23). The proponents of the trypsin-centered theory of pancreatitis argue that ER stress is a consequence of pathologic trypsinogen activation in pancreatitis. Alternately, ER stress being a trypsinogen-independent pathologic event in pancreatitis is an exciting possibility. With emerging data pointing to the existence of trypsin-independent pathogenic mechanisms in pancreatic injury (5, 6, 24–26), the clarification of the relationship of ER stress in pancreatic injury to trypsinogen activation is critical in advancing the understanding of pancreatitis.
In this study, we aim to: 1) elucidate the nature of ER stress and UPR activation in chronic pancreatitis; 2) clarify the relationship between ER stress and pathologic trypsinogen activation in pancreatic injury; and 3) evaluate the mechanism, time course, and downstream consequences of ER stress and UPR activation in pancreatitis.
EXPERIMENTAL PROCEDURES
Protocols for all experiments were approved by the Institutional Animal Care and Use Committee of the University of Minnesota. C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). All animals were housed in standard shoebox cages in a climate-controlled room with an ambient temperature of 23 ± 2 °C and 12:12-h light-dark cycle. Animals were fed standard laboratory chow, given water ad libitum, and randomly assigned to control or experimental groups. Animals were sacrificed using CO2 asphyxia.
Generation of Trypsinogen-7 Knock-out (T−/−) Mice
These mice were generated in C57BL/6 background by targeted deletion of trypsinogen-7 gene as described by us previously (5, 6).
CP
Animals were subjected to repeated episodes (twice a week, Mondays and Thursdays for 10 weeks) of acute pancreatitis induced by caerulein hyperstimulation (50 μg/kg injected intraperitoneally every hour for 6 h). Caerulein was solubilized in PBS at a final concentration of 10 μg/ml. Controls received PBS injections. Animals were sacrificed 8 days after the last cycle of injections. Entire pancreas was carefully dissected, and the tissue was then divided into three parts: 1) for RNA extraction, tissue was stored in RNAlater (Qiagen, Valencia, CA) at 4 °C; 2) for Western blot analysis, tissue was snap-frozen in liquid nitrogen and stored at −80 °C; and 3) for histological analysis, tissue was fixed in 10% buffered formaldehyde for 24 h and then transferred to 70% ethanol until processed and embedded in paraffin. Two independent experiments, separated in time, with 10 animals/group (total 20 animals/group) were conducted. The control group was composed of WT and T−/− mice. No differences were observed in the controls from WT and T−/− mice.
Human Pancreas Samples
Sections of pancreas received from the islet transplant program at the University of Minnesota transplant facility from healthy donors were used as controls. Sections of CP were obtained from surgical resection specimens. The protocol for acquisition and use of all human pancreatic samples was approved by University of Minnesota Institutional Review Board.
Acute Pancreatitis
Animals were injected with caerulein (50 μg/kg intraperitoneally) or saline. For the 30-min time point, animals were sacrificed 30 min after the first injection. For the 10-h time point, animals received injections every hour for a total of 10 injections and were sacrificed 1 h after the last injection. For resolving acute pancreatitis (AP) time point, animals were observed for 16 h after the last of eight injections.
In Vitro Experiments
Pancreatic acini were isolated from freshly harvested pancreas of C57BL/6 mice as described by us previously (5). Rat acinar cell line AR42J, maintained in our laboratory, was used for the cell death experiment. Experiments with caerulein, BAPTA-AM (Sigma), or tunicamycin (Sigma) (vehicle only for control groups) were conducted in oxygenated HEPES buffer at 37 °C. Dimethyl sulfoxide (DMSO) was used as vehicle for BAPTA-AM and tunicamycin. The manufacturer's recommended concentration, which was dependent on the ratio of various homologs (T7765 datasheet, Sigma), of tunicamycin (4 μg/ml) was used for experiments. Propidium iodide uptake was used for cell death detection (using DiOC18(3)-propidium iodide microplate assay viability kit from Invitrogen).
Trypsin and Amylase Activity
Trypsin activity in pancreatic homogenates and amylase activity were measured as described by us previously (5).
Histology and Immunohistochemistry
Hematoxylin and eosin (H&E) staining was used for evaluation of histological features. For immunohistochemistry, 4-μm-thick sections were cut from paraffin-embedded blocks. Antigen retrieval was carried out in a steamer for 30 min in high pH antigen retrieval buffer (Dako, Carpentaria, CA). After blocking in protein block (Dako) for 10 min, slides were incubated overnight in primary antibody diluted in antibody diluent (Dako). For negative controls for each antibody, slides were incubated with diluent only. Background peroxidase activity was quenched with 3% hydrogen peroxide for 10 min. Polymer-based secondary antibodies (Biocare) were used for detection with diaminobenzidine reagent (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin (Vector Laboratories). Anti-GRP78 antibody was from Cell Signaling (Danvers, MA), and anti-ATF4 antibody was from Abcam (Cambridge, MA). All imaging was done using a Nikon Microphot SA microscope fitted with a Nikon DXM 1200 F camera and captured using Nikon ACT-1 software (version 2.70).
Western Blotting
Tissue homogenates were prepared in radioimmunoprecipitation assay buffer containing protease inhibitor mixture. Protein content was quantified by the BCA method, and 30 μg of protein (10 μg for in vitro experiments) was separated by polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Blocking was done in 5% skimmed milk followed by incubation with primary antibody. HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were used and detected by chemiluminescence. Precision Plus protein standards (Bio-Rad) were used as marker. Omega 12iC imaging system (UltraLum, Claremont, CA) was used for imaging, and images were quantified using ImageJ (National Institutes of Health, Bethesda, MD) per the developer's protocol. Antibodies against ERK1/2, GRP78, CHOP, EIF2α, and pEIF2α were from Cell Signaling, antibody against IκBα was from Santa Cruz Biotechnology, and antibody against ATF4 was from Abcam.
RNA Isolation, Measurement, and Analysis
RNA was isolated from pancreatic tissue stored in RNAlater (Qiagen) using TRIzol (Invitrogen) according to the manufacturer's protocol. Following quantitation, 2 μg of RNA was used to make cDNA using Retroscript first strand synthesis kit for RT-PCR (Applied Biosystems, Grand Island, NY) according to the manufacturer's protocol. Real time PCR analysis for GRP78, CHOP, and XBP1 (Qiagen) was performed following cDNA synthesis in an ABI7300 instrument (Applied Biosystems). The data were normalized to 18 S rRNA expression levels and expressed as -fold change over controls.
Statistical Analysis
JMP 9.0 (SAS institute, Cary, NC) was used for all statistical analyses. Variables are reported as means ± S.E. and analyzed using Student t test (for normal variables) or Wilcoxon test (for nonparametric variables; specified in the text). Box-whisker plots with outliers are used for graphical depictions unless specified otherwise. Box-whisker plots were drawn using JMP 9.0. The central horizontal line in the box represents the median, the vertical edges of the box represent quantiles, and the whiskers denote range (calculated by JMP 9.0). For all analyses, α = 0.05 and two-tailed p values are reported.
RESULTS
Trypsinogen-7 Knock-out (T−/−) Mice
Details on generation and characterization of T−/− mice have been published separately (5, 6). Trypsinogen-7, which accounts for about 60% of the total trypsinogen, is deleted in T−/− mice (5). These mice demonstrate normal phenotype comparable with wild type mice, suggesting that unaltered expression of the other trypsinogen isotypes is sufficient for physiological processes (5).
Trypsinogen activation is known to peak at about 30 min after caerulein injection (6, 27, 28), which was confirmed in WT mice. Trypsinogen activation was not observed in T−/− mice (Fig. 2A). The time course of trypsinogen activation during caerulein-induced pancreatitis and its absence in T−/− as well as the absence of activation of other digestive proteases (elastase and chymotrypsinogen) in T−/− have been published separately (6).
FIGURE 2.
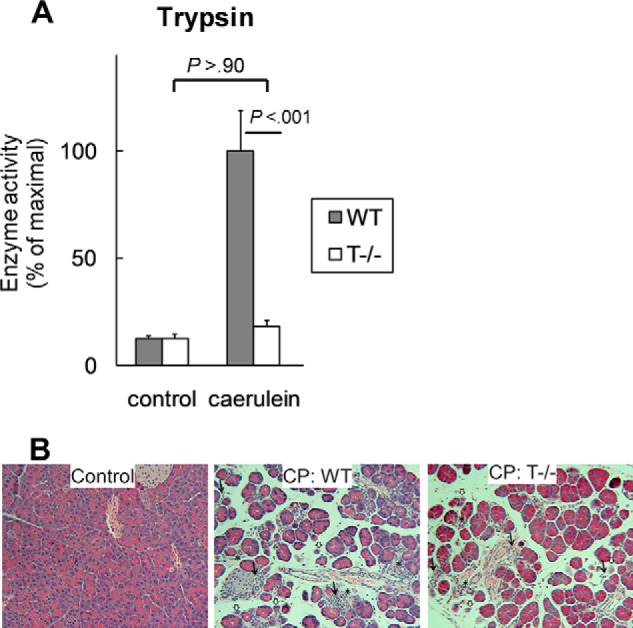
A, T−/− mice lack significant intra-acinar trypsinogen activation. Trypsin activity was measured in pancreatic homogenates 30 min after caerulein injection and reported as the percentage of maximal activity relative to the respective controls (saline injected). n = 10–14/group. B, CP was comparable in WT and T−/− mice. Representative hematoxylin and eosin-stained sections (100×) from control and CP groups demonstrating similar histomorphologic features of acinar atrophy (white arrows), ductular metaplasia (*), and chronic inflammatory infiltrate (black arrows) in WT and T−/− mice (n = 20/group in two independent experiments) are shown. A significant proportion of the empty spaces in the sections is composed of fibrotic areas that can be directly visualized with Sirius red stain as reported elsewhere (6). Error bars indicate means ± S.E.
CP in WT and T−/− Mice
Comparable features of CP were seen in WT and T−/− mice (6). Histological features are demonstrated in H&E-stained sections in Fig. 2B.
Chronic Activation of ER Stress in Pancreatic Acini during CP in WT and T−/− Mice
CP groups (WT and T−/− mice) demonstrated persistent ER stress as demonstrated by chronic activation of components of the UPR. Immunohistochemistry data localized ER stress and UPR activation to pancreatic acinar cells.
Overexpression of CHOP and ATF4 in CP
Protein analysis using Western blot revealed significantly higher levels of CHOP in WT and T−/− groups with CP as compared with controls (Fig. 3A). Further, overexpression of CHOP mRNA was seen in WT and T−/− mice with CP as compared with controls, suggesting that CHOP is up-regulated at the transcription level (Fig. 3B). Comparable expression of CHOP protein and mRNA was seen between WT and T−/− mice with CP. Overexpression of ATF4, a transcription factor expressed in the pPERK-pEIF2α pathway of the UPR, was seen in CP as compared with controls and was comparable in WT and T−/− mice with CP (Fig. 3C).
FIGURE 3.

A and B, overexpression of CHOP in CP. A, CHOP protein was up-regulated in WT and T−/− mice with CP. B, CHOP mRNA up-regulation in CP. Wilcoxon test used for analysis. n = 6/group. C, overexpression of ATF4 in CP. Representative ATF4 immunostained sections (100×) demonstrating higher ATF4 positivity in WT and T−/− mice with CP. n = 4–5/group. Error bars indicate means ± S.E. NS, not significant.
Overexpression of Chaperone GRP78 in CP
Levels of GRP78 protein were significantly higher in WT and T−/− groups with CP as compared with controls, analyzed by Western blot (Fig. 4A). Immunostaining for GRP78 revealed overexpression of GRP78 in CP groups as compared with controls (Fig. 4B). Similar expression was seen in WT and T−/− mice with CP by immunohistochemistry and confirmed by quantification of Western blots. High power fields showed maximal staining for GRP78 in the basal perinuclear region of the acinar cell where ER machinery is maximally concentrated. Further, GRP78 mRNA levels were up-regulated in CP as compared with controls (6 ± 1-fold, p = 0.04, n = 5/group).
FIGURE 4.

A and B, overexpression of GRP78 in CP. A, Western blot showing GRP78 up-regulation in WT and T−/− mice with CP. n = 5/group. C lane, control. B, immunostaining for GRP78 demonstrates up-regulation in CP, comparable in WT and T−/− mice. Representative sections are shown (n = 4–5/group). C, up-regulation of XBP1(total) mRNA in CP. Wilcoxon test used for analysis. n = 6/group. D and E, sustained ER stress in human CP. GRP78 (D) and ATF4 (E) immunostaining demonstrates up-regulation in human CP as compared with normal human pancreas confirming sustained ER stress in human CP. Representative sections (100×) are shown. Inset in D shows zoomed in (300×) views. Error bars indicate means ± S.E. NS, not significant.
Expression of XBP1 in CP
Overexpression of mRNA level of XBP1 (full XBP1 mRNA prior to splicing) was demonstrated by RT-PCR analysis in CP groups as compared with controls (Fig. 4C). Comparable levels were seen in WT and T−/− mice with CP. XBP1 is primarily up-regulated by cleaved ATF6 (50-kDa N-terminal fragment), the transcription factor in the ATF6 pathway of UPR (29) (Fig. 1). Thus, overexpression of XBP1 is suggestive of activation of the ATF6 pathway. Translation into active XBP1 occurs after XBP1 mRNA is spliced by endonuclease activity of IRE-1 resulting in removal of the 26-kb intron (8, 30). The transcription of GRP78 is regulated primarily by XBP-1 transcription factor. Thus, up-regulation of GRP78 in CP suggests activation of IRE-1 pathway.
Sustained ER Stress Is Seen in Human CP
Representative sections of normal human pancreas and sections of human CP stained with GRP78 and ATF4 (n = 2/group) are shown in Fig. 4, D and E, respectively. Overexpression of GRP78 and ATF4 in CP sections established that ER stress is sustained in pancreatic acini in human chronic pancreatitis. The subcellular distribution of GRP78 in human CP was similar to mouse CP, with maximal concentration in the basal perinuclear area of the acinar cell (Fig. 4D, inset).
ER Stress Occurs in AP Independent of Trypsinogen Activation
The components of UPR were activated in AP as shown in Fig. 5. Phosphorylation of eIF2α, which is a downstream component of the pPERK-peIF2α pathway of UPR, was seen in WT and T−/− mice to a level comparable with controls. (Fig. 5A). Similarly, up-regulation of ATF4, another component of the pPERK-peIF2α pathway, was seen in WT and T−/− mice to a level comparable with controls (2.5 ± 0.7- and 2.2 ± 0.4-fold control, WT and T−/−, respectively; p values 0.01 for WT versus control, 0.04 for T−/− versus control and not significant for WT versus T−/− groups; n = 6–7/group). Further, up-regulation of GRP78 (Fig. 5B) and CHOP (see below) were seen in AP as compared with controls, and the levels were comparable in WT and T−/− mice. These data confirm induction of ER stress and activation of UPR in AP and demonstrate convincingly that ER stress in AP is independent of intra-acinar trypsinogen activation.
FIGURE 5.

A and B, ER stress and UPR activation occurs in AP independent of trypsinogen activation. Proteins were analyzed by Western blot in pancreatic homogenates from mice with AP after hourly caerulein injections 10 times as compared with saline-injected controls. A, phosphorylation of eIF2α during AP in WT and T−/− mice. n = 4/group. B, up-regulation of GRP78 during AP in WT and T−/− mice. n = 7–10/group. C and D, ER stress induction occurred very early in the course of pancreatic injury independent of trypsinogen activation. Proteins analyzed by Western blot in pancreatic homogenates from mice 30 min after caerulein injection as compared with saline-injected controls. Phosphorylation of eIF2α is shown in C. C lane, control. Up-regulation of GRP78 is shown in D. n = 4–5/group. E, time course of CHOP activation during acute pancreatic injury. Early time point (30 min after caerulein injection), AP time point (after hourly caerulein injection 10 times), and time point of resolution of AP (mice sacrificed after 16 h of observation following hourly caerulein injection eight times) have been shown. CHOP activation was seen during the AP time point but was absent during the resolution phase as well as at the early stage of pancreatic injury. p values for WT versus control, T−/− versus control, and WT versus T−/−, respectively: < 0.0001, < 0.0001, and not significant at AP time point (n = 9–10/group); each not significant at 30 min time point (n = 5/group) and resolving AP time point (n = 5/group). Error bars indicate means ± S.E. NS, not significant.
ER Stress Is an Early Cellular Event
As early as 30 min after injection of caerulein in mice, we observed induction of ER stress as demonstrated by activation of UPR components. Phosphorylation of eIF2α and overexpression of GRP78 were noted at this time point in WT as well as T−/− mice to a similar extent (Fig. 5, C and D).
Timeline of ER Stress Induction and Activation of UPR Components
Overexpression of GRP78 and ATF4, which occurred at 30 min, were persistent at the 10-h time point of AP, but their levels were similar to controls during the resolving phase of AP (WT and T−/− groups, respectively, at the 24-h time point: GRP78, 1 ± 0.2- and 1 ± 0.4-fold control (n = 5 each group); ATF4, 1 ± 0.1- and 1 ± 0.2-fold control (n = 4 each group); p value not significant for each pairwise comparisons). Similarly, CHOP expression was comparable with controls at the 24-h time point (Fig. 5E). These data indicate that ER stress activated during AP attenuates with the resolution of AP. In contrast, mice in the CP groups, which were subjected to repeated episodes of AP, demonstrated sustained ER stress and UPR activation. The CP mice were sacrificed 8 days after the last episode of AP, which means that the observation of sustained ER stress and UPR component activation in CP was free from any confounding residual effects from the last episode of AP.
The expression of CHOP was not seen at the early time point unlike other components of UPR (Fig. 5E). As shown in Fig. 1, CHOP expression and GADD34 expression are believed to be late UPR components activated when initial UPR fails to clear ER stress. GADD34 dephosphorylates pEIF2α, serving as a feedback inhibition loop, whereas CHOP is a trigger for cell death (8). As a transcription factor, CHOP results in expression of inhibitors of apoptosis antagonists such as bcl-2 thereby promoting apoptosis (8).
Pathologic Calcium Signaling Mediates ER Stress in Pancreatic Injury
ER stress induction was confirmed with increased phosphorylation of EIF2α in mice acini in vitro within 30 min of caerulein hyperstimulation (100 nm) in HEPES buffer containing calcium chloride (1 mm) (Fig. 6A). However, no differences in phosphorylated EIF2α (pEIF2α) levels were seen after treatment with caerulein in parallel experiments in HEPES buffer lacking Ca2+ (Fig. 6A), indicating that extracellular Ca2+ is required for induction of ER stress in pancreatic acinar cells in response to caerulein hyperstimulation. Further, when cytosolic Ca2+ was chelated by pretreatment of mice acini with BAPTA-AM (10 μm for 30 min, as described by us previously in Ref. 28), pEIF2α levels remained unchanged despite caerulein treatment in HEPES containing Ca2+ (1 mm). This confirms that ER stress in pancreatic injury is dependent on pathologic Ca2+ signaling (which has been well characterized to involve sustained global rise in cytoplasmic Ca2+ levels primarily from extracellular sources triggered by signaling from ER stores (see reviews in Refs. 31–35 for details on pathologic calcium signaling)). The internal validity of these parallel experiments with caerulein, the absence of extracellular Ca2+, and BAPTA-AM pretreatment were confirmed by simultaneous measurement of amylase secretion (Fig. 6B). Our results were consistent with well established pancreatic acinar secretion physiology (31–35) known to be dependent on transient cytoplasmic Ca2+ changes involving primarily intracellular stores (thus inhibited by BAPTA-AM but not by lack of extracellular Ca2+). Further, up-regulation of CHOP (Fig. 6C) and GRP78 (Fig. 6D) mRNA was seen in mice acini treated with caerulein (100 nm, 2 h of incubation) in the presence of extracellular calcium but not in the absence of extracellular calcium, confirming that ER stress induction by caerulein hyperstimulation is dependent on pathologic calcium signaling.
FIGURE 6.

A–D, ER stress is mediated by pathologic Ca2+ signaling during early pancreatic injury. Mice acini were incubated with 100 nm caerulein (CR) or control (C) for 30 min (A and B) or 2 h (C and D) in HEPES containing 1 mm Ca2+ or without Ca2+ (no extracellular Ca2+ group). A and B, in the BAPTA-AM group, acini were preincubated with 10 μm BAPTA-AM in HEPES buffer lacking Ca2+ for 30 min prior to treatment with caerulein in HEPES containing 1 mm Ca2+. All experiments were conducted in parallel, and data shown represent n = 6–8/group pooled from two independent experiments. Amylase secretion was measured in supernatants after completion of incubation in the same experiments described above and was normalized to total protein. Amylase secretion data are intended to verify the internal validity of the experimental conditions. C and D, CHOP and GRP78 mRNA up-regulation (by quantitative PCR) indicating that ER stress induction by caerulein hyperstimulation is abrogated in the absence of extracellular calcium. n = 3/group. E–H, ER stress leads to NFκB activation in pancreatic acinar cells. E, rat AR42J cells were incubated with tunicamycin (TM) or vehicle (control, C) for 3 h. n = 3–5/group. Incubation with caerulein (CR, 100 nm) was used as a positive control. IκBα immunoblotting in whole cell lysates shows degradation of IκBα in TM group and in positive control, indicating NFκB activation. F, pancreatic NFκB activation induced by TM treatment in mice as shown by nuclear translocation of p65. Cytosolic and nuclear fractions were prepared from pancreas of TM-treated versus saline (control). Up-regulation of GRP78 in cytosolic fraction validates the effect of TM. Quantification of Western blots is shown in G and H. n = 3–4/group. I, ER stress induction leads to cell death in pancreatic acinar cells. Mice acini were incubated with 100 nm caerulein, tunicamycin, or vehicle (control) for 3 h. Cell death was measured by propidium iodide uptake viability kit. Proportion of dead cells of the total cells was measured and was expressed as -fold controls. n = 12–14/group pooled from two independent experiments. Error bars indicate means ± S.E. NS, not significant.
Pathologic Responses in Pancreatic Acinar Cells in Response to ER Stress Inducer Tunicamycin
We used tunicamycin, a widely used chemical inducer of ER stress, to study the consequences of pathologic ER stress in pancreatic acini. Rat AR42J cells showed depletion of IκBα levels after incubation with tunicamycin for 3 h (Fig. 6E), demonstrating NFκB activation as a result of pathologic ER stress in pancreatic acini. NFκB activation was also seen in the pancreas of mice injected with tunicamycin (intraperitoneal injection of 1 mg/kg, pancreas harvested after 24 h of injection) (Fig. 6, F–H). Up-regulation of GRP78 in the cytosolic fraction validates the in vivo effect of tunicamycin treatment in mice (Fig. 6, F and H), whereas nuclear translocation of p65 in tunicamycin-treated mice demonstrates activation of NFκB in the pancreas (Fig. 6, f and g). Further, mice acini treated with tunicamycin showed markedly increased propidium iodide uptake, demonstrating cell death (necrotic) resulting from pathologic ER stress in pancreatic acini (Fig. 6D). Although apoptosis and necrosis can both result from pathologic cellular processes in acinar cells (36), mice acini are known to demonstrate preponderance of necrosis during pancreatic injury (37).
DISCUSSION
We show that ER stress is chronically activated in pancreatic acinar cells in CP in mice as well as in humans. Our results establish that induction of ER stress and UPR activation in pancreatitis are independent of intra-acinar trypsinogen activation, which has been widely believed to be the primary pathologic event in pancreatic injury. We show that induction of ER stress is an early event in pancreatic injury occurring as early as the well studied key cellular events: trypsinogen activation and NFκB activation (6, 27, 38). We further establish that pathologic calcium signaling in pancreatic acinar cells mediates ER stress. Our results indicate that pathologic ER stress results in NFκB activation as well as cell death in pancreatic acini. Together, these data establish the presence of ER stress as a trypsin-independent pathologic cellular event in pancreatic injury.
Pathologic calcium signaling is a well studied and well characterized early event in pancreatic injury occurring in response to caerulein hyperstimulation (31–35). It has previously been shown to mediate pathologic trypsinogen activation and play an important role in NFκB activation during early pancreatic injury (28, 38, 39). Here, we show that pathologic calcium signaling mediates ER stress during early pancreatic injury in the caerulein model. Accumulation of unfolded and misfolded proteins and alteration of ER Ca2+ are well recognized inducers of ER stress in various cells (8, 40). Although Ca2+ responses are important for ER stress in caerulein-induced pancreatic injury, alteration of secretion and accumulation of misfolded proteins may be another mechanism (but not necessarily independent of pathologic Ca2+ signaling) of ER stress as has been demonstrated recently in in vitro studies of several CP-associated mutations in the trypsin system (41–45). In addition to hereditary mutations, alcohol has been shown to cause ER stress in the pancreas (46). The other etiologic associations in CP may also induce ER stress. Persistence of these pathologic stimuli (alcohol, expression of mutated proteins, or the other etiologic associations of CP) appears to be the likely mechanism of chronic activation of ER stress in CP.
ER membrane-bound sensor transducers PERK, IRE-1, and ATF6 (shown in Fig. 1) couple ER stress to UPR activation (8). The downstream responses (Fig. 1) triggered by these sensor transducers are aimed to restore homeostasis through: 1) increasing refolding capacity of ER by increasing ER volume, foldases, and chaperones, and increasing degradation capacity through endoplasmic reticulum-associated degradation and 2) decreasing protein load by reduction in translation (8). Limited UPR demonstrated by splicing of XBP1 but not activation of pPERK-pEIF2α pathway during pancreatic secretion (21) and failure of pancreatic development in mice with complete deletion of XBP1 (46–48) suggest that pancreatic acinar cells rely on physiologic UPR. However, if homeostasis fails, UPR leads to pathologic cellular events (8). ER stress and pathologic UPR activation led to pancreatitis in mice with partial deletion of XBP1 (46). These mice had defective initial protective UPR activation resulting in overwhelming ER stress. In this model, alcohol feeding caused ER stress in pancreatic acini, which was sufficiently cleared by intact initial UPR activation in wild type mice (46). The fine regulation of physiologic and pathologic UPR responses remains to be fully understood (8). This interesting model (46) nevertheless demonstrates that ER stress and pathologic UPR are sufficient to induce pancreatitis. Our results showing activation of several components of UPR in CP, including the late responses such as CHOP, which broadly encompasses all three sensor transduction pathways, suggest a pathologic nature of these responses as opposed to the limited UPR for homeostasis.
Our data suggest that ER stress induction in pancreatic acini results in NFκB activation as well as cell death. Persistence of ER stress likely results in cumulative effects of inflammation and cell death resulting in acinar atrophy and chronic inflammation of CP. Indeed, chronic activation of NFκB pathway in pancreatic acinar cells in experimental and human CP was reported in our previous study (6). Chronic pancreatic acinar specific overexpression of NFκB resulted in chronic pancreatitis in mice (49). Together, these results suggest that sustained ER stress in acinar cells is responsible for chronic NFκB activation in CP, which results in the pathogenesis of CP. Further studies are needed to explore this attractive possibility. In fact, ER stress is known to induce inflammation through NFκB-dependent and -independent pathways in many cell types (17, 50, 51), which are important in several chronic diseases including metabolic disorders and Alzheimer disease, as well as chronic inflammatory diseases (8).
The establishment of ER stress as a trypsin-independent early cellular event opens new avenues in our understanding of the pathogenesis of pancreatitis. The trypsin-centered theory has formed the backbone of intense research in the field of pancreatitis for the last three decades (34, 52, 53). Nonetheless, the wealth of ensuing data has so far only confirmed the existence of premature trypsinogen activation and shed light on its mechanisms (24, 34, 52, 53). Although the notion of pancreatic injury resulting from premature activation of digestive enzymes seems a natural possibility, the causality of pancreatic injury in acute and chronic pancreatitis from premature digestive enzyme activation has remained a presumption lacking direct proof (24). In fact, the interpretation of emerging data regarding cellular processes such as autophagy, lysosomal dysfunction, mitochondrial failure, oxidative stress, and bile duct dysfunction in the field of pancreatitis continues to be heavily influenced by the trypsin-centered theory (24, 34, 35, 52, 53). The relationship, time course, and roles of these cellular events have not been fully clarified at present (24). However, it is likely that these cellular events including ER stress, in addition to directly causing pancreatic damage, may be important in driving sustained NFκB activation (24, 35), which results in pancreatic injury and intense systemic inflammation of AP (35, 49, 54). Thus, trypsinogen activation, long regarded as the holy grail of pancreatitis, seems to be one among a series of pathogenic cellular events triggered during early pancreatitis. Further, at least some of these cellular events may be a direct consequence of ER stress in pancreatic injury. Although NFκB activation (17, 50, 51) and apoptosis (8) are well characterized pathologic responses resulting from unresolved ER dysfunction, the spectrum of these pathologic responses is not fully known. Growing evidence from several chronic diseases demonstrates autophagy (17, 55), oxidative stress (56, 57), and altered differentiation (58) as a result of ER dysfunction. Oxidative stress and induction of autophagy have been demonstrated in relation to ER stress in pancreatic acinar cells in one previous study (46).
Although complex genetic interactions are now increasingly being recognized in CP with genetic basis (59, 60), traditionally, increased trypsinogen activation has been postulated to be the pathogenic mechanism of CP-associated mutations in the trypsin system (PRSS1, activation peptide of PRSS1, CTRC (chymotrypsinogen C), PRSS2) (61–63) as well as in other genes thought to affect trypsin activity (SPINK1 (serine protease inhibitor Kazal type 1), CFTR (cystic fibrosis transmembrane conductance regulator), cathepsin B, and CASR (calcium-sensing receptor)) (62, 64–68). Increased trypsin activity has been seen in biochemical studies of some mutations (63, 69) although not directly confirmed in studies of in vitro (41) and in vivo (70) expression. Conversely, constitutive expression of active trypsin in the pancreatic acinar cells in mice did not cause CP (25). Thus, it is conceivable that alternate, as yet unrecognized, consequences of trypsinogen mutations could be responsible for pancreatic injury. Our data suggest that sustained ER stress resulting from misfolding of mutated trypsinogens may be one such mechanism. Such a possibility was recognized in the carboxypeptidase A1 mutations recently recognized in hereditary chronic pancreatitis (45) and is further supported by CP-associated mutations confirming ER stress induction by expression of some mutations (of PRSS1 (41, 44), activation peptide of PRSS1 (42), and CTRC (43)) in HEK or rat acinar cell lines.
In conclusion, we have established that ER stress and unfolded protein response are chronically activated in experimental and human chronic pancreatitis. ER stress is an early event in pancreatic injury mediated by pathologic calcium signaling, is independent of trypsinogen activation, and results in NFκB activation in pancreatic acini. ER stress may be an important pathogenic mechanism in pancreatitis that needs to be explored in future studies.
This work was supported, in whole or in part, by National Institutes of Health Grants DK093047, DK058694, and DK092145 (to A. K. S.) and intramural support from the Department of Surgery, University of Minnesota.
- CP
- chronic pancreatitis
- AP
- acute pancreatitis
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- PRSS1
- protease serine 1
- NFκB
- nuclear factor κB
- ATF
- activating transcription factor
- IRE
- inositol-requiring enzyme
- PERK
- double-stranded PKR-like ER kinase
- PKR
- RNA-activated protein kinase
- GRP
- glucose-related protein
- CHOP
- transcription factor C/EBP homologous protein
- C/EBP
- CCAAT-enhancer-binding protein
- XBP
- X-box-binding protein
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (acetoxymethyl ester)
- p
- phospho
- TM
- tunicamycin.
REFERENCES
- 1. Braganza J. M., Lee S. H., McCloy R. F., McMahon M. J. (2011) Chronic pancreatitis. Lancet 377, 1184–1197 [DOI] [PubMed] [Google Scholar]
- 2. Etemad B., Whitcomb D. C. (2001) Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology 120, 682–707 [DOI] [PubMed] [Google Scholar]
- 3. Guda N. M., Romagnuolo J., Freeman M. L. (2011) Recurrent and relapsing pancreatitis. Curr. Gastroenterol. Rep. 13, 140–149 [DOI] [PubMed] [Google Scholar]
- 4. Yadav D., O'Connell M., Papachristou G. I. (2012) Natural history following the first attack of acute pancreatitis. Am. J. Gastroenterol. 107, 1096–1103 [DOI] [PubMed] [Google Scholar]
- 5. Dawra R., Sah R. P., Dudeja V., Rishi L., Talukdar R., Garg P., Saluja A. K. (2011) Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141, 2210–2217 e2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sah R. P., Dudeja V., Dawra R. K., Saluja A. K. (2013) Caerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice. Gastroenterology 144, 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berridge M. J. (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249 [DOI] [PubMed] [Google Scholar]
- 8. Walter P., Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 9. Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 10. Rutkowski D. T., Hegde R. S. (2010) Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 189, 783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fonseca S. G., Gromada J., Urano F. (2011) Endoplasmic reticulum stress and pancreatic beta cell death. Trends Endocrinol. Metab. 22, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li B., Gao B., Ye L., Han X., Wang W., Kong L., Fang X., Zeng Y., Zheng H., Li S., Wu Z. (2007) Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 124, 44–49 [DOI] [PubMed] [Google Scholar]
- 13. Hasty A. H., Harrison D. G. (2012) Endoplasmic reticulum stress and hypertension: a new paradigm? J. Clin. Invest. 122, 3859–3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dickhout J. G., Carlisle R. E., Austin R. C. (2011) Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ. Res. 108, 629–642 [DOI] [PubMed] [Google Scholar]
- 15. Carrasco D. R., Sukhdeo K., Protopopova M., Sinha R., Enos M., Carrasco D. E., Zheng M., Mani M., Henderson J., Pinkus G. S., Munshi N., Horner J., Ivanova E. V., Protopopov A., Anderson K. C., Tonon G., DePinho R. A. (2007) The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 11, 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garg A. D., Kaczmarek A., Krysko O., Vandenabeele P., Krysko D. V., Agostinis P. (2012) ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol. Med. 18, 589–598 [DOI] [PubMed] [Google Scholar]
- 17. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown M. K., Naidoo N. (2012) The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 3, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salminen A., Kauppinen A., Suuronen T., Kaarniranta K., Ojala J. (2009) ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J. Neuroinflammation. 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Case R. M. (1978) Synthesis, intracellular transport and discharge of exportable proteins in the pancreatic acinar cell and other cells. Biol. Rev. Camb. Philos. Soc. 53, 211–354 [DOI] [PubMed] [Google Scholar]
- 21. Kubisch C. H., Logsdon C. D. (2007) Secretagogues differentially activate endoplasmic reticulum stress responses in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1804–G1812 [DOI] [PubMed] [Google Scholar]
- 22. Kubisch C. H., Sans M. D., Arumugam T., Ernst S. A., Williams J. A., Logsdon C. D. (2006) Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G238–G245 [DOI] [PubMed] [Google Scholar]
- 23. Lin J. H., Lavail M. M. (2010) Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 664, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sah R. P., Dawra R. K., Saluja A. K. (2013) New insights into the pathogenesis of pancreatitis. Curr. Opin. Gastroenterol. 29, 523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaiser S., Daniluk J., Liu Y., Tsou L., Chu J., Lee W., Longnecker D. S., Logsdon C. D., Ji B. (2011) Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 60, 1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halangk W., Lerch M. M., Brandt-Nedelev B., Roth W., Ruthenbuerger M., Reinheckel T., Domschke W., Lippert H., Peters C., Deussing J. (2000) Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Invest. 106, 773–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hofbauer B., Saluja A. K., Lerch M. M., Bhagat L., Bhatia M., Lee H. S., Frossard J. L., Adler G., Steer M. L. (1998) Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am. J. Physiol. 275, G352–G362 [DOI] [PubMed] [Google Scholar]
- 28. Saluja A. K., Bhagat L., Lee H. S., Bhatia M., Frossard J. L., Steer M. L. (1999) Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am. J. Physiol. 276, G835–G842 [DOI] [PubMed] [Google Scholar]
- 29. Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 30. Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 [DOI] [PubMed] [Google Scholar]
- 31. Petersen O. H. (2009) Ca2+ signaling in pancreatic acinar cells: physiology and pathophysiology. Braz J. Med. Biol. Res. 42, 9–16 [DOI] [PubMed] [Google Scholar]
- 32. Petersen O. H., Tepikin A. V. (2008) Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 70, 273–299 [DOI] [PubMed] [Google Scholar]
- 33. Petersen O. H. (2005) Ca2+ signalling and Ca2+-activated ion channels in exocrine acinar cells. Cell Calcium 38, 171–200 [DOI] [PubMed] [Google Scholar]
- 34. Sah R. P., Saluja A. (2011) Molecular mechanisms of pancreatic injury. Curr. Opin. Gastroenterol. 27, 444–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sah R. P., Garg P., Saluja A. K. (2012) Pathogenic mechanisms of acute pancreatitis. Curr. Opin. Gastroenterol. 28, 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Criddle D. N., Gerasimenko J. V., Baumgartner H. K., Jaffar M., Voronina S., Sutton R., Petersen O. H., Gerasimenko O. V. (2007) Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ. 14, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 37. Kaiser A. M., Saluja A. K., Sengupta A., Saluja M., Steer M. L. (1995) Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 269, C1295–C1304 [DOI] [PubMed] [Google Scholar]
- 38. Hietaranta A. J., Saluja A. K., Bhagat L., Singh V. P., Song A. M., Steer M. L. (2001) Relationship between NF-κB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem. Biophys. Res. Commun. 280, 388–395 [DOI] [PubMed] [Google Scholar]
- 39. Tando Y., Algül H., Wagner M., Weidenbach H., Adler G., Schmid R. M. (1999) Caerulein-induced NF-κB/Rel activation requires both Ca2+ and protein kinase C as messengers. Am. J. Physiol. 277, G678–G686 [DOI] [PubMed] [Google Scholar]
- 40. Pandol S. J., Gorelick F. S., Gerloff A., Lugea A. (2010) Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis. 28, 776–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kereszturi E., Szmola R., Kukor Z., Simon P., Weiss F. U., Lerch M. M., Sahin-Tóth M. (2009) Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum. Mutat. 30, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kereszturi E., Sahin-Tóth M. (2009) Intracellular autoactivation of human cationic trypsinogen mutants causes reduced trypsinogen secretion and acinar cell death. J. Biol. Chem. 284, 33392–33399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szmola R., Sahin-Tóth M. (2010) Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut. 59, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schnúr A., Beer S., Witt H., Hegyi P., Sahin-Tóth M. (2013) Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut. 63, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Witt H., Beer S., Rosendahl J., Chen J. M., Chandak G. R., Masamune A., Bence M., Szmola R., Oracz G., Macek M., Jr., Bhatia E., Steigenberger S., Lasher D., Bühler F., Delaporte C., Tebbing J., Ludwig M., Pilsak C., Saum K., Bugert P., Masson E., Paliwal S., Bhaskar S., Sobczynska-Tomaszewska A., Bak D., Balascak I., Choudhuri G., Nageshwar Reddy D., Rao G. V., Thomas V., Kume K., Nakano E., Kakuta Y., Shimosegawa T., Durko L., Szabó A., Schnúr A., Hegyi P., Rakonczay Z., Jr., Pfützer R., Schneider A., Groneberg D. A., Braun M., Schmidt H., Witt U., Friess H., Algül H., Landt O., Schuelke M., Krüger R., Wiedenmann B., Schmidt F., Zimmer K. P., Kovacs P., Stumvoll M., Blüher M., Müller T., Janecke A., Teich N., Grützmann R., Schulz H. U., Mössner J., Keim V., Löhr M., Férec C., Sahin-Tóth M. (2013) Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 45, 1216–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lugea A., Tischler D., Nguyen J., Gong J., Gukovsky I., French S. W., Gorelick F. S., Pandol S. J. (2011) Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140, 987–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee A. H., Chu G. C., Iwakoshi N. N., Glimcher L. H. (2005) XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 24, 4368–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harding H. P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D. D., Ron D. (2001) Diabetes mellitus and exocrine pancreatic dysfunction in Perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 49. Huang H., Liu Y., Daniluk J., Gaiser S., Chu J., Wang H., Li Z. S., Logsdon C. D., Ji B. (2013) Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 144, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kitamura M. (2011) Control of NF-κB and inflammation by the unfolded protein response. Int. Rev. Immunol. 30, 4–15 [DOI] [PubMed] [Google Scholar]
- 51. Kitamura M. (2009) Biphasic, bidirectional regulation of NF-κB by endoplasmic reticulum stress. Antioxid. Redox. Signal 11, 2353–2364 [DOI] [PubMed] [Google Scholar]
- 52. Gorelick F. S., Thrower E. (2009) The acinar cell and early pancreatitis responses. Clin. Gastroenterol. Hepatol. 7, S10–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saluja A. K., Lerch M. M., Phillips P. A., Dudeja V. (2007) Why does pancreatic overstimulation cause pancreatitis? Annu. Rev. Physiol. 69, 249–269 [DOI] [PubMed] [Google Scholar]
- 54. Rakonczay Z., Jr., Hegyi P., Takács T., McCarroll J., Saluja A. K. (2008) The role of NF-κB activation in the pathogenesis of acute pancreatitis. Gut. 57, 259–267 [DOI] [PubMed] [Google Scholar]
- 55. Jia W., Pua H. H., Li Q. J., He Y. W. (2011) Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J. Immunol. 186, 1564–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bhandary B., Marahatta A., Kim H. R., Chae H. J. (2012) An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 14, 434–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cullinan S. B., Diehl J. A. (2006) Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 38, 317–332 [DOI] [PubMed] [Google Scholar]
- 58. Tsang K. Y., Chan D., Cheslett D., Chan W. C., So C. L., Melhado I. G., Chan T. W., Kwan K. M., Hunziker E. B., Yamada Y., Bateman J. F., Cheung K. M., Cheah K. S. (2007) Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 5, e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beer S., Sahin-Tóth M. (2014) Exonic variants affecting pre-mRNA splicing add to genetic burden in chronic pancreatitis. Gut 63, 860–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rosendahl J., Landt O., Bernadova J., Kovacs P., Teich N., Bödeker H., Keim V., Ruffert C., Mössner J., Kage A., Stumvoll M., Groneberg D., Krüger R., Luck W., Treiber M., Becker M., Witt H. (2013) CFTR, SPINK1, CTRC, and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut. 62, 582–592 [DOI] [PubMed] [Google Scholar]
- 61. Chen J. M., Ferec C. (2009) Chronic pancreatitis: genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 10, 63–87 [DOI] [PubMed] [Google Scholar]
- 62. Whitcomb D. C. (2010) Genetic aspects of pancreatitis. Annu. Rev. Med. 61, 413–424 [DOI] [PubMed] [Google Scholar]
- 63. Sahin-Tóth M. (2006) Biochemical models of hereditary pancreatitis. Endocrinol. Metab. Clin. North Am. 35, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. LaRusch J., Whitcomb D. C. (2011) Genetics of pancreatitis. Curr. Opin. Gastroenterol. 27, 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mahurkar S., Idris M. M., Reddy D. N., Bhaskar S., Rao G. V., Thomas V., Singh L., Chandak G. R. (2006) Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut. 55, 1270–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Paliwal S., Bhaskar S., Mani K. R., Reddy D. N., Rao G. V., Singh S. P., Thomas V., Chandak G. R. (2013) Comprehensive screening of chymotrypsin C (CTRC) gene in tropical calcific pancreatitis identifies novel variants. Gut 62, 1602–1606 [DOI] [PubMed] [Google Scholar]
- 67. Ohmuraya M., Yamamura K. (2011) Roles of serine protease inhibitor Kazal type 1 (SPINK1) in pancreatic diseases. Exp. Anim. 60, 433–444 [DOI] [PubMed] [Google Scholar]
- 68. Ohmuraya M., Sugano A., Hirota M., Takaoka Y., Yamamura K. (2012) Role of intrapancreatic SPINK1/Spink3 expression in the development of pancreatitis. Front Physiol. 3, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Teich N., Rosendahl J., Tóth M., Mössner J., Sahin-Tóth M. (2006) Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum. Mutat. 27, 721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Archer H., Jura N., Keller J., Jacobson M., Bar-Sagi D. (2006) A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 131, 1844–1855 [DOI] [PubMed] [Google Scholar]